


Abstract
Microdosing as a regulatory concept was introduced to facilitate exploratory studies in humans. The concept involves the use of very low doses of a radionuclide-labeled compound for imaging studies or for assessing plasma pharmacokinetics using equipment that has a highly sensitive readout. The supporting principle is that use of these low doses for a limited time in well-controlled, small populations will limit exposure and have a low risk of adverse effects. Microdosing regulations specify a reduced preclinical toxicology-assessment package in order to shorten the route to human studies and reduce its cost. However, for extrapolation to therapeutically relevant doses and plasma concentrations, there are specific aspects of the use of these low doses and low plasma concentrations that require special attention. These specific aspects are reviewed in this article, with separate attention being paid to small organic molecules and protein therapeutics. The indications for microdosing in drug development are discussed in terms of the 3 pillars of survival in drug development, the first of which is characterization of tissue distribution and access to the site of action; the second, engagement of the target; and the third, induction of tissue responses relevant to a therapeutic response.
The term microdosing, which was introduced through the work of such pioneers as Garner and Lappin (1–3), indicates the potential to assay the plasma pharmacokinetics of a very low administered mass in humans using a highly sensitive means of recording the concentration–time profile. The term was additionally applied to radionuclide methods such as PET and SPECT, with their option of noninvasively recording the distribution of radiolabeled entities in organs or the entire body (4–6). For many imaging probes, use in humans was introduced as microdoses, but not always with the regulatory background of the microdosing concept. Several review articles have described microdosing in relation to accelerator mass spectrometry and PET (7,8).
The application of these new options was encouraged by regulatory agencies in Europe and the United States through documents suggesting the creation of a regulatory framework defining the preliminary toxicity and safety assessments required to take a new chemical entity into a human microdosing trial (9,10). After some important successive refinements, the regulatory framework for microdosing was accepted by the International Conference on Harmonization (11).
This regulatory framework has had great importance in facilitating human studies of new chemical entities and has been especially valuable for promoting new radionuclide imaging probes. Although microdosing has been associated predominantly with small organic molecules and site-specific labeling, there is growing interest in applying the concept to protein therapeutics and in using some of the exclusive properties of proteins for nuclear imaging purposes.
This article discusses the consequences of the facilitated regulations, the impact of performing studies at very low pharmacologic concentrations, and the differences in regulation and pharmacology between smaller (organic) molecules and larger (protein biologic) molecules.
PHARMACOLOGIC CONSIDERATIONS IN DEVELOPING A THERAPEUTIC DRUG
The microdosing study—whether a plasma pharmacokinetics study with a highly sensitive readout or an imaging study with measurement of organ distribution—may supply important information on pharmacology, but the results need to be carefully considered. A main obstacle is that the drug is intended to be used at pharmacologically relevant doses or plasma concentrations whereas the microdoses are at least 100-fold lower. Linearity in pharmacokinetics and distribution, meaning a direct dose-related scaling of the pharmacokinetics and distribution parameters, allows extrapolation from values obtained in a microdosing study to therapeutically relevant doses. However, such linearity is not always at hand.
Nonlinearity is typically introduced by processes such as binding and saturation of the target, as well as saturation of the enzyme and transport systems to which the drug is exposed. This also means that nonlinearity is typically expressed at concentration levels at which saturation occurs. Options for predicting and correcting some processes that induce nonlinearity have been proposed by Bosgra et al. (12), who suggested that nonlinearity in pharmacokinetics is relatively uncommon. However, this may not be true for compounds that target at very high affinities and for imaging studies that evaluate organ concentrations.
For example, a drug that has a high-affinity target expressed in large quantities somewhere in, or throughout, the body may clear from the plasma quickly when the plasma concentration is lower than affinity but may clear much more slowly at concentrations well above affinity. Such effects are often encountered with high-affinity proteins but are occasionally also seen with small organic molecules. Similarly, a microdosing imaging study may show a high concentration in a tissue with high target expression but may show a moderate tissue concentration if a nonradioactive drug is added at therapeutically relevant doses.
Another example is a study of blood–brain barrier penetration. Microdoses may show limited penetration due to efflux systems whereas higher doses or simultaneous exposure to another drug may saturate the efflux systems, thereby enabling blood–brain barrier penetration (13,14).
It is always advisable to perform preclinical studies with the aim of understanding whether nonlinearity in dose proportionality may exist, but proper correction for it may still be complicated in the in vivo study. The impact of binding to the specific target may, if desired and operationally feasible, be evaluated by performing two studies on the same individual: one with microdosing only and the other with microdosing of the tracer plus prior or simultaneous pharmacologic blockade of the target.
With radionuclide imaging, we can accurately and precisely define the concentration of the radiolabeled entity in tissues. In such studies, it is often suitable to define radioactivity concentration in terms of SUV with the readings (Bq/mL) as measured with the camera, divided by administered radioactivity (kBq) per body weight (kg), corrected for radioactive decay. There are two important reasons why this notation is suitable in drug-related studies, the first being that SUVs can be translated reasonably well from animals to humans, with an equal body distribution having an SUV of 1.0 in all species unless activity is lost from the body. The second reason is that drug concentration in μg/mL can be obtained as SUV times administered mass (mg) per body weight (kg), if radioactive metabolites have been properly corrected for or can be assumed negligible.
An inherent problem in radionuclide imaging studies is that even if the radioactivity concentration is measured well, the images do not reveal whether part of the signal is from radioactive metabolites of the tracer. Typically, the chemical identity of radioactivity in plasma (i.e., the native tracer and the radioactive metabolites) is evaluated through radio–high-performance liquid chromatography. This chemical identity may be evaluated in the preclinical setting, and through assumptions and modeling, reasonably good corrections may be applied to the clinical setting, depending on the magnitude of the radiometabolites and the mechanisms of their appearance.
