


Abstract
Ca2+ channels play a key role in the basic working of the heart. There is one particular type of Ca2+ channel in cardiac cells (L-type) whose gating is affected in different ways by β-adrenoceptors and 1,4-dihydropyridines. In this study, we used ex vivo studies and PET to evaluate and compare the myocardial kinetics of the enantiomers labeled with 11C (the more active: S12968, absolute configuration S; the less active: S12967, absolute configuration R) of the L-type Ca2+ channel antagonist S11568 (3-ethyl 5-methyl (±)-2-[(2-(2-aminoethoxy)ethoxy) methyl]-4-(2,3-dichlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate). Methods: [11C]S12968 was injected into the tail vein of rats (0.22 kBq–5.92 MBq) to assess the relationship between injected dose and myocardial uptake. A series of 5 rats was pretreated with 4 μmol unlabeled S12968 5 min before injection of 2.2 kBq [11C]S12968. In another series of 5 rats, unlabeled S12698 (4 μmol) was injected 5 min after injection of 2.2 kBq [11C]S12968. The animals were killed 15 min later, and the myocardial radioactivity was assessed in a γ well counter. Beagle dogs received injections of 5–15 nmol [11C]S12968 or [11C]S12967 and were imaged with PET. Presaturation and displacement experiments using 2 μmol/kg unlabeled S12968 or 6 mol/kg S12967 were performed. Results: In rats, a statistically significant relationship between myocardial uptake and injected dose of S12968 was observed. Pretreatment or displacement with unlabeled S12968 reduced myocardial radioactivity by 75% and 70%, respectively. In dogs, after injection of 5 nmol of each enantiomer, myocardial radioactivity plateaued within 3 min and the clearance from blood was rapid. Injection of 13–15 nmol [11C]S12968 led to a higher myocardial uptake and a more rapid washout, which were related to an increased coronary blood flow as shown by the linear relationship between k1—an estimate of coronary blood flow—and the mass of S12968 injected. Presaturation and displacement experiments showed that 70%–80% of S12968 binding was specific. This specificity was not observed with S12967. Plasma metabolite analysis showed that 70% of the compound was unchanged 20 min after injection. Conclusion: These results show the feasibility of imaging myocardial L-type Ca2+ channels in vivo using [11C]S12968.
Voltage-dependent Ca2+ channels are widely distributed in excitable membranes and are involved in the regulation of many cellular functions. Neurotransmitters and drugs can modulate these channels. There is one particular type of Ca2+ channel (L-type) in cardiac cells whose gating is affected in different ways by β-adrenoceptor and 1,4-dihydropyridine (DHP) agonists. β-adrenoceptors are functionally coupled to nearby Ca2+ channels by local elevations of cyclic adenosine monophosphate that affect the Ca2+ current. Expression of the DHP receptor (α-1 subunit of L-type Ca2+ channel) in the heart is regulated by differentiation and innervation and is altered in myocardial hypertrophy and congestive heart failure both in experimental models and in humans (1–7). Furthermore, regulation of cardiac Ca2+ channel expression by adrenergic pathways may have physiologic and pathophysiologic importance. Therefore, the development of a DHP receptor radiolabeled antagonist can be useful in studying human heart disease using PET. The widespread development of β-adrenoceptor antagonist therapy in congestive heart failure precludes the use of a PET ligand such as [11C]CGP12177 for the measurement of changes in myocardial β-adrenoceptor density (H. Valette et al., unpublished data, 1992). Therefore, because of the above mentioned relationship, β-adrenoceptor/L-type Ca2+ channels, and the changes in number of Ca2+ channels in pathologic circumstances, it seems of interest to have a positron emitter radiolabeled DHP ligand to use in hypertensive, myocardial hypertrophic, and congestive heart failure patients, who are frequently treated with β-adrenoceptor antagonists.
1,4-DHP derivatives are potent and specific ligands of the slow voltage-dependent L-type Ca2+ channels. The use of [3H]DHPs has resulted in the identification of high-affinity, saturable, and stereoselective binding sites in various tissues. A recently published study of the in vivo myocardial kinetics of [3H]nifedipine, [3H]nimodipine, [3H]PN200-110, and [3H]amlodipine (a racemic mixture) showed a rapid washout of the tracers from the myocardium and a low myocardial specific binding (8).
The L-type Ca2+ channel antagonist S11568 (3-ethyl 5-methyl (±)-2-[(2-(2-aminoethoxy)ethoxy)methyl]-4-(2,3-dichlorophenyl)-6-methyl-1,4-DHP-3,5-dicarboxylate) labeled with 11C has been studied in vivo using PET for myocardial imaging of L-type Ca2+ channels (9). In vitro studies have shown that among the 2 enantiomers (i.e., (−)-S11568 = S12968 and (+)-S11568 = S12967), S12968 had a higher affinity for myocardial L-type Ca2+ channels than did S12967 (10). Therefore, detailed in vivo characterization of 11C-labeled enantiomer with the highest affinity (S12968) was performed in rats as a preliminary study. Although the 2 precursors for the synthesis of each 11C-labeled enantiomer were available (11), we focused the PET studies on determining the suitability of [11C]S12968 for cardiac imaging. Only a few studies were performed using [11C]S12967. Imaging experiments were performed on dogs, and displacement of the 11C-labeled enantiomers and saturation of binding sites with the corresponding unlabeled enantiomer were evaluated.
MATERIALS AND METHODS
Radiosynthesis of 11C-Labeled Enantiomers
S12968 and S12967 were independently labeled with 11C using the corresponding enantiomerically pure carboxylate precursor (3-ethyl 2-[(2-(2-aminoethoxy)ethoxy)methyl]-4-(2,3-dichlorophenyl)-6-methyl-1,4-DHP-3,5-dicarboxylate) (11) and [11C]methyl iodide in N,N-dimethylformamide (at 40°C for 5 min). Typically, 3.7–7.4 GBq [11C]S12968 or [11C]S12967 (yield, 25%–30% decay-corrected based on [11C]CH3I) were routinely obtained within 25–30 min of radiosynthesis (including high-performance liquid chromatography [HPLC] purification), with specific radioactivities ranging from 29.6 to 44.4 GBq/μmol. Radiochemical, chemical, and enantiomeric purities were found to be >98%, >95%, and >95%, respectively.
