


Abstract
The translocator protein (18 kDa) (TSPO), formerly known as the peripheral benzodiazepine receptor, is dramatically upregulated under pathologic conditions. Activated microglia are the main cell type expressing the TSPO at sites of central nervous system pathology. Radioligands for the TSPO can therefore measure active disease in the brain. This article details the synthesis, radiofluorination, and pharmacologic evaluation of a new TSPO-specific pyrazolopyrimidine, DPA-714. Methods: The affinity of DPA-714 for the TSPO was measured in rat kidney membranes with 3H-PK11195. The in vitro functional activity of DPA-714 was measured in a steroidogenic assay in which the ability of DPA-714 to increase pregnenolone synthesis was measured with rat C6 glioma cells. The radiofluorination of DPA-714 was achieved by nucleophilic 18F-fluoride displacement of the tosylate precursor. 18F-DPA-714 was assessed in rats harboring unilateral quinolinic acid (QA) lesions. In addition, pretreatment experiments were performed with PK11195 (5 mg/kg), DPA-714 (1 mg/kg), and DPA-713 (1 mg/kg). The in vivo binding and biodistribution of 18F-DPA-714 were determined in a baboon with PET. Experiments involving presaturation with PK11195 (1.5 mg/kg) and displacement with DPA-714 (1 mg/kg) were conducted to evaluate the specificity of radioligand binding. Results: In vitro binding studies revealed that DPA-714 displayed a high affinity for the TSPO (dissociation constant, 7.0 nM). DPA-714 stimulated pregnenolone synthesis at levels 80% above the baseline. 18F-DPA-714 was prepared at a 16% radiochemical yield and a specific activity of 270 GBq/μmol. In rats harboring unilateral QA lesions, an 8-fold-higher level of uptake of 18F-DPA-714 was observed in the ipsilateral striatum than in the contralateral striatum. Uptake in the ipsilateral striatum was shown to be selective because it was inhibited to the level in the contralateral striatum in the presence of PK11195, nonlabeled DPA-714, or DPA-713. PET studies demonstrated rapid penetration and good retention of 18F-DPA-714 in the baboon brain. Pretreatment with PK11195 effectively inhibited the uptake of 18F-DPA-714 in the whole brain, indicating its selective binding to the TSPO. The injection of nonlabeled DPA-714 20 min after the injection of 18F-DPA-714 resulted in radioligand washout, demonstrating the reversibility of 18F-DPA-714 binding. Conclusion: 18F-DPA-714 is a specific radioligand for the TSPO, displaying promising in vivo properties and thus warranting further investigation.
The translocator protein (18 kDa) (TSPO) is a highly hydrophobic protein located mainly on the outer mitochondrial membrane (1,2). Originally, the TSPO was identified in peripheral tissue as an alternative binding site for diazepam (3) and, for this reason, was initially known as the peripheral benzodiazepine receptor. Later studies revealed that this receptor bound molecules other than benzodiazepines and was also present at low levels in the central nervous system. This information highlighted the misleading nature of the term “peripheral benzodiazepine receptor” and justified the need for a new name to better capture the cellular location and tissue-specific roles of this receptor. Since the main structure–function relationships of this entity involve the binding and transport of specific ligands, including cholesterol and porphyrin, the name “TSPO” was proposed (4).
The TSPO is involved in several important cellular processes, including apoptosis (5), regulation of cellular proliferation (6), immunomodulation (7), heme biosynthesis (8), and production of steroid hormones (9). The most characterized functional role of the TSPO is its ability to facilitate the transport of cholesterol from the outer to the inner mitochondrial membrane, where it is metabolized into pregnenolone by cytochrome P-450 side-chain cleavage, a process that leads to the production of various steroids (9,10). High-affinity TSPO ligands, both endogenous and exogenous, can stimulate this process, resulting in increased concentrations of steroids in the brain. For this reason, TSPO ligands are thought to hold therapeutic potential for treating memory and stress-related disorders (11,12).
Consistent with its key involvement in steroidogenesis, the TSPO is most densely distributed in steroid-producing endocrine organs (1,13). In the healthy brain, TSPO expression is negligible, but under neuroinflammatory conditions, TSPO density is markedly increased (14). In vitro and in vivo studies have proven the main cell type expressing the TSPO at sites of central nervous system pathology to be activated microglia (15,16), the resident brain macrophages (17,18). Microglial activation is known to occur during the initial stages of most neurologic disorders and even precedes neurodegeneration. Because TSPO expression reflects the extent of microglial activation, specific radioligands for the TSPO can serve as indicators of early brain disease (18).
At present, only a few TSPO-specific radioligands are available for use in PET studies. 11C-(R)-PK11195 has long been considered the standard for TSPO imaging; however, its poor signal-to-noise ratio and low brain permeability limit its accuracy and usefulness (19). Several new TSPO-specific ligands, namely, 11C-DAA1106, 18F-FEDAA1106, and 11C-PBR28, have been described and reported to display good in vivo properties in rodent brains (20,21). 11C-DAA1106 and 18F-FEDAA1106 have also been shown to display highly specific in vivo uptake in primate brains (22); 18F-FEDAA1106 was recently evaluated in human PET studies (23). Although these promising TSPO-specific ligands have newly emerged, there remains a considerable gap in information needed to completely characterize the TSPO. By synthesizing a large set of structurally and functionally diverse radioligands, we hoped to gain a better understanding of the TSPO, its therapeutic potential, and the possible existence of subtypes.