Knowledge of therapeutically relevant tissue concentrations may guide dosing in a clinical trial or suggest the need to consider safety margins. However, average tissue concentrations measured with radionuclide imaging may differ considerably from concentrations at the cellular level, for several reasons (Fig. 1). First, some compounds may not enter the cells because of physiochemical properties such as being highly hydrophilic or because of active efflux systems. In these cases, the tissue concentration is the concentration in the extracellular space multiplied by the contribution of this space to the overall tissue. Second, especially at the low concentrations associated with microdosing, a significant portion of the radioactivity in tissue may be bound to its specific target. Third, many drugs and drug candidates in microdosing studies are chosen to be highly lipophilic, possibly as a means to allow cell entry. However, such compounds may preferentially accumulate in the lipid bilayer of the cell membrane, with the overall tissue concentration thus being a combination of high concentrations in cell membranes and low concentrations in extracellular fluid and intracellular space. Additionally, some compounds, notably weak bases, may accumulate in the acidic spaces of mitochondria and lysosomes. This phenomenon of so-called lysosomotropic compounds ensues when compounds that readily pass the cellular, mitochondrial, and lysosomal membranes undergo protonation in the highly acidic intermembrane space of the mitochondria or internal space of the lysosomes, with a pH on the order of 4–5 (15,16). The protonation leads to a loss of penetration; the compound cannot leave this space and continuously accumulates to concentrations that may be up to 1,000-fold higher than in other tissue subregions. Hence, the overall signal is a composite of tracer bound to the target; possibly, tracer bound to off-targets; and tracer distributed in other compartments in the tissue. Additional experiments may be needed to elucidate the individual contribution of each.
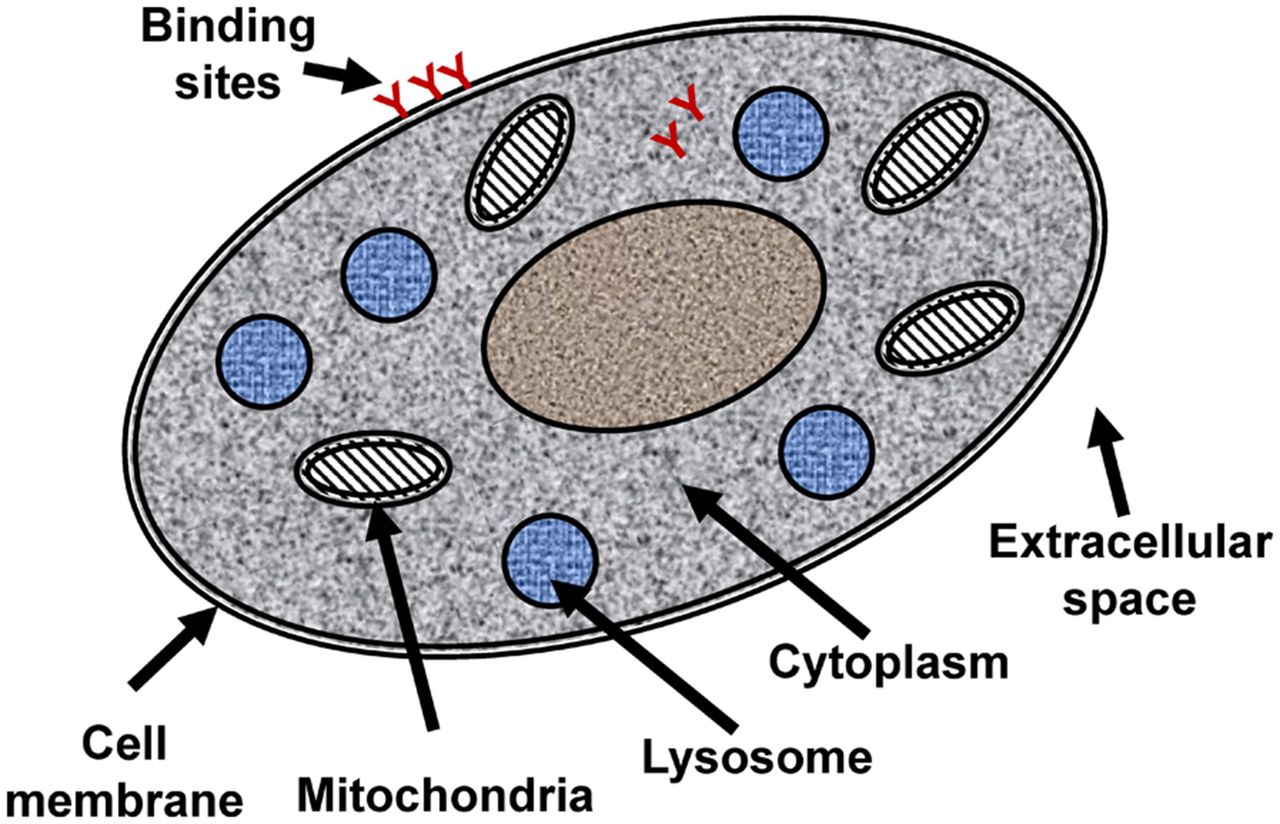
Schematic illustration of cell components that may be relevant to heterogeneous cellular distribution.
The possibility of highly heterogeneous tissue concentrations may make it more difficult to predict the degree of target engagement using tissue concentrations from an imaging study coupled with in vitro target affinity values or inhibition constants. A way to evaluate the relation between overall tissue concentration and target engagement may be to perform preclinical studies that include a heterogeneous tissue concentration, such as studies on a multicellular spheroid model or in vivo studies in animals.
To evaluate tissue concentration—especially specific target-binding parameters in a microdosing imaging study—the study should include a dynamic imaging sequence and parallel acquisition of blood samples for analysis of the fraction of radioactive metabolites in plasma. Tracer kinetic modeling supports the identification of compartments and their quantitative contribution to the tissue signal (17,18).
CONSIDERATIONS IN INTERPRETATING IMAGING STUDIES
Certain aspects of the pharmacology and pharmacokinetics of protein therapeutics, including antibodies, require special attention when imaging studies with such agents are being designed and interpreted. Because antibody clearance and distribution often span multiple days, the radionuclide must have a half-life long enough to allow assessment of time courses. Adequate radionuclides include 111In, 124I, and 89Zr, with half-lives of 2.8, 4.2, and 3.3 d, respectively. In addition, to capture images in which the signal is dominated by binding to the target, scanning needs to be delayed until a few days after the agent is administered, and to capture kinetic behavior, scanning on a few different days may be needed.
