


Abstract
Transforming growth factor-β (TGF-β) promotes cancer invasion and metastasis and is therefore a potential drug target for cancer treatment. Fresolimumab, which neutralizes all mammalian active isoforms of TGF-β, was radiolabeled with 89Zr for PET to analyze TGF-β expression, antibody tumor uptake, and organ distribution. Methods: 89Zr was conjugated to fresolimumab using the chelator N-succinyldesferrioxamine-B-tetrafluorphenol. 89Zr-fresolimumab was analyzed for conjugation ratio, aggregation, radiochemical purity, stability, and immunoreactivity. 89Zr-fresolimumab tumor uptake and organ distribution were assessed using 3 protein doses (10, 50, and 100 μg) and compared with 111In-IgG in a human TGF-β–transfected Chinese hamster ovary xenograft model, human breast cancer MDA-MB-231 xenograft, and metastatic model. Latent and active TGF-β1 expression was analyzed in tissue homogenates with enzyme-linked immunosorbent assay. Results: 89Zr was labeled to fresolimumab with high specific activity (>1 GBq/mg), high yield, and high purity. In vitro validation of 89Zr-fresolimumab showed a fully preserved immunoreactivity and long (>1 wk) stability in solution and in human serum. In vivo validation showed an 89Zr-fresolimumab distribution similar to IgG in most organs, except for a higher uptake in the liver in all mice and higher kidney uptake in the 10-μg group. 89Zr-fresolimumab induced no toxicity in mice; it accumulated in primary tumors and metastases in a manner similar to IgG. Both latent and active TGF-β was detected in tumor homogenates, whereas only latent TGF-β could be detected in liver homogenates. Remarkably high 89Zr-fresolimumab uptake was seen in sites of tumor ulceration and in scar tissue, processes in which TGF-β is known to be highly active. Conclusion: Fresolimumab tumor uptake and organ distribution can be visualized and quantified with 89Zr-fresolimumab PET. This technique will be used to guide further clinical development of fresolimumab and could possibly identify patients most likely to benefit.
The pleiotropic transforming growth factor-β (TGF-β) is excreted in low amounts by multiple cell types to prevent progression of premalignant lesions (1,2). In many tumor types the tumor-suppressive responses to TGF-β are lost in the malignant phase, in which tumor-promotive responses to TGF-β (including epithelial-to-mesenchymal transition, angiogenesis, extravasation, migration, invasion, and immune suppression) prevail. TGF-β thereby contributes to a more invasive and metastatic tumor phenotype (1,3). Mechanisms involved in this suppressor-to-promoter switch are diverse and include mutations (and epigenetic silencing) in the suppressive pathway and increased TGF-β production, release, and activation in the tumor microenvironment (1,4,5). Activity of TGF-β is locally controlled in the extracellular matrix by cleavage of active TGF-β dimers from the latent precursor (6,7).
TGF-β is a potential drug target for cancer treatment, especially in the case of highly invasive or metastatic tumors such as glioblastomas and metastatic breast cancer (8,9). Strategies in clinical development for TGF-β inhibition include antisense oligonucleotides, TGF-β–neutralizing antibodies, and small-molecule TGF-β receptor kinase inhibitors (9,10). Clinical TGF-β imaging can have an unprecedented role in the development of these TGF-β–targeted agents, because the dual functions of TGF-β in cancer make proper patient selection of crucial value. Selection seems especially important in breast cancer. Pathway analysis identified a subset of breast cancer patients with high expression of TGF-β pathway genes and an association with shorter distant-metastasis-free survival, indicating a potential benefit of TGF-β inhibition for these patients (11). In addition, others have identified a subset of patients with abrogated TGF-β signaling, which was associated with reduced relapse-free survival (12).
Improved insight into the role of TGF-β in breast cancer invasion and metastasis has recently been provided in several elegant preclinical optical imaging studies. Live intravital imaging of TGF-β signaling in tumor cells was performed in an orthotopic mouse model using rat breast cancer cells (MTLn3E) transfected with cyan fluorescent protein TGF-β–dependent reporter constructs. Here, TGF-β signaling was transiently and locally activated in single moving tumor cells. TGF-β–activated single moving cells demonstrated an increased tendency to infiltrate surrounding tissues and were consequently responsible for distant metastases (13). TGF-β bioluminescence imaging (BLI) using human breast cancer cells (MDA-MB-231) transfected with TGF-β–responsive luciferase constructs indicated a temporal TGF-β dependency of bone metastases and an antimetastatic effect of TGF-β inhibition (14,15). These TGF-β–responsive imaging approaches have increased our understanding of TGF-β signaling in metastasis but regretfully are restricted to preclinical use because of the use of transfections and the poor tissue penetration of optical techniques. Clinically applicable TGF-β imaging techniques would therefore be of value in our clinical understanding of TGF-β, in the development of TGF-β–targeting agents, and in the selection of patients most likely to benefit.
Fresolimumab (GC1008; provided by Genzyme) is a fully human IgG4 κ-monoclonal antibody capable of neutralizing all mammalian active isoforms of active TGF-β (1, 2, and 3). A phase I study with fresolimumab in 22 patients with advanced melanoma and renal cell carcinoma showed stable disease in 1 patient, a partial response in 1 patient, a mixed tumor response in 3 patients, and no dose-limiting toxicity (16). For further clinical development of fresolimumab and to identify the patients most likely to benefit, it will be helpful to know whether TGF-β is being overexpressed and activated in the tumor and whether fresolimumab reaches the target. Labeling fresolimumab with the long-lived positron emitter 89Zr should allow for noninvasive monitoring and quantification of fresolimumab tumor and organ distribution using PET. Preclinical studies, as well as ongoing clinical studies with 89Zr-bevacizumab for imaging vascular endothelial growth factor (VEGF), previously demonstrated the feasibility of PET with antibodies against soluble ligands overexpressed in tumors (17,18).