In 2001, a series of TSPO-specific pyrazolopyrimidine ligands were reported (24). Recently, one of these ligands, N,N-diethyl-2-(2-(4-methoxyphenyl)-5,7-dimethylpyrazolo[1,5-a]pyrimidin-3-yl)acetamide (DPA-713), was labeled with 11C. PET studies showed that 11C-DPA-713 selectively and specifically bound the TSPO in normal baboon brains (25). Since then, 11C-DPA-713 has been labeled more efficiently with methyl triflate (26) and evaluated in rat models of disease (27). These encouraging results prompted us to synthesize an 18F-labeled derivative of DPA-713.
The aims of this study were to synthesize the fluoroethoxy analog DPA-714, label it with 18F, and evaluate 18F-DPA-714 in a neuroinflammatory rodent model and in vivo in a normal baboon by using PET.
MATERIALS AND METHODS
General
All chemicals and solvents were of high grade and were purchased from Aldrich, except as otherwise stated. 3H-PK11195, which was used for in vitro binding, was purchased from Dupont-New England Nuclear. PK11195 used for PET studies was purchased from Tocris, and PK11195 used for ex vivo studies was obtained from Sigma. Melting points were measured with a Reichert melting point stage and are uncorrected. Proton nuclear magnetic resonance (1H NMR) spectra were recorded with a 300-MHz Varian Gemini instrument. Chemical shifts (δ) were reported in parts per million downstream from internal tetramethylsilane. Multiplicities were reported as s (singlet), d (doublet), dd (doublet of doublets), m (multiplet), and the observed coupling constant (J, Hz). Mass spectrometry was performed with a Thermo Finnigan Polaris Q gas chromatography–mass spectrometry instrument. Thin-layer chromatography was performed with aluminum-backed plates (Lomb Scientific) precoated with silica gel 60 (F254) and visualized with UV fluorescence (254 or 365 nm). Column chromatography was performed with Merck silica gel 60 (230–400 mesh). All radiosyntheses were carried out with MXFDG reagent kits from ABX Chemicals, but with the removal of the buffer solution and the aqueous NaOH syringe. The eluent vials contained 22.0 mg of Kryptofix-222, 7.0 mg of K2CO3, 300 μL of acetonitrile, and 300 μL of water. The reagent vials contained 7 mL of acetonitrile, 5 mL of ethanol, and 2 mL of ethanol.
Chemistry
N,N-Diethyl-2-(2-[4-(2-Fluoroethoxy)-Phenyl]-5,7-Dimethyl-Pyrazolo[1,5-a]Pyrimidin-3-yl)-Acetamide (DPA-714).
Diisopropyl azodicarboxylate (DIAD, 190 mg, 0.94 mmol) was added to a solution of N,N-diethyl-2-(2-(4-hydroxyphenyl)-5,7-dimethylpyrazolo[1,5-a]pyrimidin-3-yl)ethanamide (compound 1) (25) (150 mg, 0.43 mmol), triphenylphosphine (274 mg, 0.94 mmol), and 2-fluoroethanol (60 mg, 0.94 mmol) in dry N,N-dimethylformamide (DMF) (6 mL). The reaction mixture was stirred at room temperature for 48 h and concentrated with a rotary evaporator (95°C, 20 millibars). The resulting crude oil was dissolved in CHCl3 (40 mL), and the mixture was washed with water (3 × 20 mL) to remove DMF. The organic layer was collected and dried over anhydrous Na2SO4. The solvent was removed under vacuum, and the residue was purified by column chromatography (CHCl3:methanol [MeOH], 80:1, v/v, as the eluent) to yield DPA-714 (80 mg, 0.20 mmol) as pale yellow crystals at a 47% yield and with a melting point of 160°C–163°C. 1H NMR (CDCl3, 300 MHz) δ: 7.78 (d, 2H, J = 9.0 Hz), 6.52 (s, 1H), 7.01 (d, 2H, J = 9.0 Hz), 4.78 (dt, 2H, J = 4, 47 Hz), 4.26 (dt, 2H, J = 4, 28 Hz), 3.93 (s, 2H), 3.39–3.51 (m, 4H), 2.75 (s, 3H), 2.56 (s, 3H), 1.09–1.27 (m, 6H). Found: C, 66.70; H, 6.60; N, 13.14. Mass spectrum: chemical ionization (CI), m/z 399 (M + 1).
Toluene-4-Sulfonic Acid 2-[4-(3-Diethylcarbamoylmethyl-5,7-Dimethyl-Pyrazolo-[1,5-a]Pyrimidin-2-yl)-Phenoxy]-Ethyl Ester (Compound 2).
For step 1, fresh silver oxide (350 mg, 1.5 mmol), p-toluenesulfonyl chloride (210 mg, 1.1 mmol), and potassium iodide (33 mg, 0.2 mmol) were added to a stirred solution of 1,2-ethanediol (62 mg, 1 mmol) in dichloromethane (10 mL). The reaction mixture was stirred at room temperature for 2 h, filtered through a pad of celite, and washed with ethyl acetate. The solvent was removed, and the crude product was purified by column chromatography (CH2Cl2:MeOH, 40:1, v/v, as the eluent) to yield toluene-4-sulfonic acid 2-hydroxy-ethyl ester as an oil at a 46% yield. 1H NMR (CDCl3, 300 MHz) δ: 7.81 (d, 2H, J = 8.1 Hz), 7.36 (d, 2H, J = 8.1 Hz), 4.15 (d, 2H, J = 9.0 Hz), 3.82 (d, 2H, J = 9.0 Hz), 2.46 (s, 3H).