The plasma pharmacokinetics of antibodies usually comprises at least 3 phases, depending on the rate and degree of extravasation from plasma, the amount bound to targets throughout the body, and the fate of the antibody after target binding, such as internalization, recycling, and lysosomal degradation (19,20). The typical plasma pharmacokinetics for an antibody with a well-expressed target, clearance through internalization, and lysosomal degradation is illustrated in Figure 2.
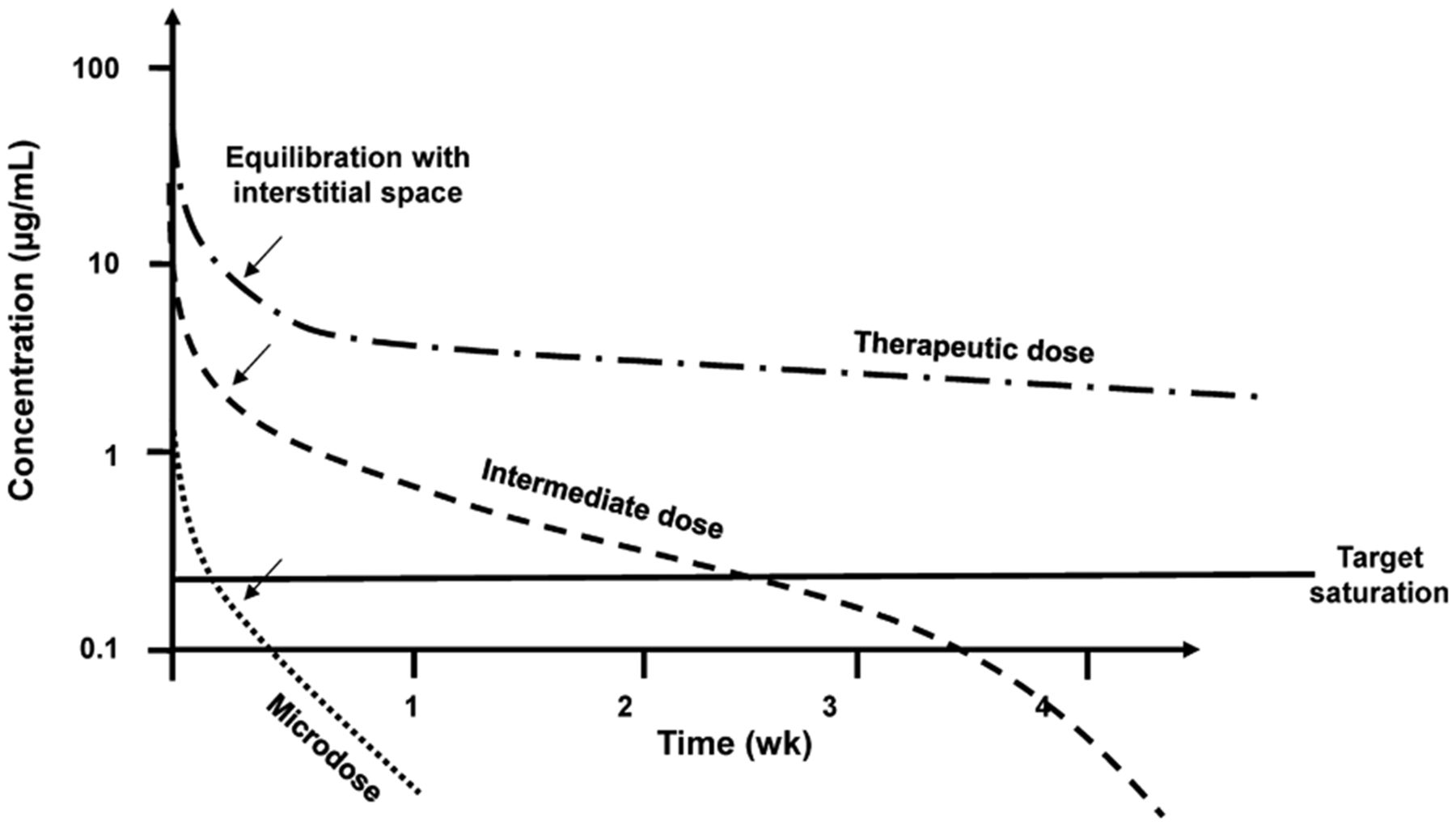
Typical plasma pharmacokinetics of an antibody.
An antibody is usually given intravenously, with an immediate distribution in the plasma space. Because this space is typically 4% of the body volume, the immediate plasma concentration is expressed as an SUV of approximately 25. Thereafter, there is typically a slow extravasation, the rate and magnitude of which depend on the character of the endothelium, the blood flow, and the overall volumes of the different organs. Typically, this phase has a collective half-life of 6–12 h, and the back-extrapolated SUV is on the order of 5 (interstitial plus plasma space, ∼20%).
The next phase includes binding and potential internalization via the target. At microdose levels, this portion of the elimination from plasma can be quite fast, on the order of a few days. However, if the mass dose is increased, target-mediated clearance is less important because of saturation by the mass dose, and the clearance half-life increases (21). At high doses, the clearance half-life may approach 2–4 wk. The generally slower clearance at higher doses is governed by the combined effect of organ clearance by nontarget mechanisms (which may be dominated by capture by muscle and liver) and prolonged residence (which is attributed to neonatal Fc receptors recycling the monoclonal antibody across the capillary endothelium). Depending on the mechanisms of clearance, a final phase of fast clearance by the specific target may come into play when the plasma concentration is reduced to levels compatible with target-mediated clearance.
An imaging study with a radiolabeled antibody typically uses a fixed amount of radioactivity, chosen to give a radiation dose in the upper range of what is acceptable for the indication. This amount of radioactivity is associated with a certain mass—the amount of which depends on the achieved specific radioactivity, with perhaps additional mass included for other reasons—and the total mass for a microdosing study should be below 30 nmol (4.5 mg for a typical antibody) (10). In turn, the plasma concentration can typically be 10 nM immediately after administration (30 nmol diluted in a plasma volume of 3 L) and the interstitial concentration can be about 3 nM (30 nmol diluted in an interstitial fluid volume of about 10 L) after equilibration with the interstitium in organs. These concentrations are typically high in comparison with target affinities, which can be in the high picomolar range, and the possibility of at least partial target inhibition cannot be excluded.