In this study, we describe the development, quality control, and preclinical validation of 89Zr-fresolimumab for noninvasive PET of TGF-β tumor expression and organ distribution of fresolimumab. We used 2 human TGF-β–transfected Chinese hamster ovary (CHO) xenograft models, 1 with intermediate and 1 with high TGF-β expression. In addition, we used an MDA-MB-231 xenograft and metastatic model of human breast cancer. The triple-negative breast cancer cell line MDA-MB-231 was selected because of the considered role of TGF-β in triple-negative breast cancer and the extensive data available concerning the role of TGF-β in this cell line (14,15).
MATERIALS AND METHODS
Cell Cultures
CHO clones were generated by transfection of DG44-CHO cells with human latent TGF-β1 (generously provided by Genzyme). Briefly, DG44-CHO cells were transfected with human latent TGF-β1 complementary DNA using the SV2DHFR vector, and stable cell lines were generated by methotrexate selection. CHO clone 11S (CHO-Cl11S) and clone 2 (CHO-Cl2) were selected because they produced intermediate (23.3 ng per 1 × 106 cells per day) levels and high (189 ng per 1 × 106 cells per day) levels of human latent TGF-β1, respectively. CHO-Cl11S and CHO-Cl2 were cultured in a humidified incubator at 5% CO2 and 37°C in minimum essential medium, supplemented with 10% dialyzed fetal calf serum and 2% l-glutamine. The triple-negative breast cancer cell line MDA-MB-231 (from American Type Culture Collection) and its luciferase-transfected bone-tropic clone MDA-MB-231-SCP2luc (provided by Dr. Yibin Kang and described earlier) (19) were cultured in a humidified incubator at 5% CO2 and 37°C in Dulbecco modified Eagle medium, supplemented with 10% fetal calf serum and 1% l-glutamine. Concentrations of active and total TGF-β1 in culture medium were assessed using an enzyme-linked immunosorbent assay (ELISA) (R&D Systems) according to the manufacturer's protocol.
Conjugation, 89Zr-Labeling, and Quality Control of Fresolimumab
Fresolimumab was conjugated and labeled as described by Verel et al. (20). Briefly, fresolimumab was first conjugated with the chelator N-succinyldesferrioxamine-B-tetrafluorphenol (N-sucDf-TFP; generously provided by VU University Medical Center) in a 5-fold molar excess. After conjugation, the product was purified by ultracentrifugation using a 30-kDa Vivaspin-2 (Sartorius) and stored at −20°C. In the second step, N-sucDf-fresolimumab was freshly radiolabeled with clinical-grade 89Zr-oxalate (IBA Molecular) on the day of use.
N-sucDf-fresolimumab and 89Zr-fresolimumab were analyzed for conjugation ratios, aggregation, and radiochemical purity by size-exclusion high-performance liquid chromatography (HPLC). The Waters size-exclusion HPLC system was equipped with a dual-wavelength absorbance detector, an in-line radioactivity detector, and a size-exclusion column (Superdex 200 10/300 GL; GE Healthcare). Sodium phosphate buffer (0.025 M Na2HPO4·2H2O/NaH2PO4·H2O) was used as the mobile phase. The retention time of fresolimumab was approximately 18 min, and 89Zr-N-SucDf and low-weight impurities were eluted at 28 min (at a flow of 0.7 mL/min).
Stability of 89Zr-fresolimumab was tested in 0.9% NaCl at 4°C and in human serum at 37°C using 20% trichloroacetic acid (Hospital Pharmacy, University Medical Center Groningen) precipitation. Trichloroacetic acid precipitation was performed in phosphate-buffered saline (PBS) (140 mM NaCl, 9 mM Na2HPO4, 1.3 mM NaH2PO4; pH 7.4) with 0.5% human serum albumin (Sanquin) and 20% trichloroacetic acid. Radioactivity in precipitate and supernatant was determined by a calibrated well-type γ-counter (LKB Wallac).
Immunoreactivity was tested in a competition assay with unlabeled fresolimumab. Recombinant human TGF-β3 (Peprotech) was used as target antigen because fresolimumab has the highest affinity (dissociation constant, 1.4 nM) for this TGF-β isoform, and therefore binding to TGF-β3 serves as a sensitive indicator for immunoreactivity of 89Zr-fresolimumab. TGF-β3 was diluted in PBS to a concentration of 4 μg/mL (pH was adjusted to 9.2–9.5 with 50 mM Na2CO3) and coated to Nunc-Immuno BreakApart ELISA plates (NUNC). Fifty microliters were added to the wells, incubated overnight at 4°C, and then blocked with 1% human serum albumin in PBS. After blocking, plates were washed with 0.1% polysorbate 80 (Sigma-Aldrich) in PBS. 89Zr-fresolimumab and fresolimumab were mixed and diluted in PBS to result in a fixed concentration of 14 nM 89Zr-fresolimumab and varying concentrations of unlabeled fresolimumab, ranging from 14 pM to 14 μM. These samples were added to the wells and incubated for 2 h. Samples were collected in 2 wash steps. Both 89Zr-fresolimumab bound to the TGF-β3–coated wells and the collected samples containing unbound 89Zr-fresolimumab were measured for radioactivity. Percentage of TGF-β3 binding was calculated as the fraction of radioactivity bound to TGF-β3–coated wells divided by the total amount of radioactivity added. These percentages were plotted using Prism software (GraphPad Software), and the concentration that resulted in 50% inhibition of the maximum binding was calculated.