For step 2, DIAD (0.48 mL, 2.4 mmol) was added to a solution of compound 1 (25) (400 mg, 1.1 mmol), triphenylphosphine (637 mg, 2.4 mmol), and toluene-4-sulfonic acid 2-hydroxy-ethyl ester (525 mg, 2.4 mmol) in dry DMF (10 mL). The reaction mixture was stirred for 20 h at room temperature and concentrated with a rotary evaporator (95°C, 20 millibars). The residue was purified by column chromatography (CHCl3:MeOH, 80:1, v/v, as the eluent) to yield compound 2 (363 mg, 0.66 mmol) as light yellow crystals at a 60% yield and with a melting point of 128°C–132°C. 1H NMR (CDCl3, 300 MHz) δ: 7.83 (d, 2H, J = 8.1 Hz), 7.74 (d, 2H, J = 9.0 Hz), 7.35 (d, 2H, J = 8.1 Hz), 6.86 (d, 2H, J = 9.0 Hz), 6.52 (s, 1H), 4.37 (d, 2H, J = 4.8 Hz), 4.19 (d, 2H, J = 4.8 Hz), 3.92 (s, 2H), 3.39–3.52 (m, 4H), 2.74 (s, 3H), 2.56 (s, 3H), 2.45 (s, 3H), 1.08–1.23 (m, 6H). Found: C, 63.37; H, 5.96; N, 9.89. Mass spectrum: CI, m/z 551 (M + 1).
Radiochemistry
Radioisotope Production.
No-carrier-added aqueous 18F-fluoride ion was produced on a PETtrace cyclotron (GE Healthcare) by irradiation of an 0.8-mL silver-bodied water target with a 25-μA current and a 16.5-MeV proton beam on 95% enriched 18O-H2O via the 18O(p,n)18F nuclear reaction.
Preparation of 18F-Labeled K–F–Kryptofix-222 Complex.
18F-Fluoride in 18O-H2O was transferred to a GE TRACERlab MXFDG synthesizer and passed through an anion-exchange resin (Waters Sep-Pak Accell Light QMA cartridge in the carbonate form, prepared by washing with 10 mL of 0.5 M K2CO3 and then rinsing with 10 mL of water) under vacuum. Trapped 18F-fluoride was eluted from the Sep-Pak cartridge and transferred to the reaction vessel with an eluent solution containing K2CO3 (7 mg in 300 μL of pure water), acetonitrile (300 μL), and 22 mg of Kryptofix-222 (4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo [8.8.8] hexacosane). Aliquots of acetonitrile were added, and the reaction mixture was evaporated to dryness after each addition (3 × 80 μL). The evaporation was performed at 95°C under nitrogen flow and vacuum.
Preparation and Formulation of 18F-DPA-714.
Compound 2 (6 mg) was dissolved in acetonitrile (3 mL), and the mixture was added to the dry 18F-labeled K–F–Kryptofix-222 complex and allowed to react at 85°C for 5 min. On completion, the reaction mixture was diluted with sterile water and passed through a tC18 cartridge (Waters Sep-Pak Accell Light tC18 cartridge, prepared by washing with 5 mL of ethanol and then rinsing with 10 mL of water). The reaction vessel was rinsed with sterile water, and the eluate again was passed through the tC18 cartridge. The trapped tC18-radiolabeled product was rinsed an additional 3 times with sterile water (total of 40 mL). The product was eluted from the tC18 cartridge with ethanol (2 mL) and sterile water (3 mL). The resulting solution was passed though a 0.22-μm Millipore CATHIVEX nonpyrogenic sterile filter to remove particulate material before high-performance liquid chromatography (HPLC) purification.
The crude mixture was injected into a Waters XTerra RP C18 10-μm (7.8 × 300 mm) semipreparative reversed-phase HPLC column. With a mobile phase of 0.1 M aqueous ammonium acetate (NH4OAc) (pH 10) and CH3CN (60:40, v/v) at a flow rate of 4.0 mL/min, the retention time (tR) of 18F-DPA-714 was determined to be 12 min. The radioactive fraction corresponding to 18F-DPA-714 was collected and evaporated under vacuum with a rotary evaporator. The residue was reconstituted in sterile water (4 mL) and filtered through a 0.22-μm Millipore GS 13-mm sterile filter into a sterile pyrogen-free evacuated vial. Under these conditions, the tR of the tosylate precursor was determined to be greater than 35 min.
Quality Control for 18F-DPA-714.
For the determination of specific radioactivity and radiochemical purity, a portion of the final solution with a known volume and radioactivity was injected into a Waters XTerra C18 5-μm (4.6 × 150 mm) analytic reversed-phase HPLC column. A mobile phase of 0.1 M aqueous NH4OAc (pH 10) and CH3CN (50:50, v/v) at a flow rate of 2.0 mL/min was used to elute 18F-DPA-714 at a tR of 2 min. The area of the UV absorbance peak at 254 nm corresponding to the carrier product was measured (integrated) on the HPLC chromatogram and compared with a standard curve relating mass to UV absorbance. The tR of the tosylate precursor was determined to be greater than 20 min.