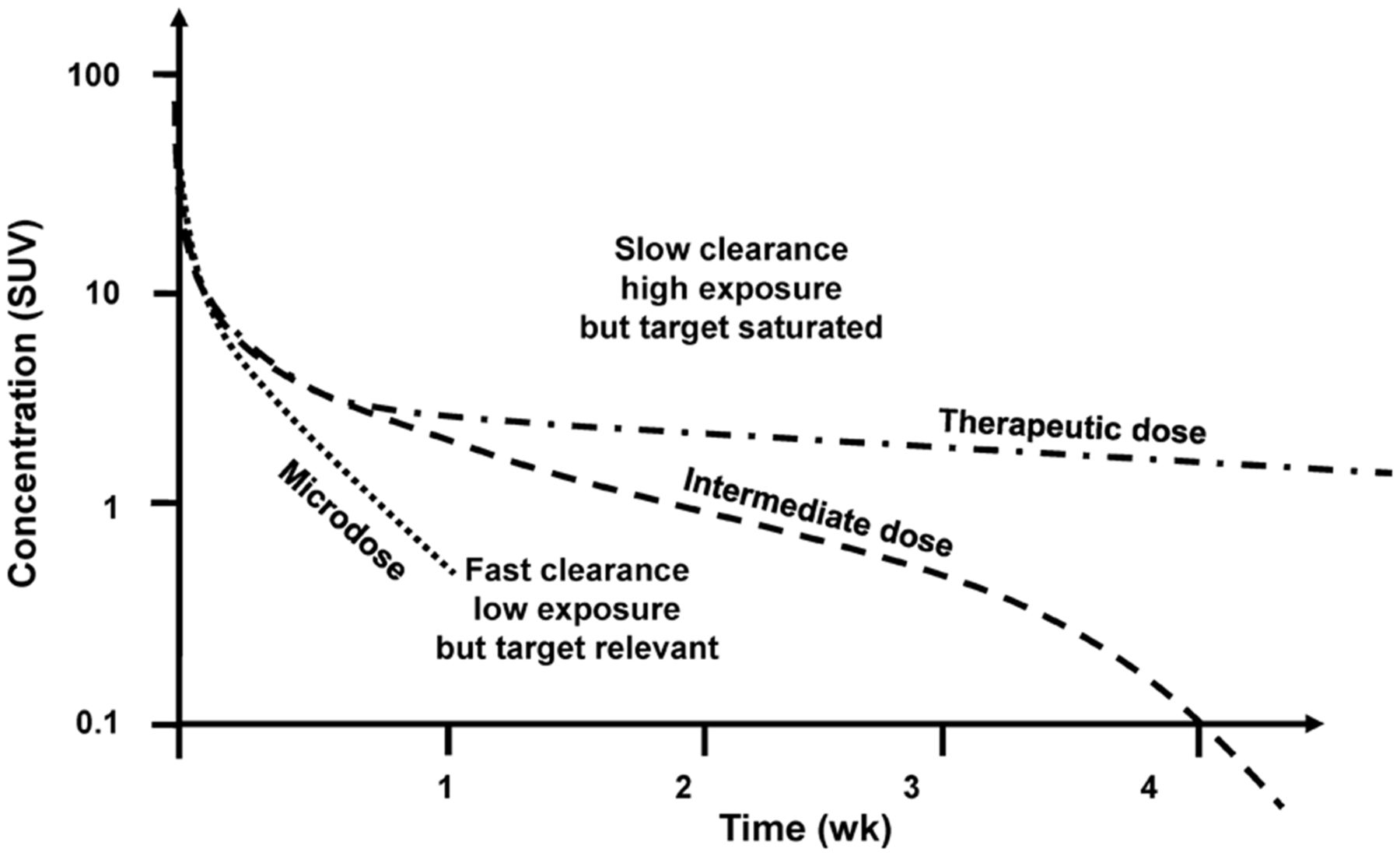
Figure 3 shows typical plasma radiopharmacokinetics profiles for a fixed amount of radioactivity but a variable amount of cold mass. The potential for the imaging study to indicate specific target interaction is greatest at low mass doses, but the signal may be low because of rapid plasma clearance. At high doses, the specific target interaction may be concealed by saturation, free tracer in tissue, or nonspecific binding but the overall signal may be high because of enhanced and prolonged plasma residence.

Typical plasma radiopharmacokinetics at fixed amount of radioactivity and different mass doses.
The Sink Effect
Imaging studies to characterize target expression and target interaction are sometimes better performed at slightly higher mass doses than the minimum set by the ability to generate a sufficient amount of radioactivity. One such case was illustrated by a study with 89Zr-labeled trastuzumab in which, through the so-called sink effect, a significant portion of the radioactivity in plasma was rapidly removed to the liver and gut, thereby minimizing uptake in tumors (22). At higher mass doses, the balance between uptake in liver and tumors shifted, allowing better identification of the tumors. The article did not clarify the exact mechanism, but presumably, access and binding of the radiolabeled antibody to soluble human epidermal growth factor receptor 2 is optimal in blood, and the antibody–receptor complex is rapidly removed from circulation by such means as Fc or scavenger receptors on Kupffer cells.
The Tissue Signal
Several key features of the target need to be considered both in planning an imaging study with a radiolabeled antibody and, more so, in understanding the tissue signal. The ability of an antibody to reach a target expressed on cell surfaces depends on the rate and magnitude of extravasation from plasma to the interstitial space. Because of the size and other properties of antibodies, they cannot readily diffuse from plasma across the endothelium, but it is assumed that the endothelium has “pores” of various sizes that allow antibodies to slowly pass the endothelial barrier (23,24). The brain, which has a tighter endothelium, seems to lack these pores, and antibodies typically do not enter the brain unless equipped with specific shuttle adaptors. The degree of pore restriction is denoted by the reflection coefficient, σ, with values of 1.0 for complete restriction and 0 for complete access (19,25,26). Extravasation is further proportional to the bulk flow of fluid across the endothelium, postulated to equal the lymph flow and approximated as proportional to the tissue blood flow. Antibodies can also pass the endothelium via specific antibody carriers called neonatal Fc receptors (27).