Animal Studies
The procedures for use of rats and dogs were in accordance with the recommendations of the European Economic Community (86/609/CEE) and the French National Committee (decree 87/848) for the care and use of laboratory animals.
Biodistribution in Rats.
Some in vivo characteristics of [11C]S12968 were assessed in male Sprague-Dawley rats (weight range, 170–190 g; IFFA Credo, Lyon, France). Before saturation-competition experiments were performed, the linearity of the relationship between uptake and amount of intravenously injected [11C]S12968 was assessed. Previous PET experiments with [11C]S11568 showed that myocardial uptake of radioactivity after intravenous injection of the radiotracer was rapid, reaching a peak value within 2 min, and was followed by a slight downslide (calculated mean [±SD] washout from dog heart anesthetized with propofol, 80 ± 5 min) (9). Radioactivity in plasma decreased rapidly during the first 3–5 min. With the assumption that the kinetics were not significantly different in rats, these animals were killed 15 min after injection of [11C]S12968. The linear relationship between the injected dose of [11C]S12968 and the uptake in various tissues was checked. [11C]S12968 (0.22 kBq–5.92 MBq in 200–300 μL saline; 3 rats for each dose; total number of rats, 21) was injected as a bolus into a tail vein. Syringes were weighted before and after injection to determine the radioactivity injected into each animal. Fifteen minutes after injection of the radiotracer, the animals were killed by decapitation. The thorax and abdomen were opened, and the heart and lungs were removed. The left ventricle was dissected. Samples of liver, kidney, striated muscle (gastrocnemius), and blood were also removed. Samples were weighted and assessed for radioactivity in a γ well counter.
A series of 5 rats was pretreated with 4 μmol unlabeled S12968 5 min before injection of 2.2 kBq [11C]S12968 and killed 15 min later. In a series of 5 other rats, unlabeled S12698 (4 μmol) was injected 5 min after injection of 2.2 kBq [11C]S12968, and the animals were killed 15 min later. After these preliminary studies, the in vivo behavior of the 2 enantiomers was assessed in dogs using PET.
PET Data Acquisition.
Female beagle dogs (mean weight, 10 kg; IFFA Credo) were anesthetized with pentobarbital; intubated; and artificially respired with 1%–1.5% isoflurane, 67% N2O, and 33% O2. The animals were positioned in a human brain scanner (model 953B/31; CTI, Inc., Knoxville, TN) that was also suitable for cardiac imaging of small animals. This scanner allowed simultaneous acquisition of 31 slices (3.37 mm apart) with an intrinsic spatial resolution of approximately 6 mm (full width at half maximum). Reconstructed images had a resolution of 8.5 mm. Transmission scans were acquired for 15 min using 3 retractable 68Ge rod sources.
PET Experimental Protocols.
Five dogs received intravenous injections of 5–15 nmol [11C]S12968. Two dogs received intravenous injections of 5–8 nmol [11C]S12967. The PET acquisition lasted 30 min. The dynamic series consisted of 17 images (6 × 30 s, 5 × 1 min, 4 × 3 min, and 2 × 5 min). Arterial blood samples were obtained from the femoral artery at designated times. In 3 dogs, a second injection of [11C]S12968 was administered 30 min after the first injection with the same PET scanning protocol.
During displacement and presaturation experiments, heart rate was continuously and carefully monitored. We observed that a dose as low as 1 μmol/kg S12698 induced severe bradycardia (a fall in heart rate of 80–90 bpm) in isoflurane-anesthetized dogs. This fact was discrepant with the previous observations (9), in which up to 6 μmol/kg S11568 (the racemic mixture) did not induce severe bradycardia. The previous experiments were performed using propofol as the anesthetic agent. Therefore, for the displacement and presaturation experiments, propofol was used again. Myocardial and blood control kinetics did not differ for the 2 different anesthetic agents.
For the saturation experiment, the dogs (n = 2 for each enantiomer) were pretreated with unlabeled S12968 (2 μmol/kg intravenously over 30 min) or S12967 (6 μmol/kg intravenously over 30 min). Then, the dogs received an intravenous injection of 185 MBq [11C]S12968 or [11C]S12967. PET acquisition lasted 45 min. The scanning protocol consisted of nine 5-min images.
For the displacement experiment, the dogs (n = 2 for each enantiomer) received an intravenous injection of 185 MBq [11C]S12968 or [11C]S12967. Twenty minutes later, a continuous infusion of S12968 (2 μmol/kg for 60 min) or S12967 (6 μmol/kg for 60 min) was administered. The PET acquisition lasted 122 min. The scanning protocol consisted of 16 images (4 × 3 min, 2 × 5 min, and 10 × 10 min).
PET Data Processing.
Myocardial and lung time–concentration curves were measured from a region of interest encompassing the left ventricular myocardium and from a region of interest placed over the periphery of the lungs. [11C]S12968 or [11C]S12967 tissue concentrations (Bq/mL) were obtained after correction for attenuation and for 11C decay and expressed as pmol/mL per nanomole injected using the value of the specific radioactivity measured at the beginning of the PET experiment. Myocardial uptake of the 2 enantiomers was compared using 1-way analysis of variance and the Bonferroni t test from small samples. The amount of radioactivity displaced by a loading dose of unlabeled S12968 was calculated by extrapolating the baseline PET time–activity curve to 120 min by simulation using a 1-compartment model, as described below. This extrapolated curve was compared with the curve obtained after the displacement.