In Vitro Binding Affinity
3H-PK11195 (specific activity, ∼322 GBq/mmol) and 3H-Ro15-1788 (specific activity, ∼311 GBq/mmol) were purchased from Dupont-New England Nuclear. For binding studies, mitochondria were prepared as previously described (28), with minor modifications as described here, from the kidneys of male Wistar rats killed by cervical dislocation. The kidneys were homogenized in 20 volumes of ice-cold 50 mM Tris-HCl (pH 7.4), 0.32 M sucrose, and 1 mM ethylenediaminetetraacetic acid (buffer A) containing protease inhibitors (benzamidine at 160 μg/mL, bacitracin at 200 μg/mL, and soybean trypsin inhibitor at 20 μg/mL) with a Teflon (DuPont) pestle in a glass homogenizer and centrifuged at 600g for 10 min at 4°C. The resulting supernatant was centrifuged at 10,000g for 10 min at 4°C. The pellet was suspended in 20 volumes of ice-cold buffer A and centrifuged at 10,000g for 10 min at 4°C. The crude mitochondrial pellet was frozen at −20°C until the time of the assay or incubated with 0.6 nM 3H-PK11195 in 50 mM Tris-HCl (pH 7.4) (buffer B) with a range of concentrations of the tested compounds (0.1 nM–10 μM) in a total volume of 0.5 mL for 90 min at 4°C. The incubation was terminated by dilution to 5 mL with ice-cold buffer B; this step was followed immediately by rapid filtration through glass fiber Whatman GF/C filters. The filters were washed (2.5 mL) with buffer B, and the amount of radioactivity retained on the filters was determined with a Packard 1600 TR liquid scintillation counter at 66% efficiency. Nonspecific binding was estimated for each sample in the presence of unlabeled 1 μM PK11195. The 50% inhibitory concentrations were determined, and the dissociation constant (Ki) values were derived according to a previously derived equation (29). The protein concentration was estimated by the method of Lowry et al. (30) with bovine serum albumin (Sigma-Aldrich) as the standard.
Cell Culture
Rat C6 glioma cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mM l-glutamine, penicillin at 100 U/mL, and streptomycin at 100 μg/mL. Cultures were maintained in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
Steroid Biosynthesis Assay
PK11195, Ro5-4864, and clonazepam were obtained from Sigma. C6 cells were seeded in 24-well plates at a density of ∼106 cells per well in a final volume of 1 mL. Before the measurement of pregnenolone production, the cells were washed 3 times with a simple salt medium consisting of 140 mM NaCl, 5 mM KCl, 1.8 mM CaCl2, 1 mM MgSO4, 10 mM glucose, and 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES)–NaOH (pH 7.4) plus 0.1% bovine serum albumin. During the experiments, the cells were incubated with this simple salt medium in an air incubator at 37°C. For the measurement of pregnenolone secreted into the medium, the further metabolism of pregnenolone was blocked by the addition of trilostane (25 μM) and SU10603 (10 μM) (inhibitors of 3β-hydroxysteroid dehydrogenase and 17α-hydroxylase, respectively) to the simple salt medium as previously described (31). The addition of DPA-714, of DPA-713, and of PK11195, Ro5-4864, or clonazepam to the C6 cells was accomplished by complete change of the simple salt medium to a medium containing the appropriate concentration (40 μM) of the compound. The final concentration of ethanol was constant for all of the wells in each experiment and did not exceed 0.5% (v/v), a concentration that, on its own, had no effect on steroid production. At the end of the incubation period (2 h), the cell medium was retained and centrifuged at 1,500g for 10 min. The amount of pregnenolone secreted into the medium was quantified by a radioimmunoassay with antipregnenolone antiserum (ICN Biochemicals Inc.) under the conditions recommended by the supplier. The cell protein concentration was measured by a previously described method (30).
Measurement of Partition Coefficient (Log D)
The log of the octanol–water partition coefficient (log D) was calculated (for DPA-713, DPA-714, and PK11195) with a previously described HPLC method (32). Phosphate buffer (0.1 M) was prepared by dissolving weighed amounts of potassium dihydrogen orthophosphate in HPLC-grade water and adjusting the pH to 7.4 with sodium hydroxide solution (0.1 M). Samples were analyzed with a C18 column (XTerra, 5 μm, 150 × 4.6 mm) and a mobile phase of MeOH:phosphate buffer (pH 7.4) (60:40, v/v) at a flow rate of 1 mL/min. The lipophilicity of each compound was estimated by comparing its tR with those of standards having known log D values. The standards used to generate a general calibration equation were aniline, benzene, bromobenzene, ethyl benzene, trimethyl benzene, and hexachlorobenzene dissolved in the mobile phase. All sample injections were performed 3 times, and the results were averaged to yield the final values. A calibration curve of log D versus tR was produced and resulted in an experimental calibration equation (y = 1.07910.7527x) with an r2 of 0.995. The exponential equation of the trend line function from the calibration graph and Excel (Microsoft) allowed the log D values to be calculated.
Ex Vivo Rodent 18F-DPA-714 Studies
Adult male Wistar rats weighing 300–320 g (Janvier) were used for ex vivo experiments. All procedures were performed in accordance with the European Community Council Directive 86/609/EEC for the care of laboratory animals. Animals were kept on a 12-h light–dark cycle (temperature, 22.4°C ± 0.5°C [mean ± SD]; humidity, 40.3% ± 7.2%), and water and food were freely available.
Quinolinic Acid (QA) Lesions.
Rats were anesthetized with isoflurane (4%, 500 mL/min), placed on a stereotactic apparatus (Stoelting; Phymep), and maintained with isoflurane (2%, 500 mL/min) during surgery. The skull was exposed, and small holes were made with the use of a dental drill. Unilateral lesions were created in the right striatum at the following coordinates (millimeters from the bregma) (33): A, 0.7; L, −3; and P, −5.5. A cannula (gauge 25; Hamilton) was inserted into the holes in the skull, and a solution of QA (300 nmol in 2 μL of phosphate buffer [pH 7.4]) was infused at a flow rate of 0.5 μL/min. The syringe was left in place for 4 min before being removed to avoid a backflow of QA. The bone was filled in with wax, and the scalp was sutured.
