


Abstract
Placental growth factor (PlGF) is a member of the proangiogenic vascular endothelial growth factor family, which is upregulated in many tumors. RO5323441, a humanized monoclonal antibody against PlGF, showed antitumor activity in human tumor xenografts. We therefore aimed to radiolabel RO5323441 and preclinically validate this tracer to study drug tumor uptake and organ distribution by PET imaging. 89Zr-RO5323441 was tested for stability and immunoreactivity in vitro. Methods: The tumor uptake and organ distribution for 10, 50, and 500 μg of 89Zr-RO5323441 was assessed in mice bearing human PlGF–expressing hepatocellular cancer (Huh7) xenografts or human renal cell carcinoma (ACHN) xenografts without detectable human PlGF expression. The effect of pretreatment with RO5323441 (20 mg/kg) on 89Zr-RO5323441 tumor uptake was analyzed in Huh7 xenografts. 111In-IgG served as a control for nonspecific tumor uptake and organ distribution. Cy5-RO5323441 was injected to study the intratumor distribution of RO5323441 with fluorescence microscopy. Results: 89Zr-RO5323441 showed a time- and dose-dependent tumor accumulation. Uptake in Huh7 xenografts at 10 μg of 89Zr-RO5323441 was 8.2% ± 1.7% injected dose (ID)/cm3 at 144 h after injection, and in ACHN xenografts it was 5.5 ± 0.3 %ID/cm3 (P = 0.03). RO5323441 pretreatment of Huh7 xenograft–bearing mice reduced 89Zr-RO5323441 tumor uptake to the level of nonspecific 111In-IgG uptake. Cy5-RO5323441 was present in the tumors mainly in the microenvironment. Conclusion: The findings show that RO5323441 tumor uptake is PlGF-specific and time- and dose-dependent.
Angiogenesis is a key feature of tumors (1). Inhibition of angiogenesis by targeting vascular endothelial growth factor (VEGF) and VEGF receptors (VEGFRs) has emerged as treatment for various tumor types (2). However, sustained clinical benefit of angiogenesis inhibitors can be hampered by compensatory mechanisms such as the upregulation of proangiogenic factors such as placental growth factor (PlGF) (2–5). Targeting PlGF could, thus, be a new strategy for tumor angiogenesis inhibition, complementary to VEGFR inhibition, and might circumvent resistance observed during current antiangiogenic therapies.
PlGF, a VEGF homolog, can be expressed by tumor cells and is expressed by several other cells (e.g., endothelial and smooth muscle cells, macrophages, fibroblasts, and leukocytes) in the tumor microenvironment (4). PlGF is present in low levels in normal tissue. It contributes to the angiogenic switch in pregnancy, wound healing, ischemic conditions, and tumor growth (6). PlGF inhibition preclinically slows down growth and metastasis of various tumors, including those resistant to VEGFR inhibitors, and enhances the efficacy of chemotherapy and VEGFR inhibitors. Additionally, PlGF inhibition reduces angiogenesis, lymphangiogenesis, and tumor cell motility. Distinct from VEGFR inhibitors, PlGF inhibition preclinically prevents infiltration of angiogenic macrophages and severe tumor hypoxia and, thus, does not initiate the angiogenic rescue program responsible for resistance to VEGFR inhibitors (7,8).
These findings led to the clinical development of RO5323441 (TB-403) (9,10). RO5323441 is a humanized monoclonal antibody against PlGF-1 and PlGF-2. In a phase I trial with RO5323441 in cancer patients, stable disease was observed in 6 of 23 patients at different dose levels. No dose-limiting toxicities were observed with doses of up to 30 mg/kg administered once every 3 wk. Because no maximum-tolerated dose could be defined (11), determination of the optimal dose for phase II studies was hampered. Rational dosing might be obtained when tumor and normal tissue uptake of the antibody is defined with 89Zr-RO5323441 PET. The feasibility of this approach for visualization of soluble angiogenic factors was already shown for the VEGF-A antibody bevacizumab (12–14). The aim of the current study was therefore to study 89Zr-RO5323441 tumor uptake and organ distribution in human tumor xenograft models with different PlGF expression.
MATERIALS AND METHODS
Cell Lines
Human hepatocellular carcinoma Huh7 (Health Science Research Resources Bank) with high human PlGF (hPlGF) expression (hPlGF messenger RNA expression 62-fold higher than HEK293 native) and the human renal cell carcinoma ACHN (American Type Culture Collection) without detectable hPlGF expression (hPlGF messenger RNA expression equal to HEK293 native) were cultured in Dulbecco modified Eagle medium containing glucose (1 g/L) supplemented with 10% heat-inactivated fetal calf serum (Bodinco BV) at 37°C in humidified atmosphere containing 5% CO2.
The Abelson murine leukemia virus–induced tumor BALB-c–derived RAW264.7 murine macrophage cell line (American Type Culture Collection) was cultured in Dulbecco modified Eagle medium containing glucose (4.5 g/L) supplemented with 10% fetal calf serum and 1% l-glutamine at 37°C in humidified atmosphere containing 5% CO2.