The eventual changes in coronary blood flow (CBF) were assessed by graphically analyzing myocardial and plasma time–activity curves. PET data were processed using a 1-compartment model (radioligand in plasma and radioligand in tissue). k1 (transfer from plasma to tissue) and k2 (transfer from tissue to plasma) were calculated for the first injections of S12968. These parameters are the first-order rate constants, with dimensions of mL min−1 mL−1 for k1 and min−1 for k2. We checked to see whether a statistically significant linear relationship existed between k1, k2, and the mass of S12968 injected. Identification of model parameters required knowledge of the plasma unchanged radioligand concentration, which was used as the input function in the modeling.
Determination of S12968 Plasma Metabolites in Dogs.
For analysis of metabolites, arterial blood samples (3 mL) were collected at 2, 5, 10, 15, and 20 min after injection of the tracer (7–10 nmol) and immediately centrifuged (5 min, 2,000g, at 4°C) to obtain cell-free plasma. For deproteinization, 0.5 mL plasma were mixed with 0.7 mL acetonitrile containing 0.001 mg/mL cold standard as the reference compound. After centrifugation at 2,000g for 5 min, the supernatant (approximately 1.1 mL) was directly used for HPLC analysis. The HPLC system consisted of 2 LC-10AS pumps (Shimadzu, Tokyo, Japan), a 2.6-mL mixing chamber, a C6W injector (Valco Instruments Co. Inc., Houston, TX) with a 1-mL loop, and a reverse-phase μBondapak C18 column (300 × 7.8 mm, 10 μm; Waters, Milford, MA) connected to an SPD-10A ultraviolet detector (Shimadzu) operated at 254 nm followed by a radioisotope detector (LB 506, 500 μL cell; Berthold Systems, Inc., Aliquippa, PA). An LB 5035 pump (Berthold) was used to add liquid scintillator (2 mL/min) to the eluent just before the radioactivity detector. Data acquisition and handling were done on a personal computer using Winflow software (version 1.21, JMBS Inc., Newark, DE). The column was eluted with application of a gradient from 30% acetonitrile in 0.01 mol/L phosphoric acid up to 90% in 5.5 min, back to 30% acetonitrile at 5.6 min, and with a total run length of 8 min. The flow rate of the eluent was maintained at 6 mL/min.
RESULTS
Biodistribution in Rats