Biodistribution Studies.
Ex vivo biodistribution studies were performed with 18F-DPA-714 6 d after creation of the unilateral striatal QA lesions. Rats were injected via the penis vein with 20–37 MBq of 18F-DPA-714 in a mixture of water and ethanol (85:15) (control group, n = 6); alternatively, rats were preinjected with PK11195 (5 mg/kg; n = 5), DPA-714 (1 mg/kg; n = 2), or DPA-713 (1 mg/kg; n = 2) 15 min before the radioligand injection. Animals were sacrificed 60 min after the injection of 18F-DPA-714. Samples of peripheral tissues (blood [taken via cardiac puncture, without anticoagulant], muscle, bone, liver, heart, and adrenal glands) and several brain areas (cerebellum, right and left striata, right and left frontal cortices, and right and left hippocampi) were removed and weighed. Radioactivity in these samples was measured with a γ-counter (2550 TR/LL; Packard). For peripheral tissues, the results are expressed as the ratio of the percentage injected dose per gram (%ID/g) in tissue to the %ID/g in blood ± SEM. For brain areas, the results are expressed as the ratio of the %ID/g in the cerebral region to the %ID/g in the cerebellum ± SEM. The Student t test was performed when the number of values was ≥5. Statistical significance was set at a P value of less than 0.05.
Baboon 18F-DPA-714 PET Studies.
A normal male Papio hamadryas baboon that was 13 y old and weighed 23.1 kg was selected for PET. The baboon was maintained and handled in accordance with the National Health and Medical Research Council code of practice for the care and use of nonhuman primates for scientific purposes. Project application was approved by the Sydney South West Area Health Service Animal Ethics Committee.
All PET data were acquired with a Siemens Biograph lutetium oxyorthosilicate PET/CT scanner. This scanner yields a reconstructed PET spatial resolution of 6.3 mm full width at half maximum at the center of the field of view. The baboon was initially anesthetized intramuscularly with ketamine (8 mg/kg). Anesthesia was maintained with an intravenous infusion of ketamine (Parnell Laboratory) in saline at a ketamine dose rate of 0.2 mg/kg/min. The baboon also received MgSO4 (2 mL intravenously) administered over 30 min as well as atropine (1 mg intramuscularly) and metoclopramide (Maxolon; Mayne Pharma) (5 mg intramuscularly). The head of the baboon was immobilized with plastic tape to minimize motion artifacts. A CT scan of the head was completed before the emission study. Acquisition of the PET data in the list mode was commenced just before the intravenous administration of 100 MBq of the radioligand in 4 mL of water for injection and was continued for 60 min. The blocking study involved pretreatment with PK11195 (1.5 mg/kg intravenously) 5 min before radioligand injection, whereas in the displacement study, nonlabeled DPA-714 (1 mg/kg intravenously) was administered 20 min after the injection of 18F-DPA-714.
At the conclusion of each study, the list-mode data were sorted into a dynamic scan comprising 54 frames (20 × 30 s, 30 × 60 s, and 4 × 300 s). The dynamic 3-dimensional PET sinograms were rebinned with Fourier rebinning and reconstructed with filtered backprojection and CT data–based corrections for photon attenuation and scatter into 47 transaxial slices, each comprising 128 × 128 voxels. Reconstructed voxel values in each frame are reported in units of Bq/mL, corrected for radioactive decay to the time of injection, and voxel dimensions were 0.206 × 0.206 × 0.337 cm. Voxel values were then converted to units of percentage injected dose per volume (mL) of brain tissue. An automated 3-dimensional registration algorithm was used to coregister the 2 reconstructed scans before region-of-interest definition. Time–activity curves representing variations in ligand concentrations versus time were constructed for the whole brain.
After the list-mode acquisition, a whole-body PET/CT scan was performed for 2 min at each of 6 bed positions to determine other sites of radioligand uptake.