Conjugation, 89Zr Labeling, and Quality Control of 89Zr-RO5323441
RO5323441 was conjugated and labeled as described by Verel et al. (15). In short, RO5323441 (25 mg/mL; Roche) was first conjugated with a 5-fold molar excess of the chelator N-succinyldesferrioxamine-tetrafluorphenol (N-sucDf-TFP; provided by Dr. Guus van Dongen, VU University Medical Center). N-sucDf-RO5323441 was purified by ultracentrifugation using a 30-kDa Vivaspin-2 filter (Sartorius), diluted in water for injection (2.5 mg/mL), and stored at −20°C. Labeling was performed with 89Zr (IBA) within 24 h before use. Radiochemical purity was evaluated by size-exclusion high-performance liquid chromatography using a Superdex 200 10/300 GL column (GE Healthcare) and showed no aggregates, fragments, free 89Zr, or other impurities (Supplemental Fig. 1A; supplemental materials are available online only at http://jnm.snmjournals.org). 89Zr-RO5323441 stability was evaluated at 4°C in solvent (0.9% NaCl) and at 37°C in phosphate-buffered saline (140 mMNaCl, 9 mM Na2HPO4, and 1.3 mM NaH2PO4; pH 7.4) and in human serum by trichloroacetic acid precipitation. 89Zr-RO5323441 was highly stable in 0.9% NaCl at 4°C and in phosphate-buffered saline or human serum at 37°C with less than 5% 89Zr release over 168 h in all tested conditions. The preservation of immunoreactivity was tested in a competition assay with unlabeled RO5323441 with recombinant hPlGF-1 (Peprotech) as the target antigen, according to the assay previously described (16). The RO5323441 concentration required for 50% reduction in hPlGF-1 binding of 5 nM 89Zr-RO5323441 was 5.1 ± 1.5 nM RO5323441, showing a fully preserved immunoreactivity (Supplemental Fig. 1B). 111In-IgG was produced as described previously (16).
Generation of Cy5-Labeled RO5323441
Purified RO5323441 was incubated in 100 mM potassium phosphate, pH 8.5, with a 5-fold molar excess of Cy5 N-hydroxysuccinimide ester (GE Healthcare Life Sciences) dissolved in dimethyl sulfoxide. The reaction was stopped after 60 min by the addition of 10 mM l-lysine, and the surplus of the labeling reagent was removed by dialysis against 20 mM histidine, 200 mM sodium chloride, 5% saccharose, pH 6.0. The Cy5 labeling degree was determined at an absorbance of 280 and 650 nm and was 2.7:1. The specificity of Cy5-labeled RO5323441 was analyzed by confocal fluorescence microscopy using recombinant Hek293 cells expressing membrane-tagged PlGF. Cy5-labeled RO5323441 was bound to the surface of the cells expressing membrane-anchored PlGF. The specificity was confirmed by competition with unlabeled RO5323441.
Animal Studies
All invasive procedures and imaging in animal experiments were performed with isoflurane inhalation anesthesia (induction, 5%; maintenance, 2%). Tumor cells were harvested by trypsinization, resuspended in culture medium and Matrigel (BD Biosciences), and inoculated subcutaneously (5 × 106 per mouse) in 6- to 8-wk-old male athymic nude mice (BALB-c/Ola HSD-fox nude; Harlan). Each subgroup consisted of 4 mice. Tumor growth was assessed 2 times per week with caliper measurement. When tumors measured 6–8 mm in diameter (∼0.3 cm3), in vivo imaging studies were started using a small-animal PET Focus 220 rodent scanner (CTI Siemens).
Huh7 xenograft–bearing mice were coinjected in the penile vein with 10, 50, or 500 μg of 89Zr-RO5323441 (5 MBq) and corresponding protein doses of 111In-IgG (1 MBq). Coinjection with 111In-IgG provides insight into behavior of an intraanimal nonspecific control for RO5323441 tumor uptake and organ distribution, improves the statistical power, and reduces animal numbers. ACHN xenograft–bearing mice and non–tumor-bearing mice were coinjected with 89Zr-RO5323441 (10 μg; 5 MBq) and 111In-IgG (10 μg; 1 MBq). In the pretreated group, mice were injected intraperitoneally with RO5323441 (20 mg/kg) 2 d before 89Zr-RO5323441 injection (10 μg; 5 MBq). Animals were imaged at 24, 72, and 144 h after injection, followed by ex vivo biodistribution analysis as described previously (16).
For microscopic analysis of tumor localization of RO5323441, Huh7 and ACHN xenograft–bearing mice were injected intravenously with 50 μg of Cy5-RO5323441, followed by tumor excision at 24 h after injection. In vivo stability of Cy5-RO5323441 was confirmed by near-infrared fluorescence imaging, which indicated accumulation in Colo205 tumors over 48 h without specific accumulation in the liver or kidneys.
All animal experiments were approved by the animal experiments committee of the University of Groningen.
Tumor Tissue Analyses
Tumors were kept on ice during biodistribution analysis and subsequently processed for histology and fluorescence microscopy. Hematoxylin and eosin staining was performed on formalin-fixed, paraffin-embedded tumors to assess tumor morphology. For fluorescence microscopy, tumor slides were stained for cell nuclei with Hoechst 33258 (Invitrogen). Macrophages were visualized by a 2-step staining with goat-antimouse CD68 (Santa Cruz) and donkey-antigoatIgG-Alexa488 (Invitrogen). Slides were analyzed with a Leica DM6000B microscope, and images were captured with a DFC360FX camera (Leica) and processed with LAS-AF2 software (Leica).
Fresh-frozen tumors were lysed and analyzed for hPlGF and murine PlGF (mPlGF) protein levels using an enzyme-linked immunosorbent assay (ELISA; R&D Systems) according to the manufacturer’s protocol.