A statistically significant linear relationship between myocardial uptake and injected dose was found: uptake (in cpm/g of wet tissue) = 353 × injected dose (in kilobecquerels) − 1,325 (r = 0.9; P < 0.01). In noncardiac striated muscle—another tissue rich in DHP binding sites—a statistically significant linear relationship between myocardial uptake and injected dose was also found: uptake (in cpm/g of wet tissue) = 159 × injected dose (in kilobecquerels) − 1,998 (r = 0.98; P < 0.01). No statistically significant relationship was found between injected dose and uptake in liver, kidney, or lung or radioactivity in blood. Pretreatment with unlabeled S12968 reduced myocardial radioactivity by 75%, and displacement by unlabeled S12968 reduced myocardial radioactivity by 70%.
PET Studies in Dogs
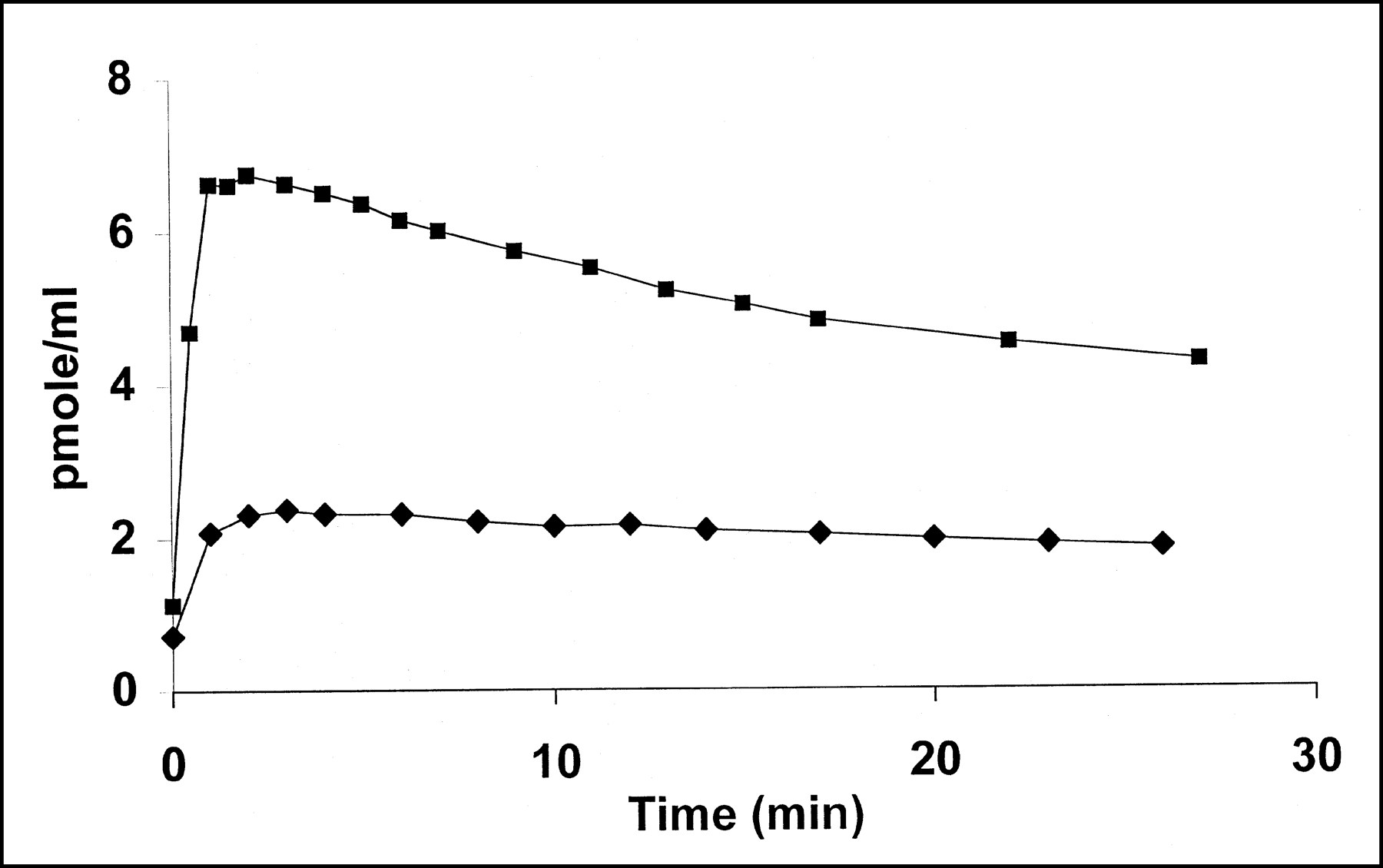
After a bolus injection of a tracer dose (5.2 nmol) of [11C]S12968 (Fig. 1) or [11C]S12967 (data not shown) myocardial concentration increased to reach a maximum in 3 min and then remained at a plateau with a slight downslope (calculated half-life, 60 min) while the blood radioactivity fell rapidly. When a higher dose of [11C]S12968 was injected (13.3 nmol; Fig. 2), the time–activity curve showed a significantly higher uptake followed by a more rapid washout of the radioactivity (calculated half-life, 30 min). A statistically significant linear relationship between k1, k2, and the mass of S12968 injected was found: k1 = 0.073 × mass (in nanomoles) + 0.018 (r = 0.93; n = 5; P < 0.05); k2 = 0.0118 × mass (in nanomoles) − 0.014 (r = 0.98; P < 0.01). A statistically significant linear relationship between k1 and k2 was also found: k2 = 0.15 × k1 − 0.0098 (r = 0.94; P < 0.01). No relationship was found between the ratio k1/k2 (the distribution volume) and the mass of S12968 injected (r = 0.22). This increase in coronary blood flow likely explains both the increased uptake and the more rapid washout. After the injection of 5- to 15-nmol doses of [11C]S12968 or [11C]S12967, vital signs did not change.
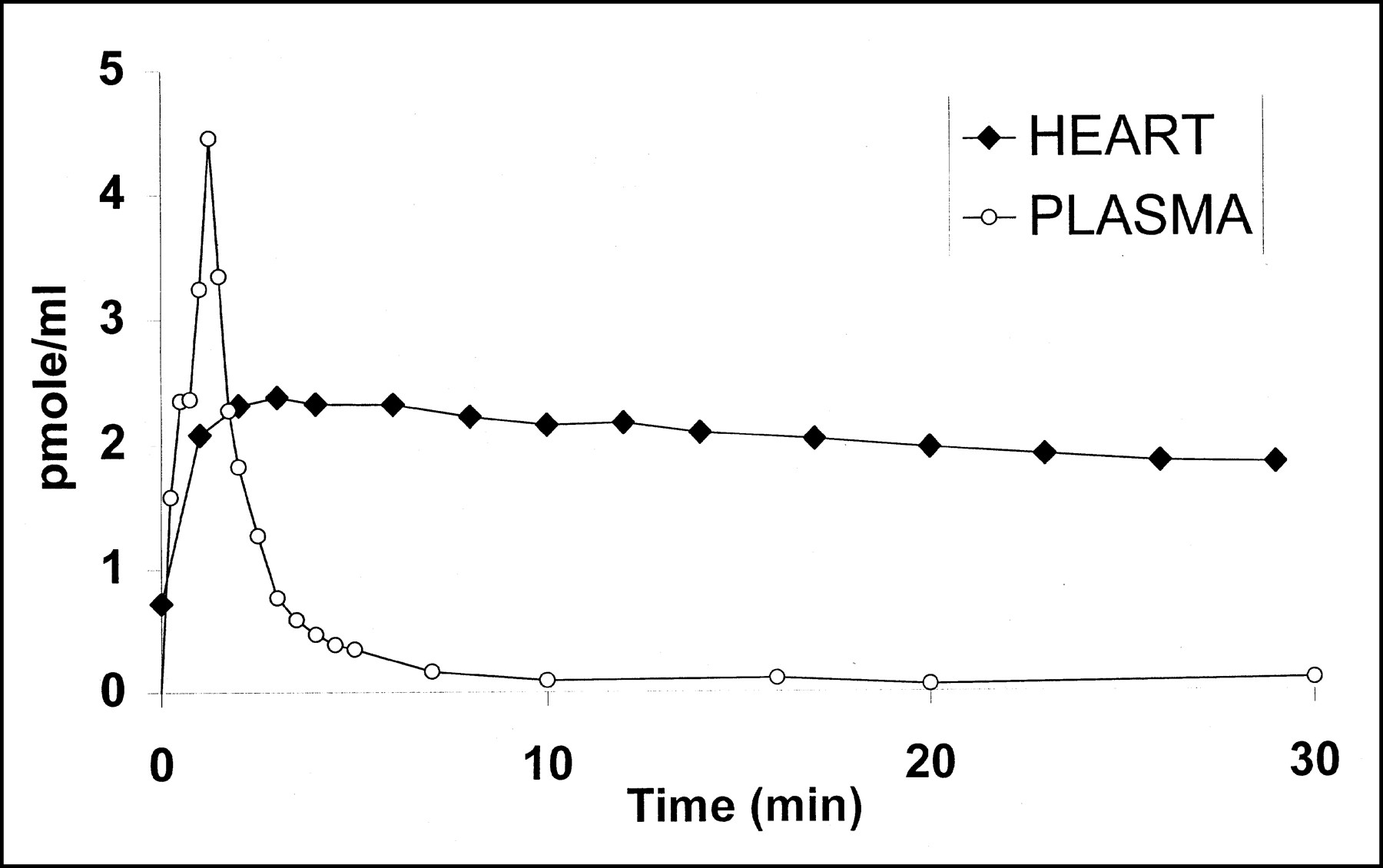
Time course of myocardial and plasma radioactivity (uncorrected for metabolites) in dog after injection of tracer dose (5.2 nmol) of [11C]S12968.