Conjugation and 111In Labeling of Control Human IgG
Human IgG (Sanquin) was conjugated and labeled according to Ruegg et al. (21). Briefly, IgG was first conjugated to the bifunctional conjugating agent 2-(4-isothiocyanatobenzyl)-diethylenetriaminepentaacetic acid (p-SCN-Bn-DTPA) (Macrocyclics). After conjugation, the product was purified by ultracentrifugation using a 30-kDa Vivaspin-2 and stored at −20°C. Conjugated human IgG was radiolabeled with 111InCl3 (Covidien) on the day of use.
Animal Studies
In vivo imaging and biodistribution experiments were conducted using male athymic mice (BALB/C nude; Harlan). All experiments were approved by the animal ethics committee of the University of Groningen. Tumor cell inoculation, BLI, small-animal PET, and micro-CT were performed with isoflurane inhalation anesthesia (induction, 3%; maintenance, 1.5%).
For the CHO xenograft model, mice were injected subcutaneously with 2 × 106 CHO-Cl2 or CHO-Cl11S cells suspended in Hank buffered salt solution (Invitrogen). 89Zr-fresolimumab (5 MBq; 10, 50, or 100 μg) and 111In-IgG (3 MBq; 10, 50, or 100 μg) were administered via the penile vein. For the MDA-MB-231 xenograft model, mice were injected subcutaneously with 2 × 106 MDA-MB-231 cells mixed equally with Matrigel (BD Bioscience). 89Zr-fresolimumab (5 MBq; 10 μg) and 111In-IgG (3 MBq; 10 μg) were administered via the penile vein. For the MDA-MB-231 metastatic model, mice were injected intracardially (left ventricle) with 105 MDA-MB-231-SCP2luc cells suspended in PBS. Metastatic tumor growth was measured twice weekly with BLI. BLI was performed for 30–45 min after intraperitoneal administration of ds-luciferin (150 mg/kg) with an IVIS100 (Xenogen). When metastatic tumor growth was measurable, approximately 2–4 wk after inoculation, 89Zr-fresolimumab (5 MBq; 10 μg) and 111In-IgG (3 MBq; 10 μg) were administered via the penile vein.
All animals were imaged using a Focus 220 rodent scanner (CTI Siemens) for small-animal PET and a MicroCAT II scanner (CTI Siemens) for subsequent micro-CT. Static images of 15- to 45-min acquisition time were obtained at 24, 72, and 144 h after injection. After image reconstruction, in vivo quantification was performed with AMIDE (A Medical Image Data Examiner) software (version 0.9.1; Stanford University) (22), and tumor accumulation was calculated as standardized uptake value (SUV). Animals were sacrificed after the last scan and organs were excised, rinsed for residual blood, weighed, and counted for radioactivity. Tissue activity was expressed as percentage injected dose per gram of tissue (%ID/g). Subsequently, organs of interest were split and partly formalin-fixed and paraffin-embedded for histologic analysis and partly stored at −80°C for ex vivo TGF-β1 measurement.
Ex Vivo Analyses on Organ-of-Interest Tissue
Ex vivo TGF-β1 measurement was performed in organs of interest using an ELISA according to the manufacturers’ protocol. This ELISA measures active TGF-β1 quantitatively, and latent TGF-β1 was measured after activation by acidification to discriminate between latent and active TGF-β1. Measurement of TGF-β1 was performed because CHO cells were transfected with this isoform. Formalin-fixed, paraffin-embedded organs of interest were stained with hematoxylin and eosin and for phospho-Smad2 (pSmad2) (Cell Signaling). Staining for pSmad2 served as a surrogate for active TGF-β, since it is currently not possible to stain for active TGF-β itself.
Statistical Analysis
Data are presented as means ± SD from at least 3 individual experiments or animals, unless stated differently. Statistical analysis was performed using the Mann–Whitney test for nonparametric data and the unpaired t test for parametric data. A P value of 0.05 or less was considered significant.
RESULTS
89Zr-Fresolimumab Labeling and Quality Control
HPLC analysis showed an aggregation of 1.4% ± 1.1% after conjugation of fresolimumab with sucDf-TFP and an effective conjugation of 62% ± 9%. N-sucDf-fresolimumab could be labeled with 89Zr to a specific activity of up to 1,000 MBq/mg, with a radiochemical purity of 97.0% ± 1.2% over all experiments, not requiring further purification. A typical representative HPLC analysis of 89Zr-fresolimumab is shown in Supplemental Figure 1A (supplemental materials are available online only at http://jnm.snmjournals.org).