RESULTS
Chemistry
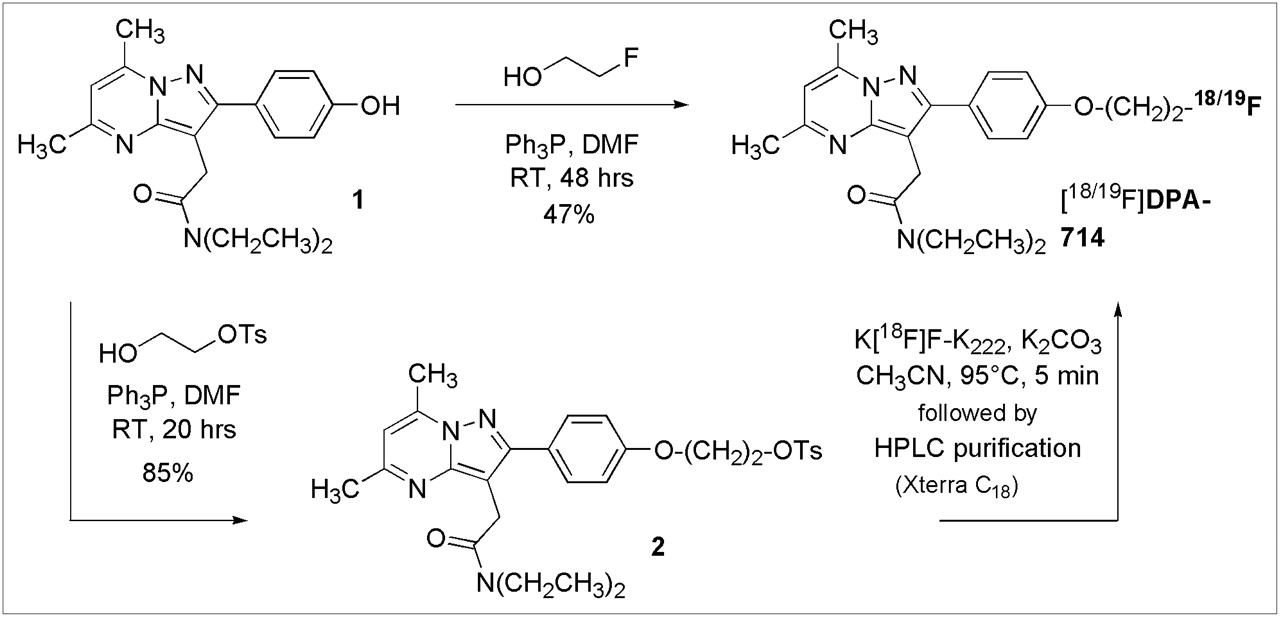
DPA-714 and its tosylate analog (compound 2) were prepared as shown in Figure 1. Both compounds were synthesized for use in the radiolabeling process: DPA-714 as a nonlabeled standard to confirm correct product formation and the tosylate analog as a precursor for 18F labeling. In summary, Mitsunobu coupling of the phenolic pyrazolo[1,5-a]pyrimidine (compound 1) (25) and 2-fluoroethanol in the presence of DIAD and triphenylphosphine in dry DMF afforded DPA-714 at a 47% yield. Initial attempts with tetrahydrofuran were unsuccessful because of the limited solubility of compound 1 in this solvent. Analogously, Mitsunobu coupling of compound 1 and toluene-4-sulfonic acid 2-hydroxy-ethyl ester (the latter prepared by sulfonylation of 1,2-ethanediol in the presence of fresh silver oxide and a catalytic amount of potassium iodide) in the presence of DIAD and triphenylphosphine in dry DMF afforded the tosylate analog (compound 2) at an 85% yield.
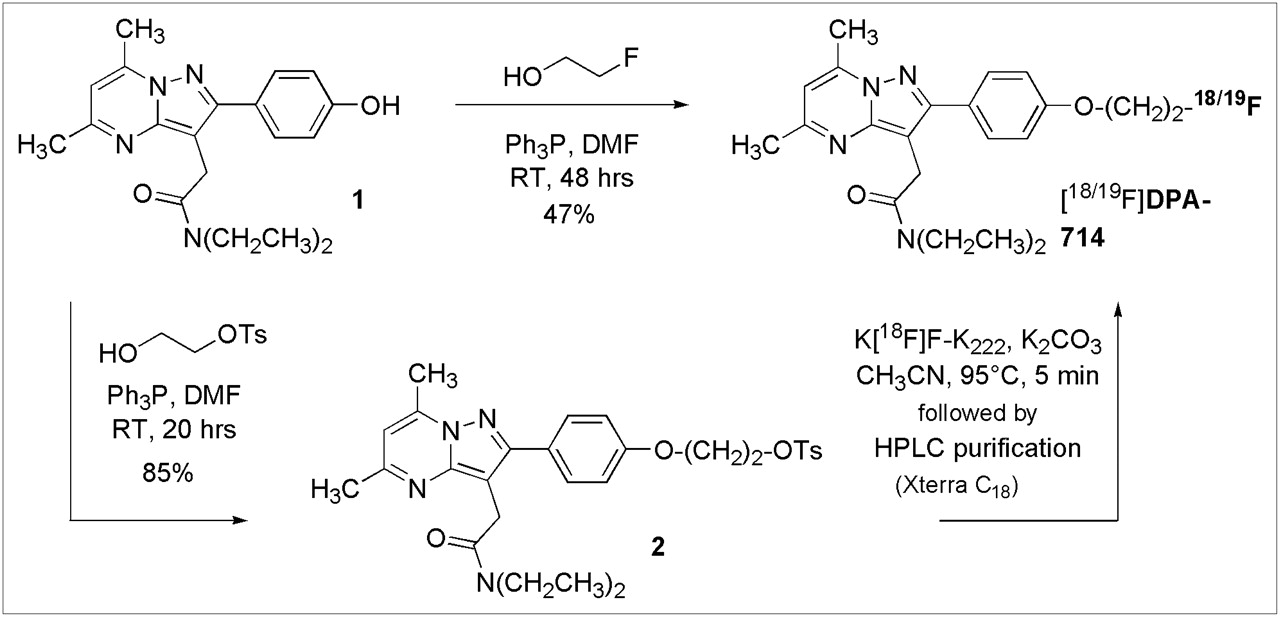
Chemical synthesis of DPA-714 and its tosylate derivative. K222 = Kryptofix-222; OTs = −OSO2(C6H4-pCH3); Ph3P = triphenylphosphine; RT = room temperature.
Radiochemistry
DPA-714 was labeled with the positron emitter 18F (half-life: 109.8 min) by nucleophilic aliphatic substitution (Fig. 1) with a GE TRACERlab MXFDG synthesizer. 18F Fluorination was achieved by reaction of the tosylate precursor (compound 2) with cyclotron-produced 18F-fluoride as an 18F-labeled K–F–Kryptofix-222 complex in acetonitrile at 85°C for 10 min. 18F-DPA-714 was purified by semipreparative reversed-phase HPLC. Total radiosynthesis time for the preparation of 18F-DPA-714 was 40 min. Initial attempts to produce 18F-DPA-714 with only 2.5 mg of the tosylate precursor (compound 2) afforded the pure product at a 6% non–decay-corrected radiochemical yield. Increasing the amount of starting tosylate up to 12 mg improved the process. The highest yields, 16% non–decay-corrected, were obtained with 6 mg or more.