89Zr-RO5323441 Cell-Binding Studies
Binding of 89Zr-RO5323441 to the human Huh7 and ACHN cell lines and the murine RAW264.7 macrophage tumor cell line was studied in the presence or absence of hPlGF-1 (Peprotech) or mPlGF-2 (R&D Systems). 89Zr-RO5323441 (0.22 nM; 10 kBq) was incubated for 24 h at 37°C with 1 × 106 cells per well. During incubation, PlGF (human or murine) was absent or added in a 10:1, 1:1, and 1:10 molar ratio with RO5323441. An Fc receptor blocking reagent (20 μL; no. 130-059-901 [MiltenyiBiotec]) was used to block the Fcγ receptors (FcγR) in RAW264.7. After incubation, cells were washed before harvesting, and the cell-bound 89Zr was measured in a well-type γ-counter (LKB Wallac). All conditions were tested in 3 independent experiments.
Statistical Analysis
Data are presented as mean ± SD. For statistical analysis, GraphPad Prism (version 4.00 for Windows; GraphPad Software) was used. Statistical analysis was performed using the 2-tailed unpaired t test. A P value of less than 0.05 was considered significant.
RESULTS
89Zr-RO5323441 Uptake in hPlGF-Expressing Huh7 Xenografts
In Huh7 tumors, 89Zr-RO5323441 uptake was seen at all dose cohorts, and uptake increased over time, indicating specific tumor uptake. The 144-h scan for the 10-μg 89Zr-RO5323441 dose cohort showed the best tumor visualization, with a higher tumor-to-background ratio than the 50- and 500-μg 89Zr-RO5323441 dose cohorts (Fig. 1A).

(A) Representative small-animal PET examples of Huh7 xenografts injected with 10, 50, or 500 μg of 89Zr-RO5323441 and scanned at 24, 72, and 144 h after tracer injection. (B and C) Small-animal PET quantification of 89Zr-RO5323441 blood-pool activity (B) and tumor uptake (C).
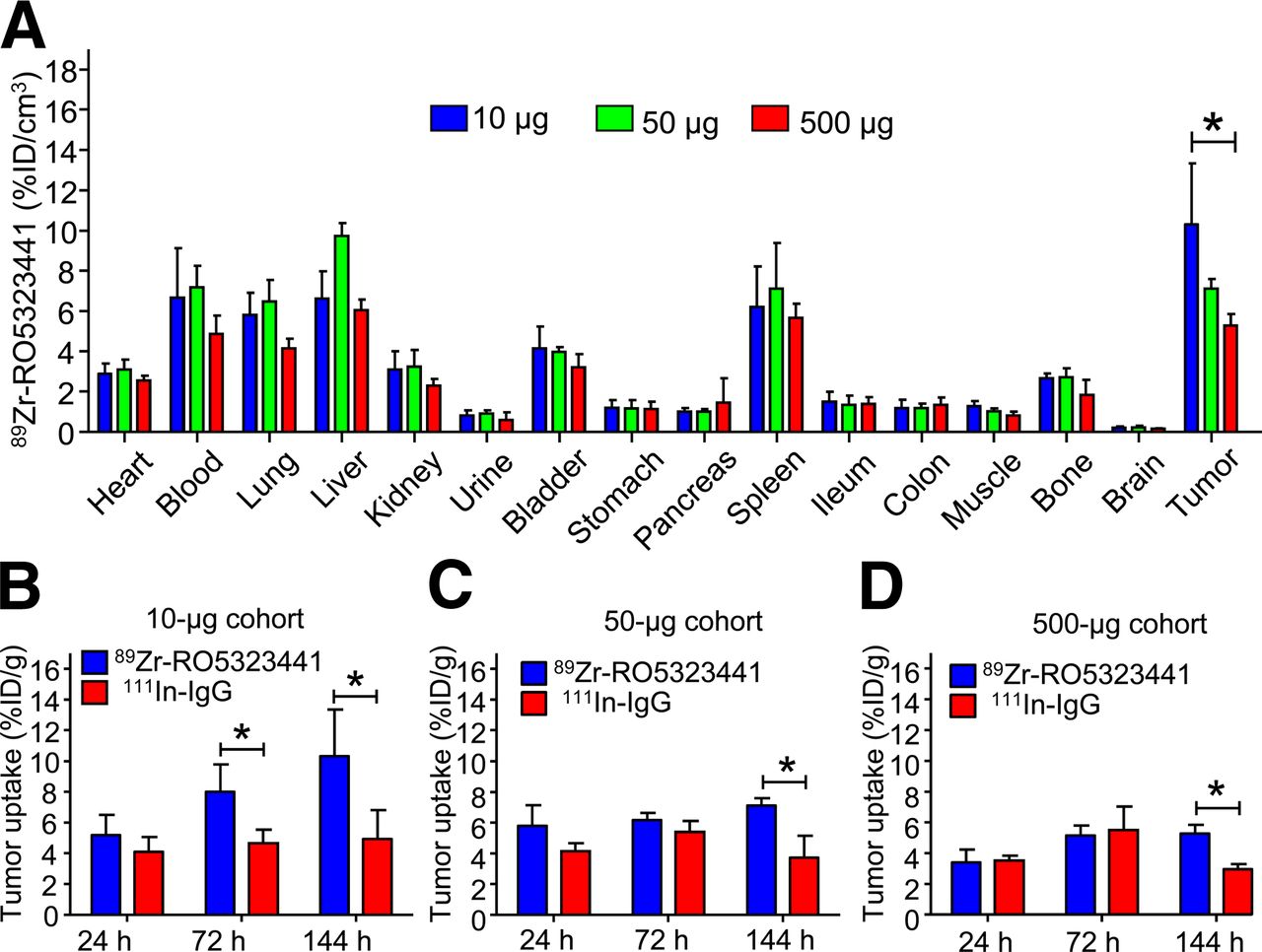
Quantification of the 89Zr-RO5323441 presence in the small-animal PET scans showed that 89Zr-RO5323441 blood-pool levels were similar for the 3 dose cohorts, with a decrease between 24 and 144 h (Fig. 1B). 89Zr-RO5323441 tumor uptake increased over time at all protein dose cohorts; the highest percentage injected dose per centimeter cubed (%ID/cm3) in tumors occurred in the 10-μg cohort at 144 h. For the 50- and 500-μg cohorts, maximal tumor uptake was observed at 72 h and remained the same at 144 h (Fig. 1C). The absolute 89Zr-RO5323441 tumor uptake levels were 0.8 ± 0.2, 3.1 ± 0.2, and 27.4 ± 3.98 μg/cm3 for, respectively, the 10-, 50-, and 500-μg injected dose cohorts. For 89Zr-RO5323441, the quantified small-animal PET data show a non–dose-dependent blood clearance and a time- and dose-dependent tumor uptake.