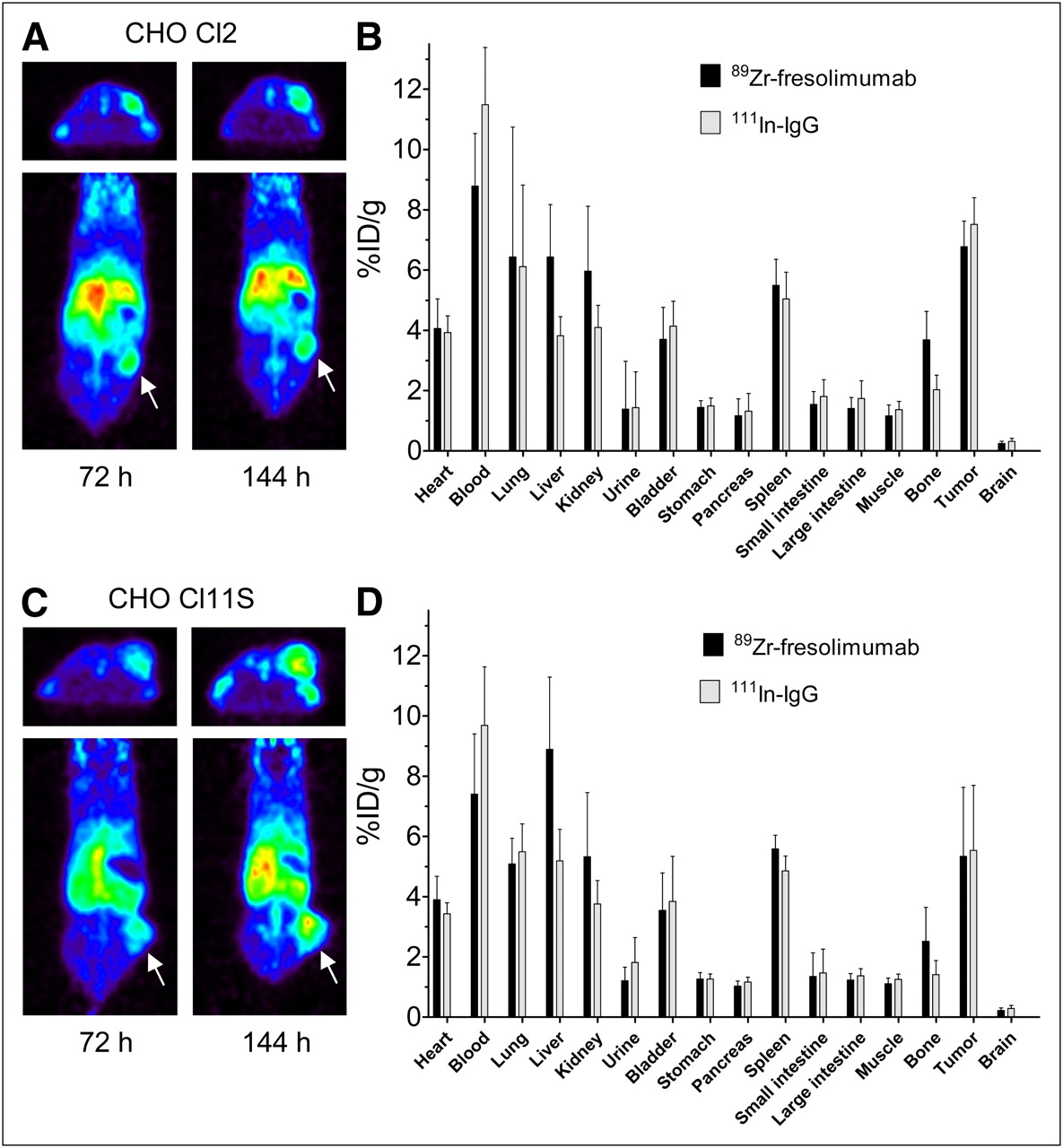
Small-animal PET with 89Zr-fresolimumab showed tumor uptake in both CHO-Cl2 and CHO-Cl11S (A and C, respectively; arrow indicates tumor). Tumor uptake and organ distribution of 89Zr-fresolimumab and control 111In-IgG as quantified ex vivo (B and D, respectively).
89Zr-fresolimumab was highly stable in solution (0.9% NaCl) at 4°C and in human serum at 37°C over time (>168 h). The average decrease in radiochemical purity of 89Zr-fresolimumab per day in human serum at 37°C was 0.44% ± 0.13%, 0.84% ± 0.11%, and 1.21% ± 0.09% for a specific activity of 250, 500, and 1,000 MBq, respectively. The average decrease in radiochemical purity of 89Zr-fresolimumab per day in solution (0.9% NaCl) at 4°C was 0.42% ± 0.05%.
To prove that labeling fresolimumab did not alter the activity of fresolimumab, a competitive binding experiment was performed with unlabeled fresolimumab in competition with 89Zr-fresolimumab. This experiment resulted in an average 50% inhibition of the maximum binding of 18 nM fresolimumab (95% confidence interval, 12–28 nM) for the competition of TGF-β3 binding of 14 nM 89Zr-fresolimumab, indicating fully preserved immunoreactivity (Supplemental Fig. 1B).