In Vitro Binding
The affinity of DPA-714 for the TSPO was evaluated by use of a membrane-binding assay with 3H-PK11195 as the radioligand and rat kidney tissue. To ensure the selectivity of DPA-714, its binding to the central benzodiazepine receptor (CBR) was assessed with 3H-Ro15-1788 and rat brain tissue. The affinity of DPA-714, Ki = 7.0 ± 0.4 nM, was not as high as that of DPA-713, Ki = 4.7 ± 0.2 nM, but was slightly higher than that of PK11195, Ki = 9.3 ± 0.5 nM, in the same assay (Table 1). All 3 TSPO ligands, DPA-714, DPA-713, and PK11195, displayed negligible affinities for the CBR.
Affinities of Ligands for TSPO and CBR with 3H-PK11195 and Rat Kidney Membranes or 3H-Ro15-1788 and Rat Brain Tissue as Receptor Sources, Respectively
In Vitro Steroid Biosynthesis Assay
DPA-714 was assessed for its ability to increase pregnenolone synthesis by use of a well-developed steroidogenic assay (24). DPA-714 stimulated pregnenolone synthesis at levels 80% above the baseline, displaying significantly greater potency than PK11195. DPA-713 had no effect on steroidogenesis in the same assay (Fig. 2).
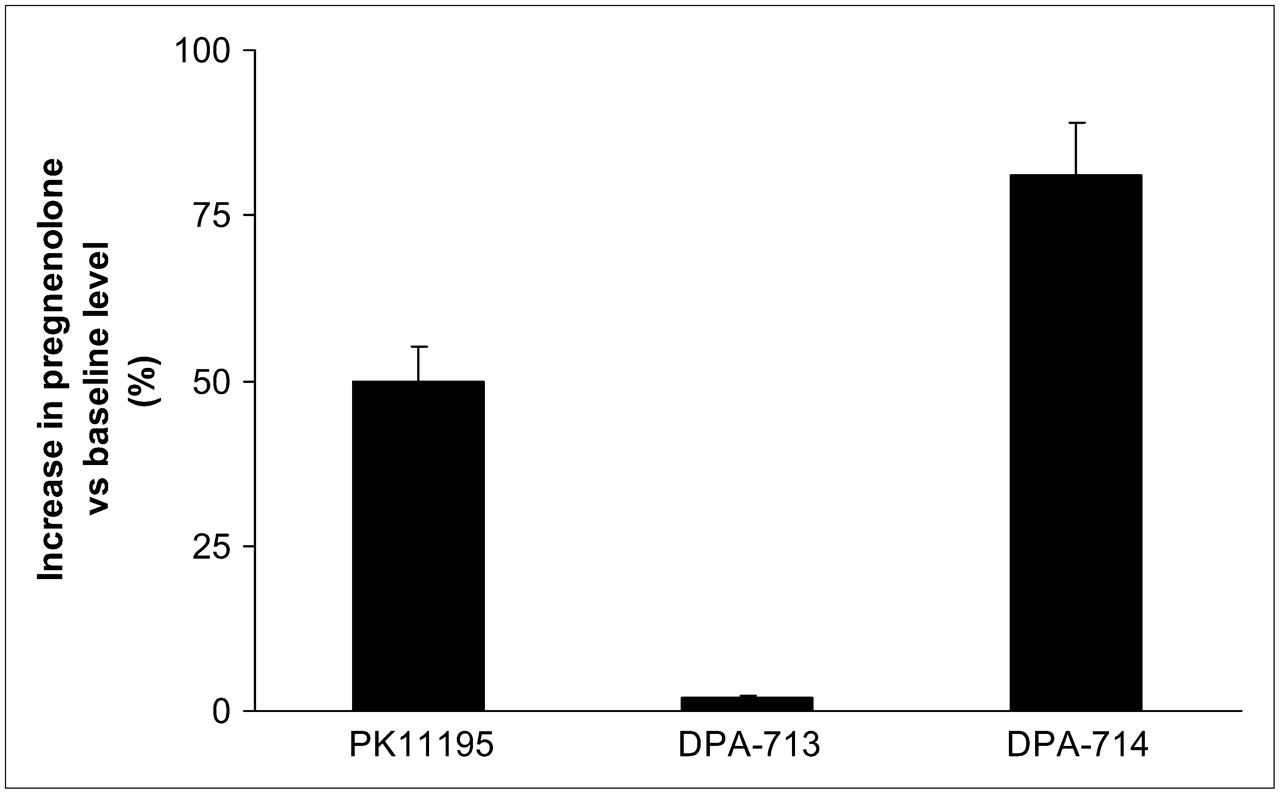
Effect of TSPO ligands on pregnenolone accumulation in rat C6 glioma cells. All compounds were used at same concentration (40 μM). At end of incubation period (2 h), amount of pregnenolone was quantified by radioimmunoassay.
Log D Determination
The lipophilicities of DPA-714, DPA-713, and PK11195 were evaluated by use of HPLC. No column deterioration was observed for the duration of the experiments, and all measurements were obtained on the same day to minimize error. The tR values were recorded for the reference compounds, and a calibration graph was generated. DPA-714 and DPA-713 had the same log D value (2.44), which was lower than that of PK11195 (3.35) (Table 1).