Comparison of time course of myocardial radioactivity in dog heart after injection of 2 doses of [11C]S12968. With lower injected dose (5.2 nmol [⧫]), time–activity curve remained at plateau with slight downslope. With higher injected dose (13.3 nmol [▪]), higher uptake followed by more rapid washout of radioactivity was observed.
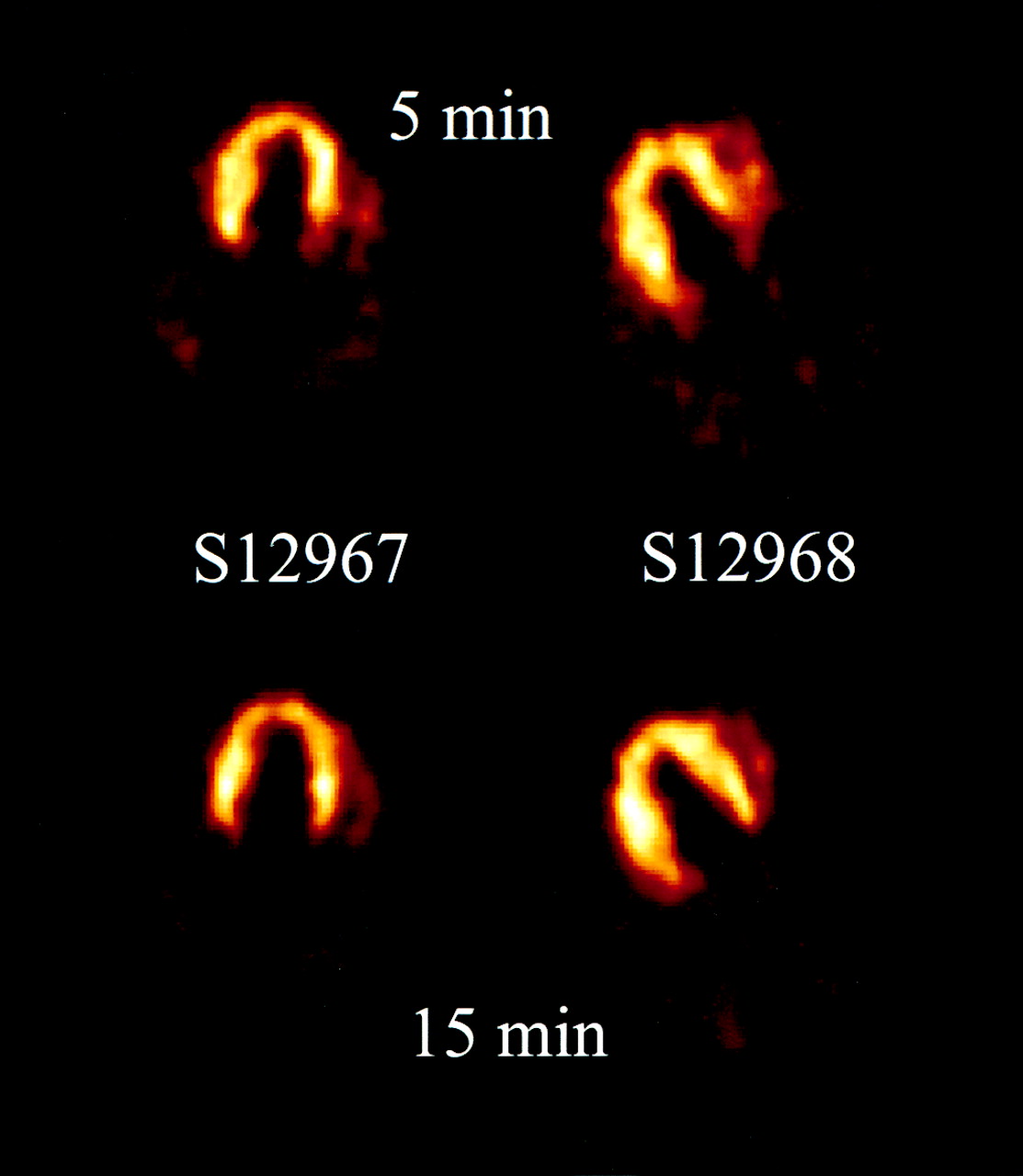
The time course of the myocardium-to-lung ratio remained high, approximately 2:4, after the first 5 min and was stable until the end of the experiment. Therefore, PET images showed good contrast between heart and lung (Fig. 3). Mean (±SD) myocardial uptake of radioactivity 5 min after injection was not statistically different for either enantiomer after a tracer injection (5 nmol): 0.36 ± 0.09 pmol/mL per nanomole injected for S12968; 0.49 ± 0.07 pmol/mL per nanomole injected for S12967 (P = 0.17). The corresponding value for a second tracer injection (5 nmol, injected 30 min after the first tracer injection) was 0.32 ± 0.1 for S12968 (P = 0.5 vs. the first tracer injection). These similar values were observed only between the 2 injections and only if the 2 injections contained the same number of nanomoles. This experiment was not performed with S12967.

Images of dog heart after injection of 5 nmol [11C]S12967 or [11C]S12968, 5 and 15 min after injection.