Ex vivo biodistribution analysis in Huh7 xenograft–bearing mice confirmed a normal IgG distribution of 89Zr-RO5323441 in nontumor organs. This distribution was hardly RO5323441 protein dose-dependent. In contrast, relative 89Zr-RO5323441 tumor uptake showed a clear RO5323441 dose-dependent decrease in %ID per gram (%ID/g) of tumor (Fig. 2A). In the 10-μg cohort, 89Zr-RO5323441 tumor uptake was, respectively, 72% (P = 0.016) and 109% (P = 0.023) higher than 111In-IgG at 72 and 144 h after tracer injection (Fig. 2B). At 144 h, tumor uptake of 89Zr-RO5323441 in the 50-μg cohort was 91% (P = 0.0041) and in the 500-μg 79% (P = 0.0004) higher than for 111In-IgG (Figs. 2C and 2D). These biodistribution data confirm the time- and dose-dependent 89Zr-RO5323441 tumor uptake in Huh7 xenografts observed with the small-animal PET quantification.

(A) 89Zr-RO5323441 biodistribution in Huh7 xenografts at 144 h after tracer injection in all 3 dose cohorts. (B–D) 89Zr-RO5323441 and 111In-IgG tumor uptake of 10- (B), 50- (C), and 500-μg (D) cohorts.
On the basis of the high tumor-to-nontumor contrast on small-animal PET images, the tumor accumulation over time shown by small-animal PET scan quantification and biodistribution data, and the high ratio with 111In-IgG observed in our biodistribution analysis, 10 μg was chosen as the 89Zr-RO5323441 protein dose for further experiments.
89Zr-RO5323441 Biodistribution in Non–Tumor-Bearing Mice
For non–tumor-bearing mice, organ distribution of 10 μg of 89Zr-RO5323441 was similar to that of 111In-IgG (Supplemental Fig. 2). There was no specific accumulation of 89Zr-RO5323441 over time in most nontumor organs, except for the liver (+26% from 24 to 144 h after injection; P = 0.027), and a nonsignificant trend in bone. Liver and bone uptake were possibly the result from binding to scavenging receptors in the liver or could indicate in vivo tracer metabolism to 89Zr-labeled antibody fragments and release of free 89Zr, which could accumulate in bone. Organ distribution of 89Zr-RO5323441 was similar in non–tumor- and tumor-bearing mice.
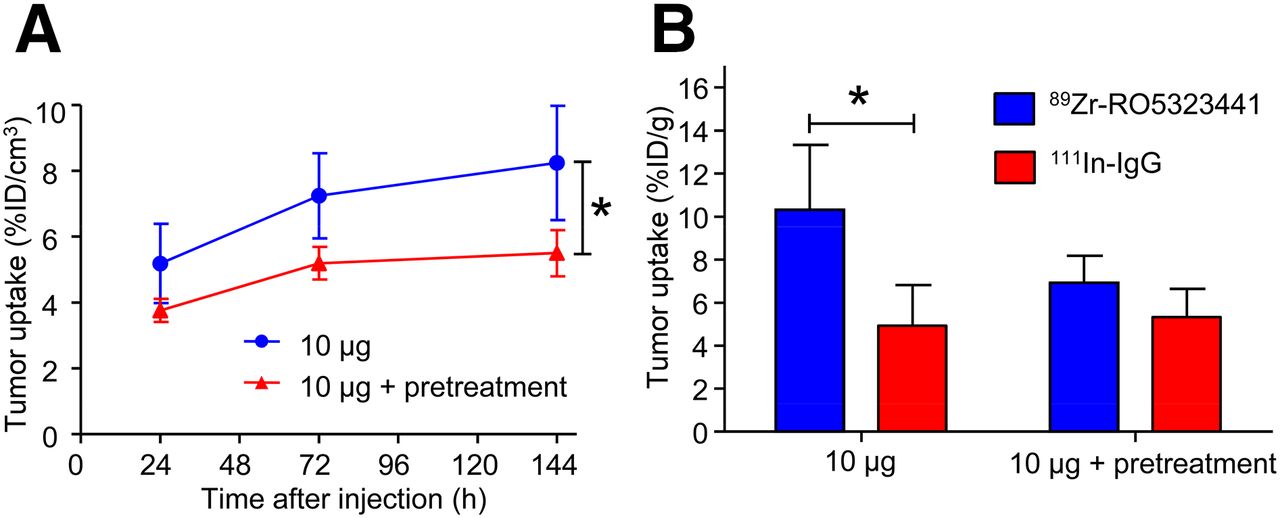
RO5323441 Pretreatment Reduces 89Zr-RO5323441 Uptake in Huh7 Tumors
Small-animal PET quantification showed that a 20 mg/kg pretreatment of RO5323441 reduced the tumor uptake of 10 μg of 89Zr-RO5323441 by 33% (P = 0.026) (Fig. 3A). 89Zr-RO5323441 tumor uptake in RO5323441-pretreated mice was comparable to the tumor uptake in the 500-μg 89Zr-RO5323441 dose cohort. Biodistribution data showed that pretreatment with RO5323441 in Huh7 xenografts reduced 89Zr-RO5323441 tumor uptake to background levels—that is, not higher than 111In-IgG (Fig. 3B)—confirming the small-animal PET quantification data. Biodistribution of 89Zr-RO5323441 in nontumor organs was not influenced by RO5323441 pretreatment (data not shown).