89Zr-Fresolimumab Small-Animal PET and Biodistribution in CHO Xenografts
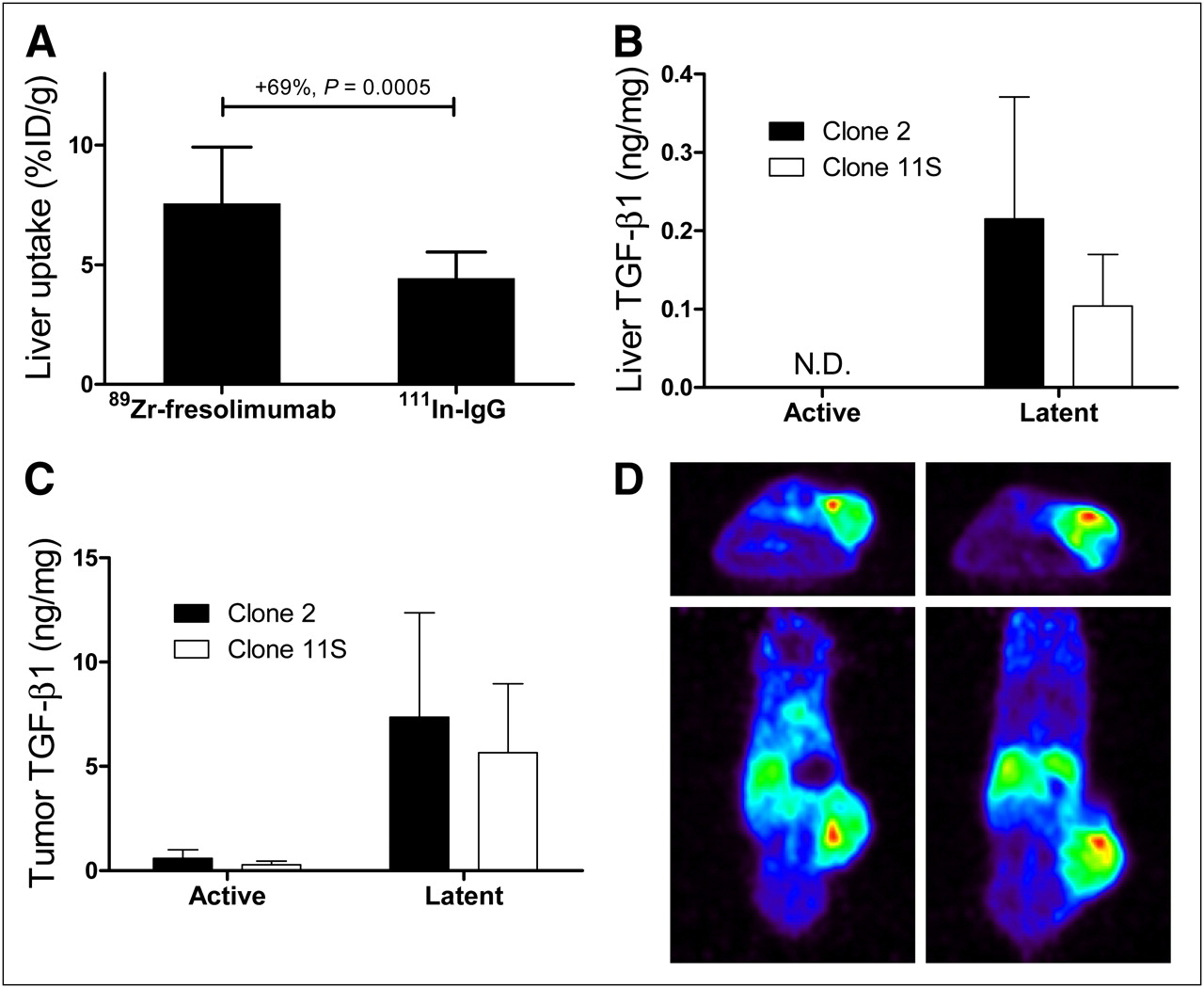
89Zr-fresolimumab small-animal PET was first performed on mice harboring xenograft tumors with CHO clones expressing intermediate and high levels of human latent TGF-β1. Assessment of in vitro expression of human latent TGF-β1 in culture medium samples with ELISA confirmed the intermediate and high expression of CHO-Cl11S and CHO-Cl2, respectively (data not shown). Small-animal PET of mice bearing CHO-Cl11S and CHO-Cl2 tumors with 89Zr-fresolimumab indicated clear tumor accumulation and visualization in both models at 72 and 144 h after injection, with a slightly higher tumor uptake at 144 h after injection (Figs. 1A and 1C). No visual difference in 89Zr-fresolimumab tumor uptake between both CHO clones could be detected, and also quantification of the tumor uptake as assessed by SUV did not show a difference: SUV in CHO-Cl2 was 2.0 ± 0.1 and 2.3 ± 0.2 at 72 and 144 h after injection, respectively, and SUV in CHO-Cl11S was 2.0 ± 0.4 and 2.2 ± 0.6 at 72 and 144 h after injection, respectively. Ex vivo analysis of 89Zr-fresolimumab and 111In-IgG biodistribution indicated a similar uptake of both tracers in all tumors and comparable distribution over most organs (Figs. 1B and 1D). Organ uptake of 89Zr-fresolimumab was higher than 111In-IgG in liver and bone of both groups of mice. Subgroup analysis of the different protein doses of 89Zr-fresolimumab showed similar tumor uptake of 10, 50, or 100 μg of 89Zr-fresolimumab, with a nonsignificant trend toward lower liver uptake in the 10-μg dose group (Table 1). In the 10-μg group only, there was a 92% ± 28% higher uptake of 89Zr-fresolimumab than 111In-IgG in kidneys (P = 0.0079). Liver uptake of 89Zr-fresolimumab was 7.6 ± 2.4 %ID/g and was higher than 111In-IgG in all individual mice, with a mean difference of 69% ± 28% (P = 0.0005; Fig. 2A). To assess whether this was caused by high TGF-β levels in the liver, we determined TGF-β1 levels in liver homogenates with ELISA. Levels of active TGF-β1 were below the detection limit in all samples, and levels of latent TGF-β1 were 0.22 ± 0.16 and 0.10 ± 0.07 ng/mg of protein in livers of mice with CHO-Cl2 and CHO-Cl11S xenografts, respectively (Fig. 2B). TGF-β1 levels in tumor homogenates also showed the same pattern of higher levels in tissue from CHO-Cl2 xenografts than from CHO-Cl11S xenografts, although this difference was not significant (Fig. 2C). The highest TGF-β1 levels were found in homogenates of tumors in 2 mice with skin ulceration at the tumor site. These tumors showed a focally increased 89Zr-fresolimumab uptake at the site of ulceration with small-animal PET (Fig. 2D), correlating with TGF-β1 levels in homogenates of the ulcerations of 26- and 21-ng latent TGF-β1 per milligram of protein, which were the highest levels of all tissue samples measured (highest latent TGF-β1 level in tumors without ulcerations was 17 ng/mg). High 89Zr-fresolimumab uptake was also present in sites with scar tissue in 2 mice that were injured by their dominant congener cage mate before 89Zr-fresolimumab injection (data not shown).