Pretreatment with unlabeled S12968 (2 μmol/kg) reduced myocardial uptake (0.09 ± 0.01 pmol/mL per nanomole injected) of [11C]S12968 by 75%. This pretreatment induced significant bradycardia in all dogs (heart rate dropped by an average value of 56 bpm). In contrast, pretreatment with unlabeled S12967 (6 μmol/kg) did not affect myocardial uptake of [11C]S12967 (0.4 ± 0.06 pmol/mL per nanomole injected) or heart rate.
A similar pattern was observed during displacement experiments: 80% of the [11C]S12968 myocardial radioactivity was displaced by unlabeled S12968 (2 μmol/kg), and this displacement was always accompanied by significant bradycardia (heart rate dropped by 65 bpm). In contrast, infusion of unlabeled S12967 (6 μmol/kg) did not affect the [11]CS12967 radioactivity in the heart and did not induce bradycardia.
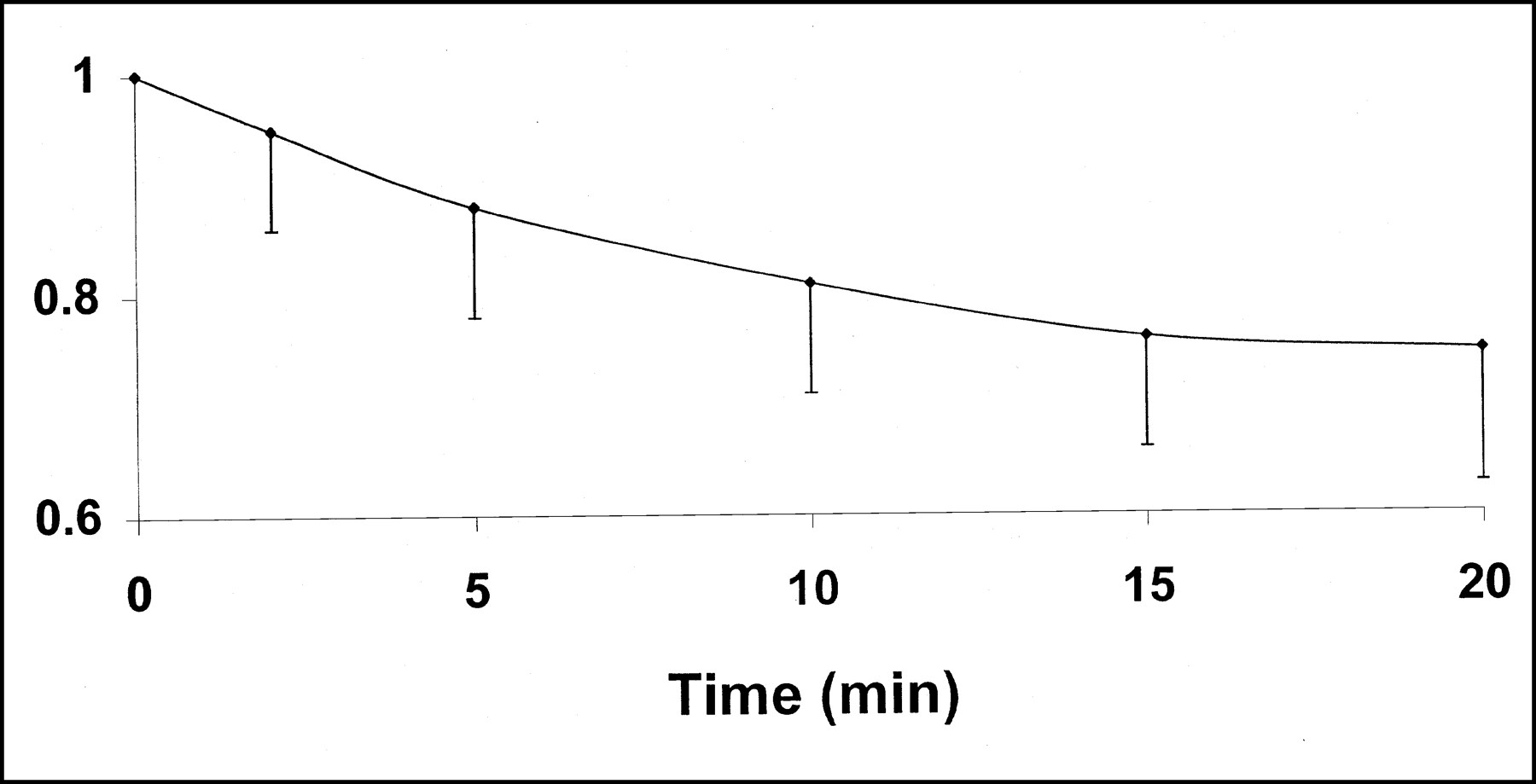
The metabolic profile in dogs is shown in Figure 4. Seventy percent of unchanged tracer was found in the plasma 20 min after injection. A lipophilic metabolite was present at 20 min after injection (15% of the radioactivity). Three hydrophilic metabolites were detected (15% of the radioactivity). The chemical structure of these metabolites remains to be determined.

Metabolic profile of S12968 in dogs (n = 4). Average of 70% of unchanged tracer was found in plasma 20 min after injection.
DISCUSSION
This study used PET to describe the myocardial kinetics of the 2 enantiomers of the Ca2+ channel inhibitor S11568. Saturability of binding sites and displacement of the ligand are the usual criteria that validate use of a ligand. These criteria were satisfied for S12968 but not for S12967. In vitro studies have shown that the 2 enantiomers have different affinities for vascular and myocardial L-type Ca2+ channels. In pig aorta, both enantiomers displaced [3H]PN200-110 in a competitive manner, with Ki values of 50.7 nmol/L for S12968 and 317 nmol/L for S12967 (10). In rat heart microsomes, the corresponding values obtained with the same tritiated ligand were 5.6 and 96 nmol/L (10). Displacement curves indicated that the compounds interact with a single site that is the same for both compounds. Specific binding of S12968 on cardiac microsomes was found to be 80% of total binding at Kd (0.2 nmol/L) (10). Inhibitory concentrations of 50% (inhibition of L-type Ca2+ current measured by patch clamp technique in guinea pig ventricular myocytes) were 0.08 μmol/L for S12968 and 2 μmol/L for S12967 (10). All these in vitro data suggested that S12968 could be a better candidate for PET studies. The in vitro affinity of S12967 appeared to be too low for the compound to be a good candidate for PET studies. The current study confirmed that the affinity is too low.