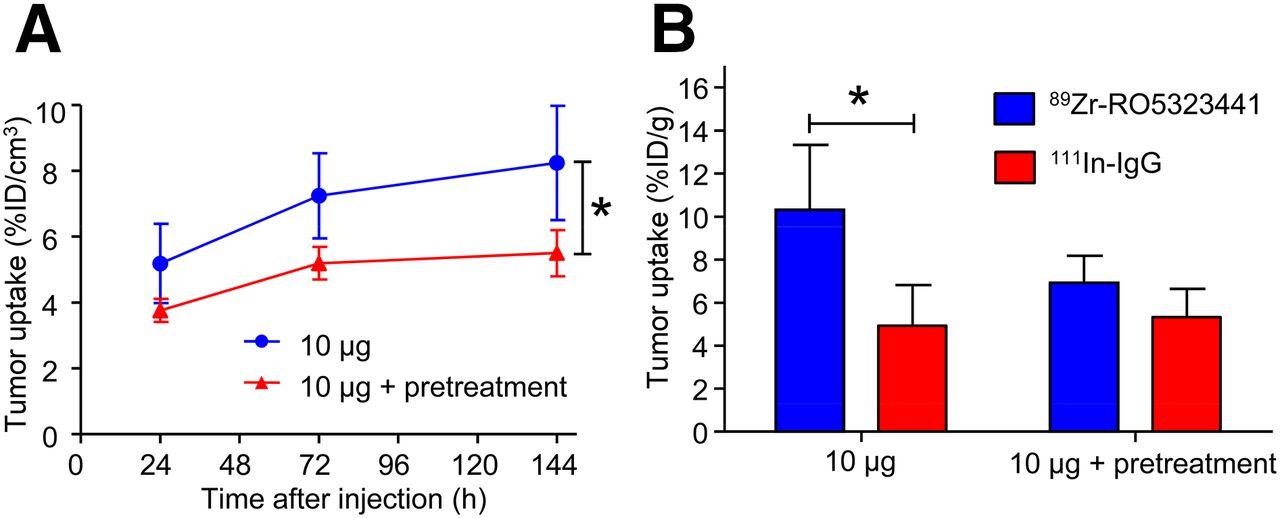
Influence of RO5323441 pretreatment on 89Zr-RO5323441 tumor uptake in Huh7 xenografts quantified from small-animal PET data (A) and biodistribution data at 144 h after tracer injection (B).
89Zr-RO5323441 Uptake in ACHN Tumors That Do Not Express hPlGF
Small-animal PET scans showed that 89Zr-RO5323441 tumor uptake in ACHN tumors was 33% lower (P = 0.021) than in Huh7 tumors at 144 h after injection (Fig. 4A). Biodistribution results revealed that 89Zr-RO5323441 uptake in ACHN tumors was comparable to 111In-IgG uptake. The ratio of 89Zr-RO5323441 to 111In-IgG in Huh7 tumors was higher than the ratio in ACHN tumors at all time points: 46% at 24 h (P = 0.0003), 61% at 72 h (P = 0.023), and 89% at 144 h (P = 0.0005) after tracer injection (Fig. 4B). These results indicate PlGF tumor level specific uptake in Huh7 tumors.
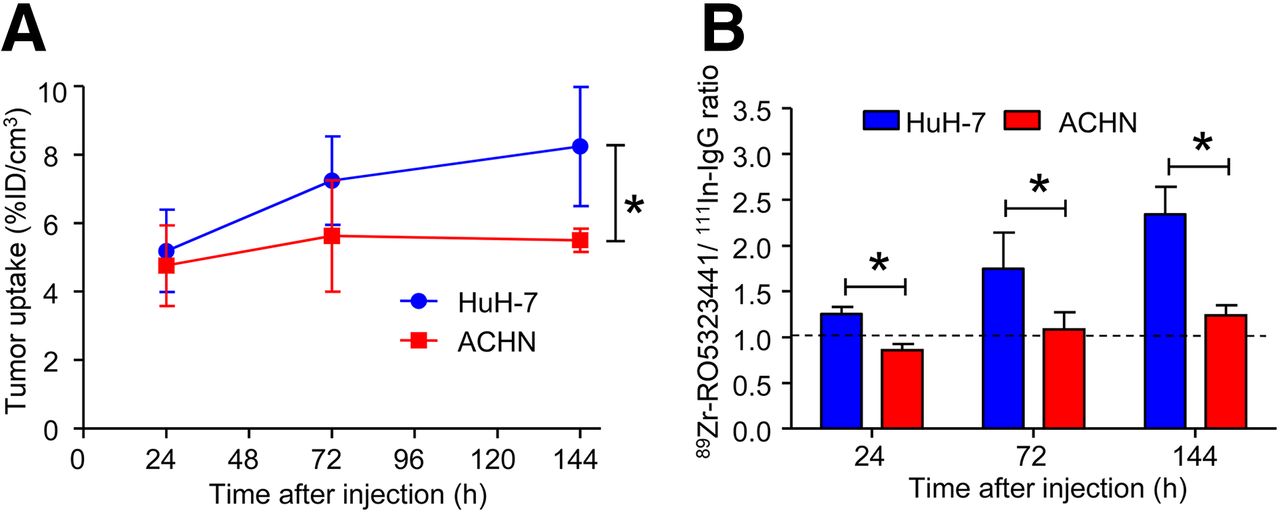
89Zr-RO5323441 uptake in ACHN and Huh7 tumors as assessed by small-animal PET (A) and biodistribution (B).