The number and sizes of pores, the bulk flow, and the expression of neonatal Fc receptors differ in different tissues and may differ under pathologic conditions. We therefore have important tissue-specific factors that, together with plasma pharmacokinetics, affect time course and degree of access to the interstitium. However, these factors are assumed not to be dependent on dose. If a tissue lacks target expression or the target is saturated, there is the potential that a microdosing imaging study can assess the rate and degree of extravasation and, with correction for interstitial space and exclusion space (28), obtain a reasonably correct value for the antibody concentration in the interstitium, a value that can complement our understanding of therapeutically relevant dosing. Using a range of in vivo preclinical and clinical data, Shah and Betts (29) estimated non–target-related tissue distributions and defined tissue-to-plasma ratios, or “antibody biodistribution coefficients,” for various tissues.
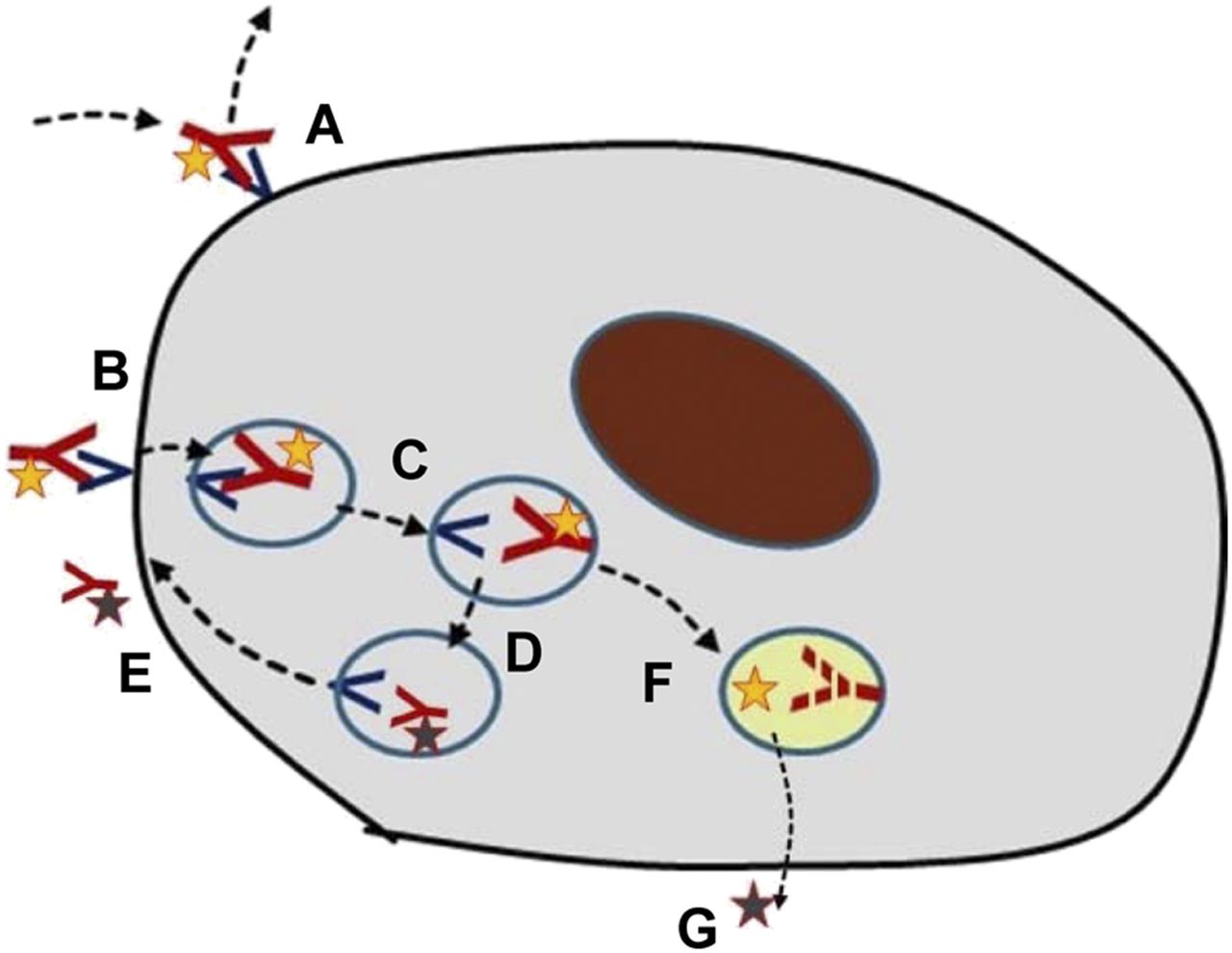
A critical issue in the targeting of antibodies is knowledge of the mechanistic aspects of target binding and the ensuing processes (Fig. 4). After being distributed to the space where the target is expressed, the antibody may bind reversibly to the target, often with a relatively slow off-rate. As in typical receptor binding, the amount bound will be proportional to the number of free receptors and be related to the on–off rate constants and the concentration in the space where the target exists (typically interstitium). Over a longer time, semiequilibrium will occur, with the specific binding being reduced in parallel with plasma pharmacokinetics. Therefore, in a microdosing study, multiple scanning time points are recommended unless an optimal time point and a mode of analysis have already been defined and verified.