Dose Escalation of 89Zr-Fresolimumab and Control 111In-IgG

Liver uptake of 89Zr-fresolimumab and 111In-IgG in CHO xenograft mice (A). TGF-β1 levels were determined by ELISA in homogenates of liver (B) and tumor (C) tissue. Two mice with skin ulceration at tumor site showed locally increased uptake of 89Zr-fresolimumab (represented by red areas) at site of ulceration (D). N.D. = not detectable.
Histologic Analysis of CHO Xenograft Tumors and Livers
Hematoxylin and eosin staining showed no obvious difference in morphology between tumors from xenografts of either of the CHO clones (Supplemental Fig. 2). All tumors largely consisted of vital vascularized tissue with tumor and stromal cells and minor areas of necrosis. Hematoxylin and eosin staining of livers from CHO xenograft mice showed a normal morphology. All tested samples showed nuclear staining for pSmad2. Concurring with the small-animal PET data, there was no difference in pSmad2 staining in tumors and livers from xenograft mice of either CHO clone, although liver tissue showed a more intense staining than the CHO tumors.
89Zr-Fresolimumab Small-Animal PET and Biodistribution in MDA-MB-231 Xenografts
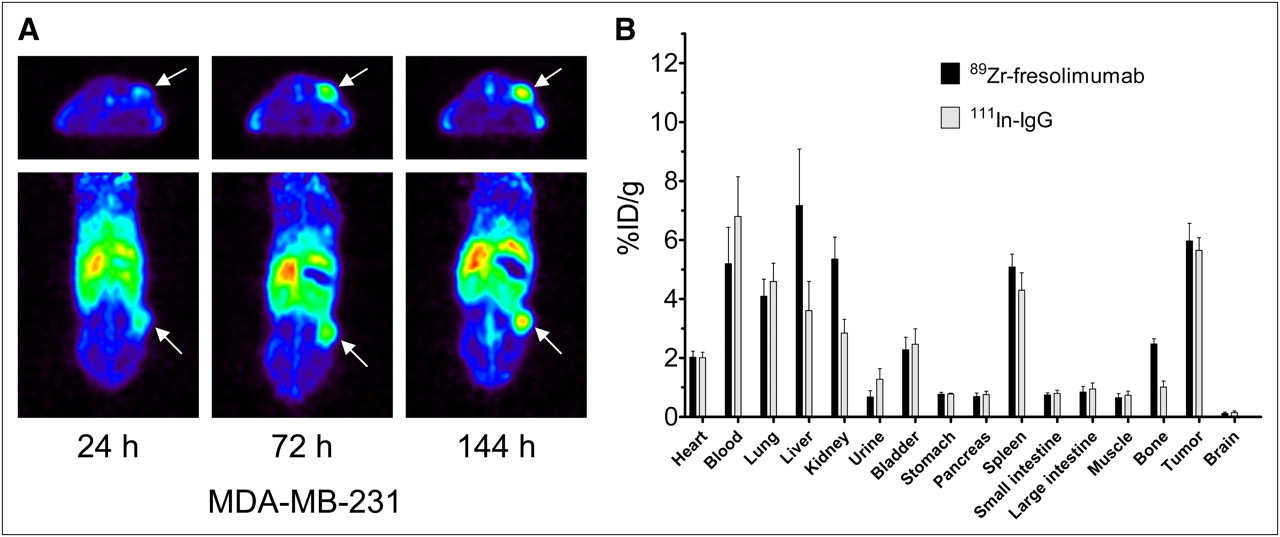
To further investigate the organ and tumor distribution of 89Zr-fresolimumab, we used the MDA-MB-231 human breast cancer xenograft model. Small-animal PET showed a clear 89Zr-fresolimumab tumor accumulation over time (Fig. 3A). Tumor accumulation was also shown by SUV quantification: 1.5 ± 0.2, 2.3 ± 0.3, and 2.6 ± 0.3 at 24, 72, and 144 h after injection, respectively. Ex vivo biodistribution analysis again showed a similar tumor uptake of nonspecific control 111In-IgG at 144 h after injection (Fig. 3B). Organ uptake of 89Zr-fresolimumab was higher than 111In-IgG in liver (P = 0.0459), kidneys (P = 0.0078), and bone (P = 0.0007) and reflected the biodistribution seen in CHO xenografts.
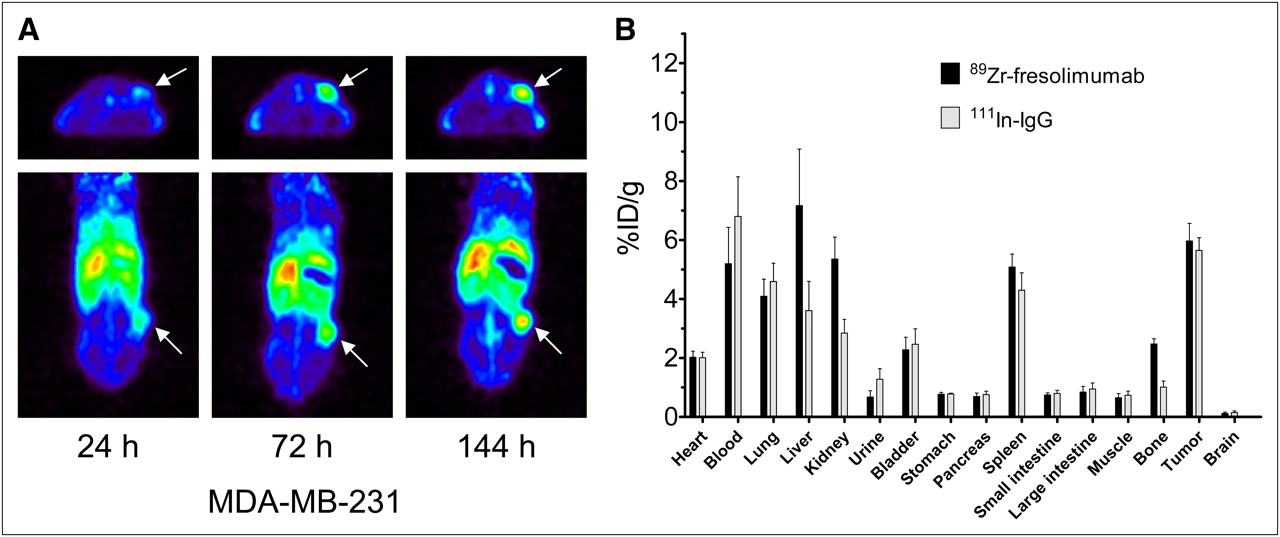
Small-animal PET (A) and ex vivo biodistribution (B) of 89Zr-fresolimumab in MDA-MB-231 xenografts.