Ex Vivo Analysis of Huh7 and ACHN Tumors
Tumor histology of Huh7 and ACHN xenografts obtained from the 10-µg 89Zr-RO5323441 cohorts at 24, 72, and 144 h after injection showed areas of tumor cells, stromal cells, vasculature, and some necrotic areas (Fig. 5A shows representative hematoxylin and eosin staining).
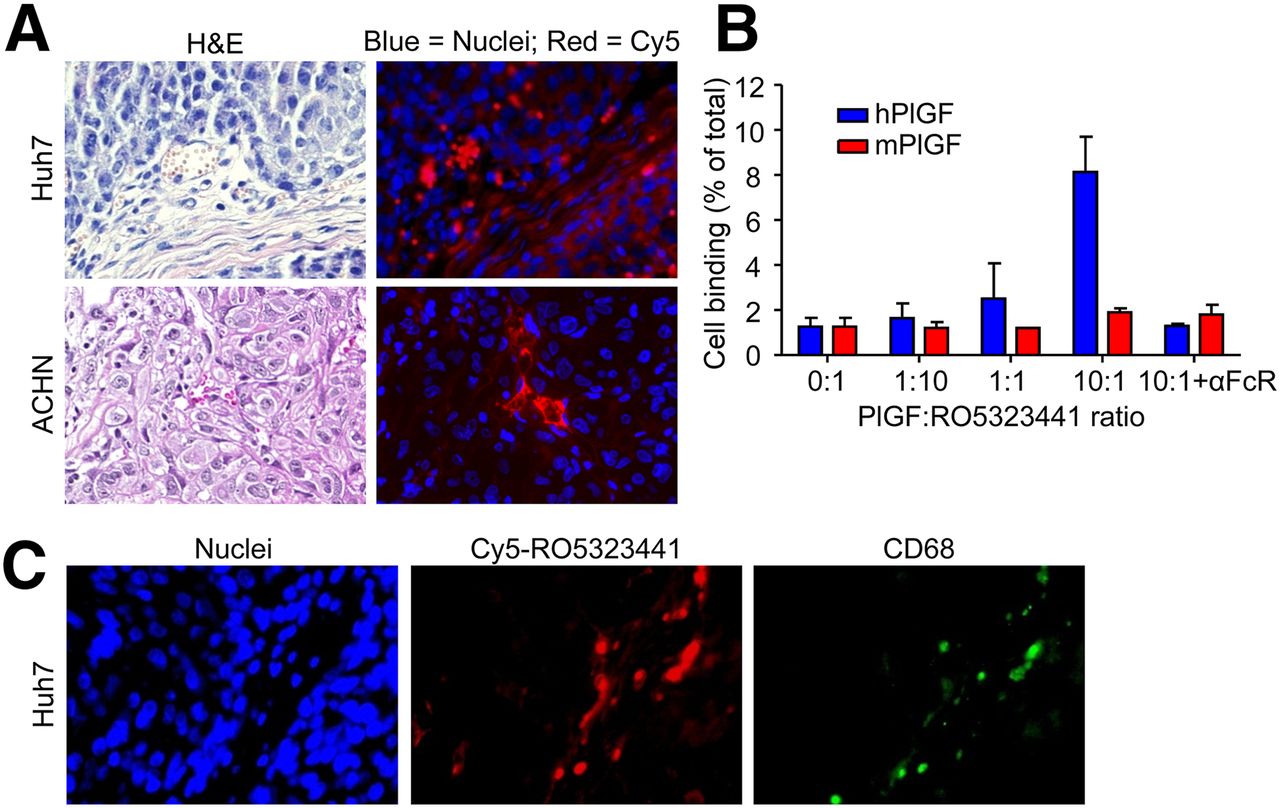
(A) Representative examples of Huh7 and ACHN histology, as assessed by hematoxylin and eosin (H&E) staining, and Cy5-RO5323441 microscopic tumor distribution, as assessed by fluorescence microscopy. (B) 89Zr-RO5323441 binding to RAW264.7 macrophages after 24 h incubation with hPlGF or mPlGF and an Fc receptor blocking reagent (αFcR). (C) Hoechst staining visualizes cell nuclei. Costaining of Huh7 tumor with anti-CD68 antibody to visualize macrophages.
Fluorescence microscopy revealed that in Huh7 tumors, Cy5-RO5323441 accumulated in areas of tumor cells and in necrotic regions, vasculature, and the surrounding connective tissue (Fig. 5A). In ACHN tumors, Cy5-RO5323441 accumulated only in the surrounding connective tissue, necrotic regions, and vasculature but not in the areas of tumor cells. Costaining of the Huh7 tumor slides with an anti-CD68 antibody to visualize macrophages indicated that Cy5-RO5323441 was present in the CD68-positive regions and in the vasculature (Fig. 5B).
hPlGF protein levels measured with ELISA were 153 ± 62 pg/mg of protein in Huh7 xenografts and nondetectable in ACHN xenograft tissue. Murine PlGF levels were 253 ± 177 pg/mg of protein in Huh7 and 1,063 ± 207 pg/mg of protein in ACHN xenograft tissue.