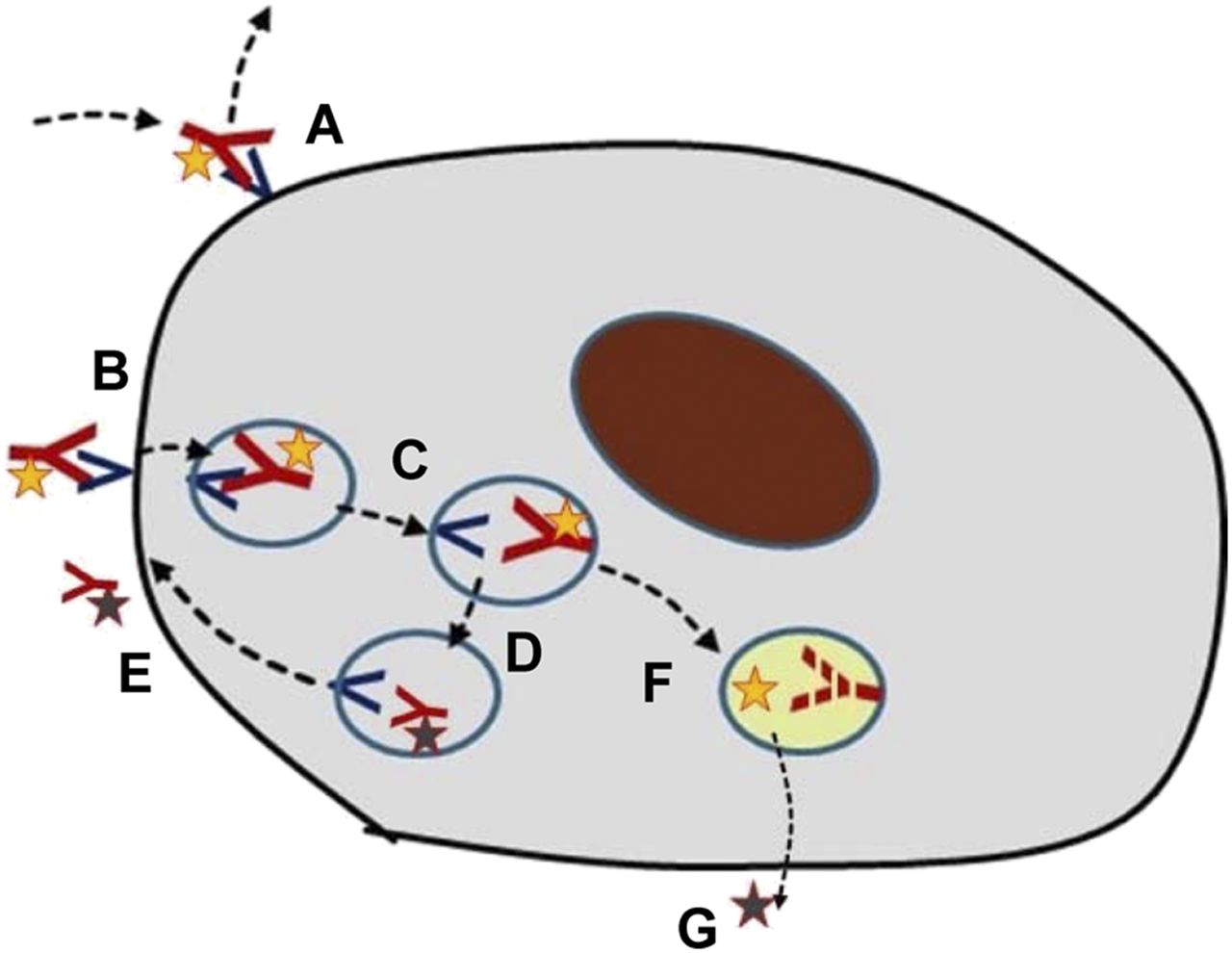
Different possible cellular fates of radiolabeled antibodies. (A) Binding to and dissociation from extracellular receptor. (B) Binding to extracellular receptor followed by internalization. (C and D) Transfer among lysosomal compartments. (E) Release of receptor and ligand from cell. (F) Lysosomal degradation of receptor or ligand. (G) Release of radionuclide from cell.
In many cases, binding to a surface receptor leads to internalization of the antibody–receptor complex. This, in turn, may be associated with lysosomal degradation of both the antibody and the receptor. A second possibility is that the receptor may be recycled to the cell surface, typically in about 10 min, whereas the antibody is degraded. A third is that the antibody and the receptor may both be recycled. Partial combinations of these fates are also possible.
The mechanism of internalization is fundamentally important for the kinetics of the radiolabel and the selection of the radionuclide. 111In and 89Zr are so-called residualizing radionuclides whereby the radioactivity remains for extended periods in the cell if the antibody undergoes lysosomal degradation (30,31). Hence, the cellular radioactivity is increasing continuously in proportion to the amount being internalized, and a high signal can often be seen. 124I, on the other hand, is released from the antibody during degradation and is actively transported from the cell within a few minutes. Except for retention of radioactivity, the residualizing radionuclides are better suited for quantitative analysis of receptor binding and internalization.
The potential for recycling or lysosomal degradation of the receptor has other important implications: receptor recycling means that cell surface expression is retained throughout exposure to the antibody mass, whereas lysosomal degradation leads to a successive loss of surface receptors during exposure, especially if the mass dose is increased beyond microdosing levels. Because of the importance of the different paths of receptor interaction, any new project with radiolabeled antibodies, whether in a microdosing study or as a tool to explore dose dependence, should investigate the mechanistic features of receptor binding and internalization preclinically in cell assays or in vivo animal studies.
MODELING OF UPTAKE BY TISSUE
Proper analysis of an immuno-PET study requires several assumptions and considerations regarding various factors of plasma pharmacokinetics, extravasation, target interaction, and fate after internalization. Because it is generally not possible to extract tissue and evaluate radioactivity concentrations in different tissue compartments, a full elucidation of these factors would require a range of tissue radioactivity measurements at different time points and under different pharmacologic conditions, such as dose levels and times after dosing. Additionally, such data would need to be evaluated using a mathematic–pharmacologic model incorporating the pharmacologic- and tissue-specific factors.
Because of the slow extravasation and slow plasma clearance of antibodies, their course must be captured over prolonged times, but patient compliance typically restricts the number of scanning visits to no more than four. Acquiring information at only a single time point can be adequate if there is sufficient information to interpret the data, particularly key parameters such as receptor binding and internalization, or if nonspecific phenomena can be estimated and subtracted. The experience of my group is that with some reasonable assumptions, relevant parameters can be extracted from 2–4 scans at suitable time points after tracer administration combined with a few blood samples. Performing 2 radiolabeled antibody studies on the same patient, each of which has at least 2 scans, is cumbersome but doable, as indicated by a recent study that included dose findings for a human epidermal growth factor receptor 3 antibody (32).