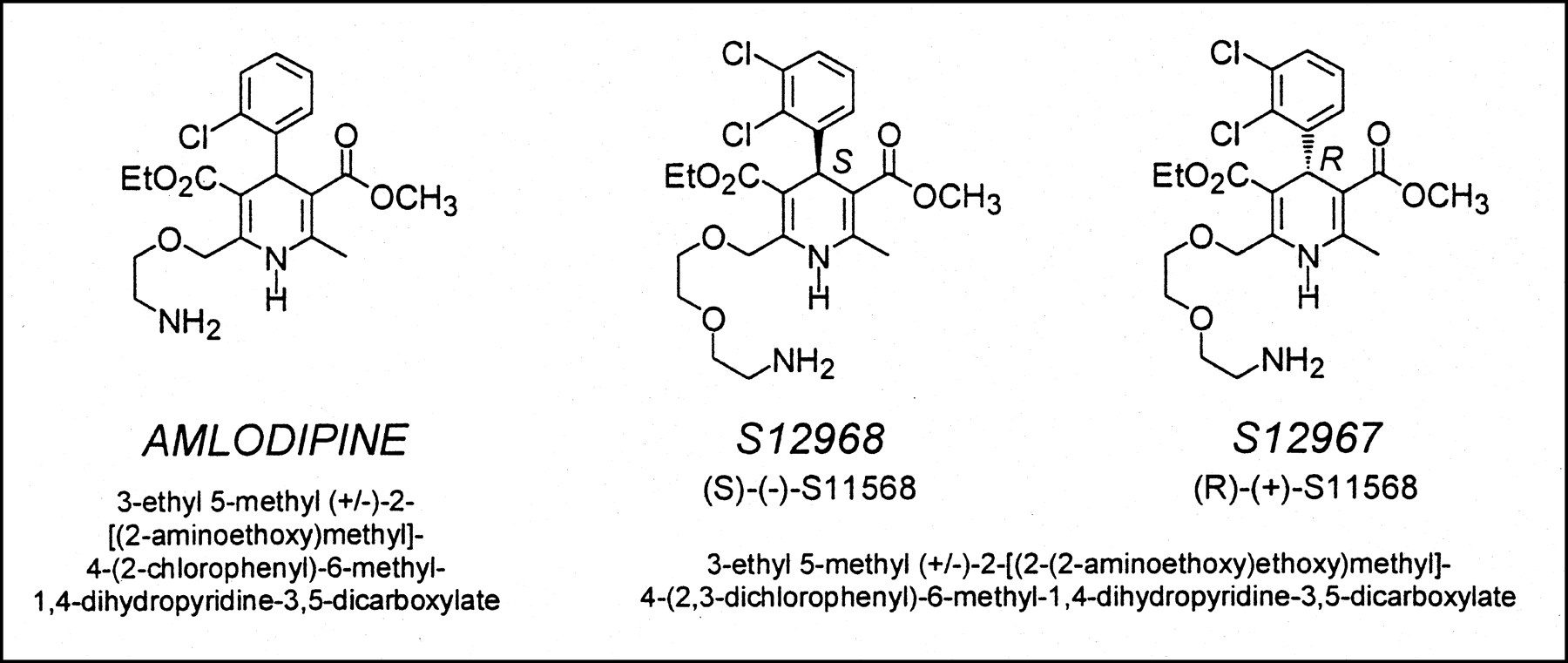
Because of the frequently observed trapping of DHPs in the membrane bilayer, several facts concerning S12968 should be pointed out. Trapping (12)—that is, binding to a nonspecific site different from the Ca2+ channel itself—of DHPs in the lipid membrane bilayer has been extensively studied for several DHP compounds, such as amlodipine, nimodipine, nisoldipine, and lacidipine (13–16). The observation that, after injection of a tracer dose of S12968, radioactivity in the heart reached a plateau that was maintained despite a rapid decrease in blood radioactivity is consistent with binding of [11C]S12968 to myocardial tissue. In fact, this finding could also be observed when a compound was trapped in the membrane. However, the major part (approximately 70%) was binding (and not trapping) of [11C]S12968 to Ca2+ channels, because presaturation or displacement by unlabeled S12968 significantly reduced myocardial radioactivity in both rats and dogs. Although coronary blood flow was not specifically measured, a part of the decreased radioactivity during the displacement experiment could be explained by an increased washout caused by an increased CBF. In tissues with a high density of DHP binding sites (heart and muscle), the linearity of the relationship between uptake and injected dose observed in rats also suggests a specific binding because this relationship was not found in nonexcitable cells. Increased washout from dog myocardium was observed when the injected dose was increased, meaning that the radioligand can escape from its binding sites. The case is different when a compound is trapped in the lipid bilayer, because it constitutes a very large container (e.g., the partition coefficient of amlodipine in biologic membranes is 19,000 (16)). The observed bradycardia during the competition experiment also favors an action of the compound on the channel itself. Combined, these facts are against a significant trapping of S12968 in the lipid bilayer of cardiac membrane. The chemical structure of amlodipine is close to that of S12968 (Fig. 5), as is its lipophilicity (octanol/buffer partition coefficient, 30 for amlodipine (16) and 35 for S12968 (17,18)). The in vivo myocardial and plasma kinetics of (±)-amlodipine in mice (8) are different from the kinetics of S12968 in rats. In contrast to amlodipine, S12968 does not cross the blood–brain barrier (8,17). Therefore, in spite of chemical and physical similitude, the reasons that S12968 and (±)-amlodipine behave differently in vivo remain unclear. Possibly, the use of a racemic mixture of amlodipine is a drawback for a clear determination of the in vivo behavior of the drug. The myocardial kinetics of the active enantiomer of amlodipine may be different from those of the racemic mixture.