89Zr-RO5323441 Binding to RAW264.7 Macrophages
To further study the mechanism by which cells, such as macrophages, that are in a microenvironment might be involved in RO5323441 tumor uptake, we studied the uptake of 89Zr-RO5323441 by RAW264.7 murine tumor macrophages in vitro. The binding of 89Zr-RO5323441 to RAW264.7 concentration dependently increased in the presence of hPlGF, with a 6.4-fold increase (P = 0.0018) at a 10-fold molar excess of hPlGF (Fig. 5C). The hPlGF-induced macrophage binding of 89Zr-RO5323441 was completely blocked by coincubation with a Fc receptor blocking reagent. mPlGF was not able to induce binding of 89Zr-RO5323441 to RAW264.7 macrophages, indicating the higher affinity of RO5323441 for hPlGF.
The binding of 89Zr-RO5323441 to Huh7 cells was 2-fold higher than binding to ACHN cells (P = 0.0030, Supplemental Fig. 3), possibly due to the autocrine production of heparin-binding hPLGF by Huh7. 89Zr-RO5323441 binding to Huh7 and ACHN was not affected by the non–heparin-binding hPlGF-1 isoform. The addition of mPlGF, which also binds heparin, induced a slight increase in both cell lines (1.6- and 1.9-fold, respectively).
DISCUSSION
In this study, we determined 89Zr-RO5323441 tumor uptake and organ distribution in human tumor–bearing mice. We used 3 different tracer doses and 3 different imaging and biodistribution time points, 2 tumor models with different target expression levels, an unspecific IgG control, and a RO5323441 pretreatment dose. All together, these data showed that 89Zr-RO5323441 tumor uptake was time-, dose-, and PlGF-dependent, supporting the feasibility of implementing the use 89Zr-RO5323441 with PET scanning for clinical studies.
Because this is the first study, to our knowledge, on in vivo visualization of PlGF, the best possible comparison is with results obtained with 89Zr-labeled bevacizumab. For optimal comparison, the antibody tracer dose should be considered. 89Zr-bevacizumab showed a SKOV-3 tumor uptake of 6.8 ± 1.8 %ID/g with 100 μg of antibody (12), which is similar for 89Zr-RO5323441 tumor uptake. Higher levels of 89Zr-RO5323441 tumor uptake, expressed at %ID/g, might have been obtained when lower antibody doses would have been used. 111In-bevacizumab biodistribution studies showed uptake levels of up to 25 %ID/g for a 1-μg bevacizumab dose (17). In the present study, we used 10 μg of 89Zr-RO5323441 because this was the lowest dose that allowed sufficient counts for PET quantification at 6 d after injection. It would have been possible to perform a biodistribution-only experiment with a lower dose (e.g., 1 µg), and this would likely have resulted in a higher tumor uptake when expressed as %ID/g. However, ex vivo biodistribution studies are nonsequential and not translational, and we aimed to validate 89Zr-RO5323441 as an imaging tool. Overall, 89Zr-RO5323441 PlGF PET preclinical performance is comparable to that of 89Zr-bevacizumab VEGF PET.
Fluorescence-labeled RO5323441 was present in the tumor microenvironment at 24 h after injection. Our mouse models were engrafted with human Huh7 and ACHN tumors, and therefore the microenvironment of these human tumor xenografts consisted of human tumor cells and murine stroma. This microenvironment, as shown with ELISA, contained both hPlGF and mPlGF. RO5323441 binds both hPlGF and mPlGF, with a Biacorekoff rate of 80 and 35 min, respectively, indicating a more tight binding to hPlGF than mPlGF (data not shown). Additionally, RO5323441 binding experiments with RAW264.7 cells support the higher affinity for binding to hPlGF. This higher affinity explains the PlGF-specific uptake of 89Zr-RO5323441 in the hPlGF-overexpressing Huh7 tumors. As expected, binding of 89Zr-RO5323441 to Huh7 and ACHN tumor cells in vitro was low. mPlGF induced a small increase in 89Zr-RO5323441 binding to Huh7 and ACHN cells. One explanation could be that we used hPlGF isoform 1 as hPlGF and mPlGF isoform 2 as mPlGF, because mice only express this isoform. One of the differences between PlGF1 and PlGF2 is that PlGF1 lacks a heparin-binding domain, which is necessary for binding to the neuropilin-1 coreceptor (18). mPlGF2 therefore could have been bound to Huh7 and ACHN cells via heparin, allowing binding of 89Zr-RO5323441.
The in vitro experiments with RAW264.7 macrophages suggest that 89Zr-RO5323441 forms immune complexes preferably with hPlGF. The 89Zr-RO5323441 and hPlGF complex in turn binds to the Fcγ receptors on macrophages, followed by phagocytosis of the immune complex; this is especially supported by the finding that 89Zr-RO5323441 itself shows little interaction with macrophages. Only in the presence of hPlGF is there hPlGF concentration–dependent 89Zr-RO5323441 binding. Because hPlGF is the target of RO5323441, competition of RO5323441 binding would intuitively be expected when hPlGF is added and thus decreased cellular binding. However, our results and those of others (19) provide evidence for the Fcγ receptor–mediated macrophage uptake of IgG immune complexes, such as the complex of RO5323441 with hPlGF. This evidence can be explained by the fact that macrophages express Fcγ receptor subtypes II and III (FcγRII and FcγRIII), which have a low affinity for monomeric IgG. However, FcγRII and FcγRIII interact with high avidity with IgG immune complexes, with consequential selective phagocytosis of IgG immune complexes (19). The relevance of Fcγ receptor interactions of antibody–antigen immune complexes in the tumor accumulation of soluble antigen-targeted antibodies such as RO5323441 is not exactly clear yet. Our Cy5-RO5323441 ex vivo data and 89Zr-RO5323441 in vitro data, however, indicate that PlGF-specific tumor uptake of RO5323441 might be modulated by macrophages. The Fcγ receptor–mediated uptake of antibody–antigen complexes is rather generic. It is therefore unlikely that this mechanism is specific for the RO5323441–PlGF complex. Tumor uptake of other soluble antigen-targeted antibodies such as the VEGF-A antibody bevacizumab might therefore also be dependent on immune complex formation. Bevacizumab–VEGF-A165 immune complexes can interact with platelets via FcγRIIA, potentially playing a role in the thrombotic events often seen during bevacizumab therapy (20).