Immuno-PET with an antibody directed toward a soluble target may show much higher uptake in tumors than in normal tissues, as indicated by published studies with 89Zr- and 111In-labeled bevacizumab (33,34). Intuitively, one could assume that a complex of labeled antibody and vascular endothelial growth factor, a soluble target, would be prone to leave the tissue where it was formed, but imaging studies suggest a sustained residence. One explanation is that a portion of the vascular endothelial growth factor is bound to the extracellular matrix and that the complex is thereby retained (35). Another explanation, as suggested by a preclinical study of radiolabeled antibody complexed with placental growth factor (36), is that the complex is captured by resident immune cells in the tissue because the Fc receptor affinity of the antibody is enhanced when bound to its ligand.
INDICATIONS FOR MICRODOSING STUDIES IN DRUG DEVELOPMENT
Imaging is being increasingly used in early drug development, and molecular imaging has special features attractive for guiding further development.
When a project is being planned and then advancing through its phases of development, specific hurdles requiring special attention may appear, potentially affecting further development or even calling for termination of the project. Morgan et al. (37) reviewed the reasons for failure of a range of drug development programs and concluded that there are 3 key components to consider: whether the drug reaches the site of action (i.e., can we expect to reach a concentration at the site of action that is compatible with expected therapeutic levels?), whether the drug interacts with the target (i.e., can we expect a degree and duration of target engagement that is suggestive of therapeutic potential?), and whether the drug induces a functional effect relevant to assumptions of therapeutic benefit (i.e., can we induce these effects at relevant doses and without major effects on normal-organ physiology?). These questions point toward quantitative assessments; qualitative assessments are not sufficient.
In light of adequate quantitation, it is essential to approach the limitations of microdosing in pharmacology as indicated in these questions. For example, in microdosing with a small organic molecule, some understanding of how cellular heterogeneity affects the distribution is required in order to estimate the drug concentration at the target and extrapolate to assumed therapeutic concentrations. A radiolabeled antibody may show high uptake in a tumor, but if the scan was obtained at too high a mass dose or at an unsuitable time point, the uptake may still be high and allow good visualization but be dominated by nonspecific phenomena.
Imaging Studies to Show How the Site of Action Is Accessed
The physiochemical properties of small organic molecules are a starting point for predicting the tissue and cellular distribution that can be expected. For example, a very low or very high lipophilicity is predictive of a restricted distribution across cell membranes, including passage across the blood–brain barrier (38). Hence, it is likely that drugs of this type will be rejected at an early stage for such indications. The potential that a drug may be a substrate for the more common efflux systems can also be evaluated early in its development. Additional factors, some not easily predicted, also affect the extent to which a drug can cross the blood–brain barrier.
If the target is of a type—such as cell surface receptors—that is accessible from the interstitial space, the drug can be assumed to reach the target quickly by being rapidly distributed to the interstitial space and equilibrating with plasma. More complicated situations may exist with agents that bind avidly to plasma protein, but the free fraction in plasma and tissue may be evaluated preclinically (39). Hence, for many drugs under development, a microdosing study for the sole purpose of understanding passive distribution may not be needed. The tissue distribution and availability at the site of action can be relatively well predicted by the properties of the compound, preclinical experiments, and knowledge of plasma pharmacokinetics.
Compounds with more complex tissue behavior might benefit from a microdosing study, preferably at an early stage of development. In some cases, the clinical and laboratory results from a drug trial may raise questions about access to the target that, in retrospect, might have been prevented by a microdosing study.
If the target is intracellular, the concentration and time course to be expected may be difficult to predict—in view of the confounding factors mentioned above—and a microdose imaging study may help support progression of the drug through development. On the one hand, a microdosing imaging study may be indicated but prior scrutiny may reveal a poor potential to get an answer that can be extrapolated to a concentration at relevant doses. On the other hand, if an agent has a suitable binding affinity in relation to target expression, a microdosing imaging study may show specific binding to the target, which in turn would prove that the drug is reaching the site of action.
In the case of protein therapeutics such as antibodies, there is still insufficient knowledge about the rate and degree of distribution to the site of action in humans if the target is expressed in the interstitial space. The assumptions are that distribution from plasma to interstitium is governed predominantly by the reflection coefficient and bulk fluid flow, factors that are not well known, especially in pathologic tissue. Additionally, we can expect large interindividual variability, heterogeneity within tissues, and heterogeneity among different areas of pathology, just as there is among different lesions in the same individual. Therefore, imaging distribution studies for new protein therapeutics are indicated in many cases, at least until we have a broader knowledge of such agent’s distribution in both healthy humans and disease states.
It is important that such studies be performed with sufficient time points and with plasma pharmacokinetics evaluations. An adaptive study design is often more effective, allowing for modifications as data emerge from previously scanned subjects. In addition, for antibodies, specific binding to the target may be demonstrated and thereby prove access to the site of action. However, nonspecific binding may be enhanced in pathologic tissue, with mere observation of a high signal not being proof of specific targeting.
Imaging Studies to Prove Engagement of the Target
Demonstration of target engagement, including an understanding of doses adequate to act on or inhibit the target’s further biologic actions, is key in the development of a new drug. This information is usually not available through systemic biomarkers, and evaluations using biopsy are hampered by logistic limitations and sometimes technical hurdles. This is a prime field in which molecular imaging can contribute essential information. However, imaging probes with suitable properties and preclinical and clinical demonstration of utility are required. The microdosing concept has become important to the development of such probes, especially since the establishment of regulations to facilitate this development.
If the drug scaffold under development can serve as the imaging probe, the drug’s preclinical toxicity assessments may be sufficient to begin human imaging studies. However, it is advisable to begin development of an imaging tool, including its human qualification, well ahead of development of the drug components, and hence the microdosing regulatory path can still be followed to shorten the timelines. In many cases, the drug candidate may be found not to have optimal imaging properties, such as a suitable plasma half-life or affinity matching the target density, and development of another scaffold in the same or a different chemical series may be considered instead. There is also the option of exploring more than one scaffold in parallel to find the one with the best targeting performance. Here, also, the microdosing concept facilitates development.