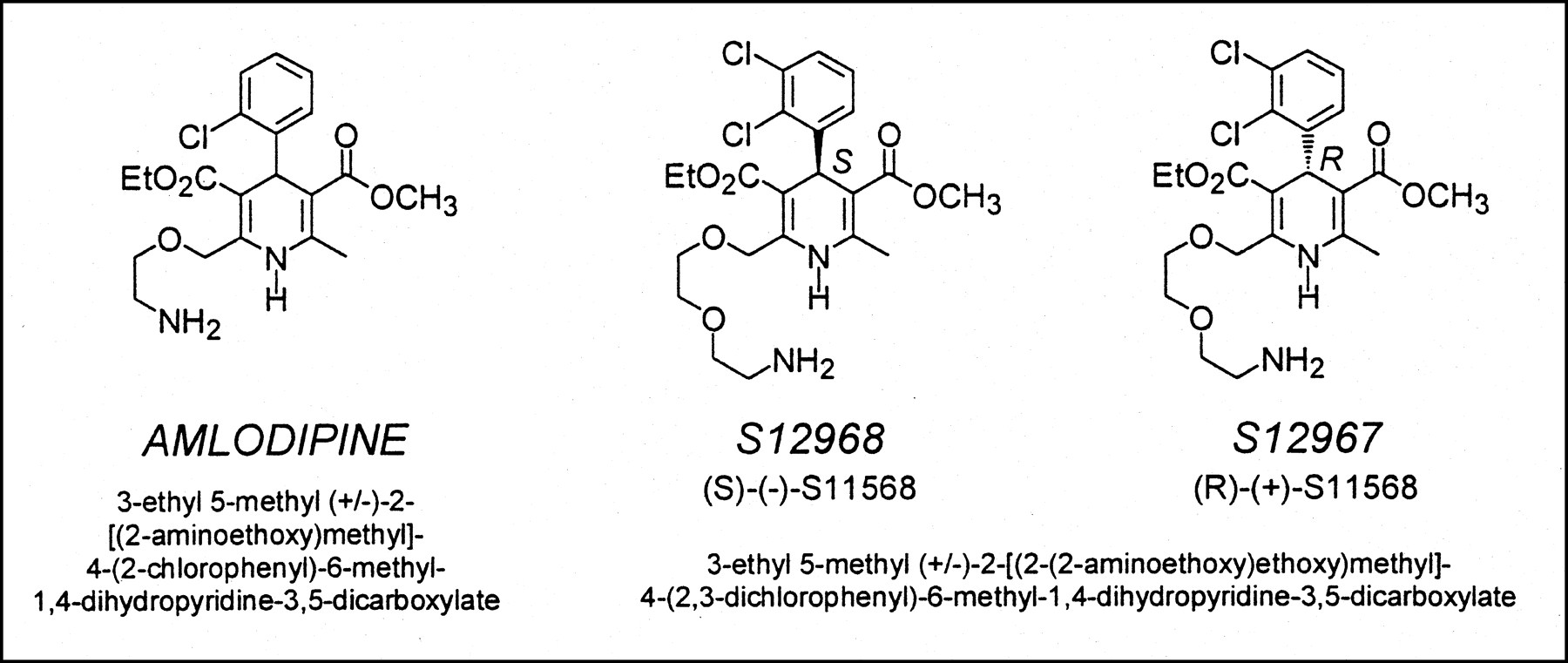
Comparison of chemical structures of S12968 and (±)-amlodipine. In spite of similarity of chemical structures, in vivo behaviors of the 2 compounds are extremely different (8).
The lethal dose of S12968 in dogs has been found to be 70 μmol/kg intravenously (Institut de Recherche Servier, unpublished data, 1992). This dose is several times higher than that needed for myocardial imaging (5 nmol for a dog weighing 10 kg). The findings of both the Ames test and the micronucleus test were negative (Institut de Recherche Servier, unpublished data, 1992). Like several other DHPs, S12968 was shown to have mildly negative inotropic effects, which were counterbalanced by systemic vasodilatation (19). This profile was observed in anaesthetized open-chest pigs using a high dose of S12968 (total cumulative dose, 581 μg/kg) and therefore cannot be extrapolated to humans. In the same study, the effects of S12968 on left ventricular transmural blood flow were statistically significant for the highest dose tested (20 μg/kg/min for 15 min). In this study, we found a relationship between an estimate of coronary blood flow (k1) and the amount of S12968 injected. This relationship can be a drawback for the use of S12968 for quantification of DHP binding sites, because these changes in CBF have to be taken into account during mathematic modeling of PET data. But this problem is not insurmountable, because it was successfully solved for methyl-quinuclidinyl benzylate, a compound that increases heart rate and, therefore, CBF (20). The coronary vasodilatory effects of S12968 indicate its action on vascular smooth muscle DHP binding sites. PET cannot differentiate between binding to these cells and to myocytes. However, the density in DHP binding sites is 10 times higher in myocytes than in vascular cells (21), and binding to coronary vascular smooth cells therefore appears negligible for PET imaging.
CONCLUSION
Our data show the ability of [11C]S12968 to image myocardial L-type Ca2+ channels. Although the activity of the channel cannot be measured with this ligand, its use can provide new insights in the study of myocardial hypertrophy and congestive heart failure both in experimental models and in humans.
Footnotes
Received Oct. 4, 2000; revision accepted Feb. 15, 2001.
For correspondence or reprints contact: Héric Valette, MD, Service Hospitalier Frédéric Joliot, DSV-DRM-CEA, 4 Place du Général Leclerc, 91401 Orsay, France.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}