Conflicting opinions exist on the significance of PlGF as a target for treatment in oncology (21). In preclinical models, PlGF plays a role in resistance to antiangiogenic therapies. PlGF inhibition showed antitumor activity in tumor models resistant to VEGFR inhibitors (7). Additionally, elevated serum levels of PlGF were reported in patients after antiangiogenic therapy with bevacizumab or sunitinib (22–24). In a preclinical study with the VEGF- and PlGF-neutralizing decoy receptor sFLT01, increased serum PlGF levels reflected a systemic host response instead of a tumor response. In addition, circulating PlGF was upregulated in mice with responding as well as progressing tumors (25). These findings suggest limited potential and utility of serum PlGF as a biomarker for antiangiogenic therapies. 89Zr-RO5323441 tumor uptake was found to be PlGF-dependent. Therefore, 89Zr-RO5323441 PET imaging might well be superior to measuring circulating PlGF levels to monitor PlGF tumor expression during antiangiogenic treatment. When the angiogenic rescue program is activated in the tumor, this will coincide with a local PlGF upregulation in the tumor microenvironment and an increased 89Zr-RO5323441 uptake and possibly support the rationale for combining RO5323441 with VEGFR inhibitors. The vasculature normalization effect of bevacizumab can hamper the tumor penetration of simultaneously injected large molecules (e.g., antibodies such as RO5323441) (26). PlGF PET with 89Zr-RO5323441 might therefore also be of value in the optimization of scheduling combined antiangiogenic treatments.
The therapeutic potential of PlGF inhibition recently has drawn additional attention. In contrast to the initially published antitumor effects of an anti-PlGF antibody, other PlGF-neutralizing antibodies had no significant effect on tumor growth and angiogenesis in several preclinical tumor models (27). There is no unifying explanation for these conflicting results; however, differences in physicochemical properties, posttranslational modifications, or other alterations may have influenced the efficacy of the different PlGF antibodies (8). Moreover, it was found that a functional VEGFR-1 in tumor cells is required for an antitumor effect of anti-PlGF antibodies (28). It is as of yet unclear how the antibody dose affects the efficacy (9). In the clinical phase I dose-escalation studies with RO5323441, no dose-limiting toxicities were found with doses of up to 30 mg/kg (10,11). It is possible that the maximum effective dose was already reached with optimal target saturation of its target. Insight in biodistribution of RO5323441 by quantification of 89Zr-RO5323441 tumor uptake may offer an interesting support for this dosing dilemma by serving as readout for target saturation by different doses of RO5323441. Our results showed that a 20 mg/kg dose of RO5323441 reduced the human tumor uptake of the 89Zr-RO5323441 tracer dose in mice to background 111In-IgG levels. These results likely reflect PlGF saturation by the treatment dose but could also be a consequence of RO5323441-induced vessel normalization. Vessel normalization, which is now widely acknowledged for bevacizumab, also occurs after PlGF inhibition (8,29). Dynamic contrast-enhanced MR imaging to monitor changes in tumor perfusion in the clinical evaluation of 89Zr-RO5323441 and RO5323441 could potentially be used to distinguish PlGF saturation effects and vessel normalization.
CONCLUSION
The performance of 89Zr-RO5323441 PET for PlGF detection is of interest because 89Zr-bevacizumab and 111In-bevacizumab have already proven to be valuable VEGF-A imaging tracers in clinical studies. The 89Zr-bevacizumab uptake in tumor lesions in renal cell cancer and melanoma patients is beyond even the expectations based on preclinical results (13,14). Given the extensive similarity between VEGF-A and PlGF, an enhanced 89Zr-RO5323441 tumor-to-background ratio versus findings in the xenograft model is expected also in the human setting. Because in the human setting both tumor cell– and stromal cell–derived PlGF are of human origin and will thus be recognized by 89Zr-RO5323441, it is possible that our preclinical findings with 89Zr-RO5323441 PET underestimate the tumor uptake that will be seen in cancer patients. In addition, the antibody tracer dose of 10 µg in a mouse of 25 g versus a typical dose of 5 mg in a human of 70 kg favors the antibody–antigen ratio in humans. To determine RO5323441 human tumor uptake and how this is affected by bevacizumab, we will quantify the tumor uptake of RO5323441 by serial 89Zr-RO5323441 PET scans in patients with recurrent glioblastoma (EudraCT no. 2011-004974-27).
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This study was supported in part by an ESMO Translational Research Fellowship, a Roche Post-Doc Fellowship, and grant RUG2007-3739 of the Dutch Cancer Society. K. Michael Weidner, Frank Herting, and Jean Tessier are employees at Roche. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
Mats Bergstrom, Celine Pallaud, and Martin Steegmaier contributed to the design, analysis, and interpretation of this study.
Footnotes
↵* Contributed equally to this work.
Published online Apr. 26, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 1, 2012.
- Accepted for publication December 11, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}