89Zr-Fresolimumab Small-Animal PET and Biodistribution in MDA-MB-231-SCP2luc Metastatic Model
Because TGF-β is involved in breast cancer metastasis (13,14), we evaluated 89Zr-fresolimumab imaging in a metastatic breast cancer model as well. All mice had developed multiple (bone) metastases 3–5 wk after intracardiac injection of MDA-MB-231-SCP2luc cells, as was visualized with BLI (Fig. 4), corresponding with results from others with this model (19). Metastases were mainly localized in jaws, skull, sternum, spine, shoulders, hips, and lower limbs. Neither micro-CT nor 89Zr-fresolimumab small-animal PET visualized any of the metastases detected by BLI. Only 1 mouse showed 89Zr-fresolimumab uptake at a site suggestive of metastasis (Fig. 4). Ex vivo 89Zr-fresolimumab biodistribution was similar to that found in CHO and MDA-MB-231 xenograft models, with high liver uptake of 89Zr-fresolimumab (data not shown). No toxicity of 89Zr-fresolimumab was seen in any of these mice.

Representative example of bioluminescence and small-animal PET/CT images of mouse in which bone metastatic MDA-MB-231-SCP2luc cells were injected into left ventricle of heart in model of disseminated metastasis. Metastases were visible with BLI in jaws, skull, sternum, spine, shoulders, hips, and lower limbs.
DISCUSSION
In the present study we describe, for what is to our knowledge the first time, the development, quality control, and preclinical validation of 89Zr-fresolimumab for noninvasive PET of tumor and organ distribution of fresolimumab.
Development and quality control of 89Zr-fresolimumab provided results similar to what we had seen earlier with the 89Zr labeling of antibodies directed at other targets (17,23), indicating the robustness of this labeling method. Small-animal PET with 89Zr-fresolimumab showed tumor uptake in CHO xenografts and in MDA-MD-231 xenografts. Remarkably high 89Zr-fresolimumab uptake was seen in sites of tumor ulceration in 2 mice and in scar tissue of 2 other mice—processes in which TGF-β is involved (24).
Our study showed for 89Zr-fresolimumab a distribution comparable to 111In-IgG in most organs, except for a higher uptake in liver and kidneys. This increased uptake in nontumor organs was not seen previously with VEGF-directed 89Zr-bevacizumab and human epidermal growth factor receptor-2–directed 89Zr-trastuzumab (17,23). 89Zr-fresolimumab liver uptake was especially increased when higher fresolimumab doses were used. The higher 89Zr-fresolimumab kidney uptake was seen only in the low-dose 89Zr-fresolimumab group of 10 μg. High liver uptake of 89Zr-fresolimumab likely is the result of a specific, TGF-β–driven, interaction between 89Zr-fresolimumab and TGF-β in the liver and would thus indicate high levels of active TGF-β in the liver. Our analysis of liver homogenates did not show high levels of the active TGF-β1 form. However, immunohistochemical staining for pSmad2 of liver tissues indicated that active TGF-β was present in the liver within hours before tissue collection (25). This presence of active TGF-β has probably caused the accumulation of 89Zr-fresolimumab in the liver and can be the consequence of rapid hepatic clearance of active TGF-β from the circulation and subsequent lysosomal degradation (26). This might mean that human TGF-β from CHO or MDA-MD-231 tumors will on activation be rapidly cleared by the liver, in which it accumulates; is recognized by 89Zr-fresolimumab; and results in 89Zr-fresolimumab accumulation in the liver. In addition to tumor-derived human TGF-β, activated mouse TGF-β from nontumor origin likely will accumulate in the liver and result in high 89Zr-fresolimumab liver uptake, because fresolimumab also binds to mouse TGF-β with high affinity. Another reason for the high liver uptake might be complex formation of 89Zr-fresolimumab with active TGF-β in the tumor microenvironment and in the circulation, subsequently followed by hepatic clearance of this complex. Altogether, these results indicate a TGF-β–specific biodistribution of 89Zr-fresolimumab. Additionally, the high 89Zr-fresolimumab liver uptake also matches strikingly the available preclinical data on the biodistribution of 111In-decorin. Decorin, a small proteoglycan from the extracellular matrix, binds TGF-β with high affinity. Intravenous injection of 111In-decorin into mice showed rapid hepatic clearance, especially by accumulation in nonparenchymal cells (27). The high liver uptake of 89Zr-fresolimumab reported in this preclinical study indicates the relevance of clinical 89Zr-fresolimumab imaging studies to explore not only fresolimumab tumor uptake but also its organ distribution. Increased kidney uptake of 89Zr-fresolimumab likely does not represent 89Zr-fresolimumab uptake caused by high TGF-β kidney levels but uptake of 89Zr-fresolimumab catabolites from hepatic processing. Hepatic processing is often saturable and thus dose-dependent, thereby explaining why increased kidney uptake of 89Zr-fresolimumab catabolites was seen only in the lowest 89Zr-fresolimumab dose of 10 μg. The high bone uptake of 89Zr-fresolimumab, compared with 111In-IgG, can be the result of high TGF-β levels in bone (28). However, we cannot exclude that this is an artifact due to bone uptake of dissociated 89Zr because we have also seen high bone uptake of 89Zr-bevacizumab when compared with 111In-bevacizumab (17).