For small organic molecules, especially in the field of neurologic diseases, there are abundant examples of using imaging probes to verify expression of the target and explore the degree to which a drug engages the target. The use of probes labeled with 11C or 18F is especially valuable since they may allow multiple assessments of drug effects within the same individual while keeping radiation doses within acceptable limits. For example, they may be used for intraindividual dose escalation studies or for evaluating the time course of target engagement (40–43). An adaptive trial design guided by a pharmacokinetics/pharmacodynamics model is often the most effective and can supply important dosing information in a limited number of subjects (44).
The development of an imaging probe based on protein scaffolds such as antibodies is more complicated than that based on small organic molecules. One limitation is the slow kinetics, which requires the studies to be extended over multiple days. Another is that the amount of radioactivity that can be administered is much lower for longer-lived radionuclides such as 89Zr or 111In, leading to noisier images. 89Zr-labeled antibodies are typically given at a dose of 37 MBq in oncology studies, with a resulting radiation exposure of 20 mSv (45). However, we recently performed a biodistribution study in which an 89Zr-labeled protein was given to healthy volunteers at a dose of 12.5 MBq, with a resulting radiation exposure below 10 mSv, an acceptable level (46). Although the images were rather noisy, the quantitative values were fully adequate and gave important information on the drug distribution.
Finally, the issue of immunogenicity needs to be carefully considered when proteins are given in microdosing studies. Immunologic reactions are typically accentuated if there is more than one administration, but they are also of concern for a single administration. Although experience with radiolabeled proteins developed under the microdosing concept is limited, we believe microdosing imaging studies should be considered in the development of protein therapeutics, particularly to supply information on target interaction. Furthermore, such imaging studies may reveal the degree of heterogeneity in target expression within and between patients (47), information that can be crucial in selecting patients for clinical trials and in suggesting indications for the drug.
Imaging Studies to Prove a Functional Response by the Tissue
Beyond proof of target engagement, a critical component in the early development of novel drugs is to ensure that there is a biologic or physiologic tissue response that can be assumed to be mechanistically relevant to a clinical response. In early clinical studies, especially phase I trials, a positive clinical readout is not expected, partly because patient selection often is not optimal. For example, oncology trials often include patients who are in the final stage of disease and were previously heavily treated. Additionally, phase I trials are powered for the key objective of evaluating adverse events from a dose in order to suggest the doses to be used in phase II trials. Methods of verifying tissue response may be included, but some of the present imaging methods are relatively blunt, requiring long treatment periods and large cohorts to verify a response such as a reduction in the rate of liver fibrosis or in the rate of degeneration of the striatal dopaminergic terminals.
18F-FDG PET is often included to monitor antitumoral effects in oncology. For some types of treatments and some types of tumors, this is fully adequate and may indicate responses very early with a precision that allows for conclusions even in an individual patient. However, beyond this use of 18F-FDG PET and the recording of angiogenesis, imaging today can offer relatively little to the early determination of treatment response in tissue biology and physiology. Additional imaging tools need to be found for this purpose, and the microdosing concept is an important path to the development of these tools. Avenues that have been scouted include monitoring of immune cells in inflammatory diseases and oncology and monitoring of myofibroblast activity, the active component of fibrosis development.
CONCLUSION
The large investments in personnel and materials, ensuing high costs, and long timelines needed to develop novel drugs, along with an awareness that the situation is getting worse, has prompted pharmaceutical companies and state agencies to seek ways to increase efficiency. A key manifest was the Critical Path Initiative, which encouraged greater flexibility in clinical trials and a search for new endpoints. For the development of molecular imaging, the preclinical requirements for entry into humans were facilitated by the regulatory framework of microdosing (9–11). Furthermore, an impressive development of radiolabeling options using primarily 11C and 18F for small organic molecules and smaller proteins, together with improved methods and increased experience in labeling proteins with longer-lived radionuclides such as 89Zr, has created options for wider use of radiolabeled scaffolds.
There is an increased awareness by pharmaceutical companies that imaging studies can supply important information early in drug development and give credibility to further development. Key points in this process have been summarized by Morgan et al. (37) as the 3 pillars of drug development: ensuring distribution to the site of action, engaging the target, and inducing a relevant physiologic response. However, questions about the value of such early studies are still arising concerning initiation hurdles, planning, and—with respect to the reliability of the results—performance.
This article has tried to explain the options for imaging, as well as the mechanistic aspects and specific factors behind it, so as to engender a realistic understanding of what imaging is able to provide to early drug development. Pharmaceutical and biotechnology companies should take advantage of the technical and regulatory facilitation that has taken place in the development of imaging probes and should consider the option of molecular imaging early in the planning of drug development, preferably before candidate selection. This is necessary because the timelines are still long for validation of an imaging probe in humans.
Regulation of the microdosing concept was an important step forward, but there are still several hurdles impeding the path from concept to human study: finding the financial means for academia to perform preclinical toxicity assessments; clearing the ambiguity in what pharmaceutical companies and academia consider to be good manufacturing practice for imaging probes in toxicity evaluations and the clinical setting; and specifying how much effort should be spent on preclinical validation of an imaging probe before it becomes acceptable to proceed to a human study.
Molecular imaging can and should supply quantitative data on drug behavior in any part of the human body, and these data should be placed in the context of translation: from animals to humans and back, and from low microdosing levels to expected therapeutic concentrations. The key promise of microdosing studies, performed and analyzed properly, is that they make it possible to obtain this information.
DISCLOSURE
Mats Bergstrom is an external consultant in molecular imaging to pharmaceutical and biotechnology companies. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online May 25, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 28, 2017.
- Accepted for publication May 22, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Development of a PET Tracer for OGA with Improved Kinetics in the Living Brain
- 11C-Sorafenib and 15O-H2O PET for Early Evaluation of Sorafenib Therapy
- Swarming behavior and in vivo monitoring of enzymatic nanomotors within the bladder
- Monitoring the collective behavior of enzymatic nanomotors in vitro and in vivo by PET-CT
- 89Zr-Immuno-PET: Toward a Noninvasive Clinical Tool to Measure Target Engagement of Therapeutic Antibodies In Vivo
- Immuno-PET Imaging to Assess Target Engagement: Experience from 89Zr-Anti-HER3 mAb (GSK2849330) in Patients with Solid Tumors