To study the specificity of 89Zr-fresolimumab tumor uptake, results were compared with 111In-IgG, showing that tumor uptake of 89Zr-fresolimumab was similar to 111In-IgG tumor uptake. Lack of specific, that is, TGF-β–driven, tumor uptake of 89Zr-fresolimumab in our models could be a consequence of the fact that fresolimumab binds selectively to the active form of TGF-β. Our ELISA analysis of TGF-β1 levels in tumor homogenates, as well as clinical ELISA data on tumor homogenates of gastric cancer patients (29), show that more than 90% of total TGF-β is present in its latent form, leaving little antigen for fresolimumab binding. Our CHO models were generated to produce latent TGF-β1, as this would more closely resemble natural conditions than would cells that produce active TGF-β1, because all normal and tumor cells produce TGF-β only in its latent form. However, specific accumulation of 89Zr-fresolimumab requires local activation of TGF-β1 in the tumor, and not all TGF-β–activating mechanisms necessarily result in the release of free, active TGF-β (30). In addition to the low levels of free, active TGF-β, also the biologic half-life of active TGF-β is within 2–3 min, much shorter than that of latent TGF-β (110 min) (31), making imaging of TGF-β with an antibody recognizing only the active form even more challenging. Obviously, this does not exclude the therapeutic potential of fresolimumab. Our imaging data clearly show tumor accumulation of fresolimumab, likely because of the enhanced permeability and retention effect (32), indicating that fresolimumab reaches the target site in high concentrations (6.1 ± 1.6 %ID/g of tumor over all mice). The presence of fresolimumab in the tumor microenvironment inhibits local activation of latent TGF-β, thereby reducing the stimulatory response of the tumor. Therefore, the amount of fresolimumab in the tumor, as visualized and quantified with 89Zr-fresolimumab PET, could be a predictor for outcome.
With respect to absence of specific preferential tumor uptake of 89Zr-fresolimumab, this tracer differed from other targeted antibody-based tracers we developed. During imaging of other soluble tumor targets, such as VEGF, we found a 2-fold increased uptake of 89Zr-bevacizumab in SKOV-3 xenografts versus control IgG (17). With 89Zr-trastuzumab human epidermal growth factor receptor-2 imaging in the same model, tumor uptake of 89Zr-trastuzumab was 5-fold higher than control IgG (33). However, we also now know that preclinical results can underestimate clinical findings, because with clinical imaging studies, we found with 89Zr-bevacizumab a higher tumor uptake than with 89Zr-trastuzumab (18,34). These superior differential results in the clinical setting may prove to be the case for 89Zr-fresolimumab as well, therefore supporting the further pursuit of TGF-β–specific clinical imaging. Furthermore, the lack of 89Zr-fresolimumab visualization of metastases in our model of breast cancer bone metastases is likely caused by the subresolution size (<2 mm) for small-animal PET of these lesions. These considerations further illustrate the potentials of clinical evaluation of 89Zr-fresolimumab for a complete understanding of fresolimumab distribution in cancer patients and addresses the value of fresolimumab in the treatment of metastatic cancer. The clear visualization of tumor ulcerations with 89Zr-fresolimumab, together with the high levels of TGF-β1 we measured with ELISA in these ulcerations, encourages the further investigation of the use of fresolimumab in inflammatory diseases such as pulmonary fibrosis.
CONCLUSION
89Zr-fresolimumab small-animal PET was shown to be preclinically feasible for imaging and quantification of fresolimumab tumor uptake and organ distribution. 89Zr-fresolimumab PET is ready for clinical evaluation and might contribute to the clinical development of fresolimumab. We will use this technique to quantify the tumor uptake of fresolimumab in patients with high-grade gliomas.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Guus van Dongen at the VU University Medical Center Amsterdam for providing N-sucDf-TFP. This study was supported by grants 2007-3739, 2009-4273, and 2010-4739 of the Dutch Cancer Society. Richard C. Gregory is an employee of Genzyme Corporation. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Nov. 9, 2011.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication May 6, 2011.
- Accepted for publication July 14, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Colocalized targeting of TGF-{beta} and PD-L1 by bintrafusp alfa elicits distinct antitumor responses
- TGF-{beta} Antibody Uptake in Recurrent High-Grade Glioma Imaged with 89Zr-Fresolimumab PET
- PET/CT-Derived Whole-Body and Bone Marrow Dosimetry of 89Zr-Cetuximab
- Interrogating Tumor Metabolism and Tumor Microenvironments Using Molecular Positron Emission Tomography Imaging. Theranostic Approaches to Improve Therapeutics
- Placental Growth Factor (PlGF)-Specific Uptake in Tumor Microenvironment of 89Zr-Labeled PlGF Antibody RO5323441
- Advances in Immuno-Positron Emission Tomography: Antibodies for Molecular Imaging in Oncology
- Molecular Imaging of Tumors with Radioactive Labeled Antibodies from Laboratory to the Clinic