Rodent Studies
Peripheral Biodistribution.
Figure 3 shows the peripheral distribution of 18F-DPA-714 in each of the 4 groups of rats with QA lesions (control and 3 pretreatment groups: PK11195, DPA-713, and DPA-714). The results for the control group showed that the highest accumulation of 18F-DPA-714 was in the heart and adrenal glands, with only minimal accumulation in the bone, liver, and muscle. Rats pretreated with PK11195 (5 mg/kg) demonstrated significant inhibition of radioligand uptake in the heart, bone, and liver but not in the adrenal glands or muscle. In contrast, pretreatment with either DPA-714 (1 mg/kg) or DPA-713 (1 mg/kg) resulted in the inhibition of 18F-DPA-714 uptake in the heart, liver, bone, and muscle and in partial blockade in the adrenal glands.
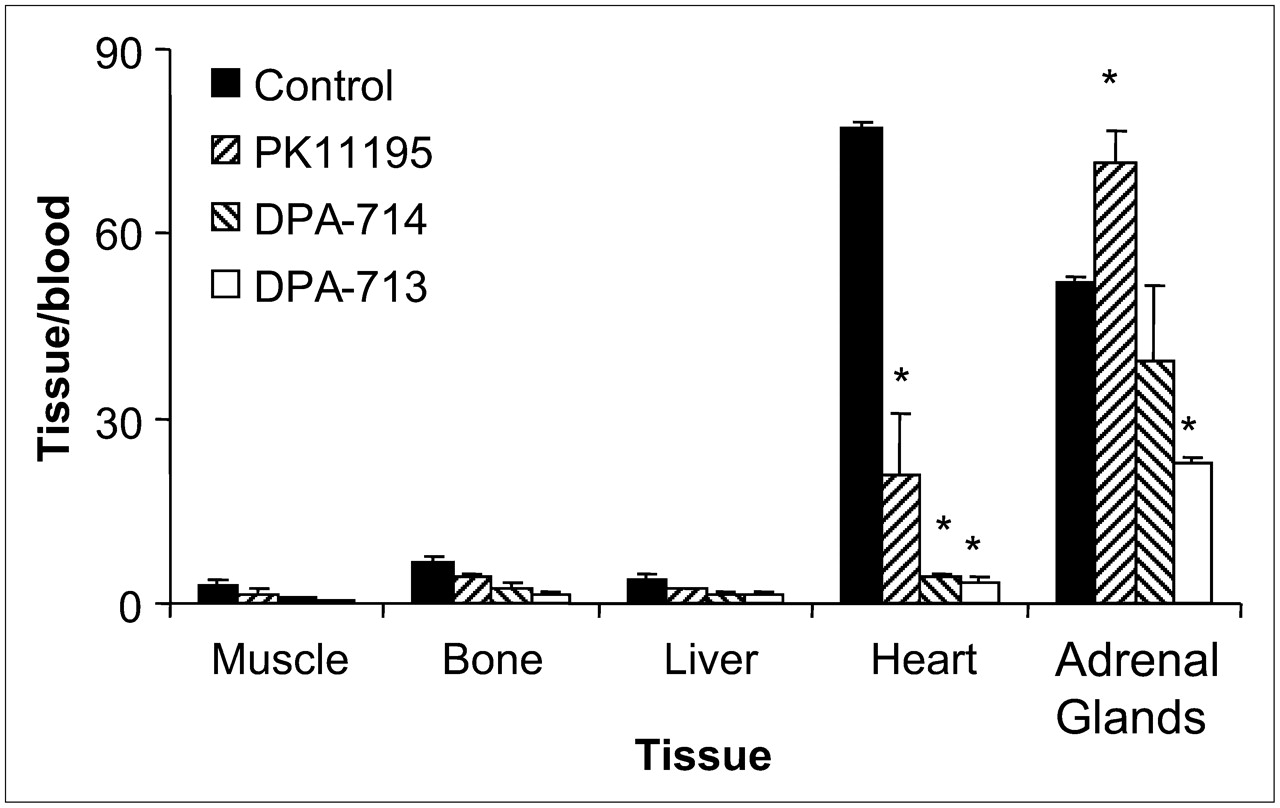
Uptake of 18F-DPA-714 (control) and effects of pretreatment with DPA-714, DPA-713, and PK11195 in various peripheral tissues of rats with QA lesions. 18F-DPA-714 uptake in each tissue is expressed relative to uptake in blood. *P < 0.05.
Cerebral Biodistribution.
Figure 4 shows the cerebral distribution of 18F-DPA-714 in each of the 4 groups of rats with QA lesions. The results for the control group indicated a statistically significant increase in 18F-DPA-714 uptake in the ipsilateral striatum (4.81 ± 0.47 vs. 0.61 ± 0.14; P < 0.05). The accumulation of 18F-DPA-714 in the ipsilateral striatum was decreased by preinjection with PK11195 (1.90 ± 0.59 vs. 1.18 ± 0.16; P < 0.05). A visible decrease was also seen after preinjection with either DPA-714 (2.00 ± 0.23 vs. 0.76 ± 0.17) or DPA-713 (1.08 ± 0.08 vs. 1.06 ± 0.05).

Uptake of 18F-DPA-714 (control) and effects of pretreatment with DPA-714, DPA-713, and PK11195 in various cerebral regions of rats with QA lesions. 18F-DPA-714 uptake in each region is expressed relative to uptake in cerebellum. *P < 0.05. Cx = cortex; Hipp = hippocampus; Str = striatum.
Baboon PET Studies
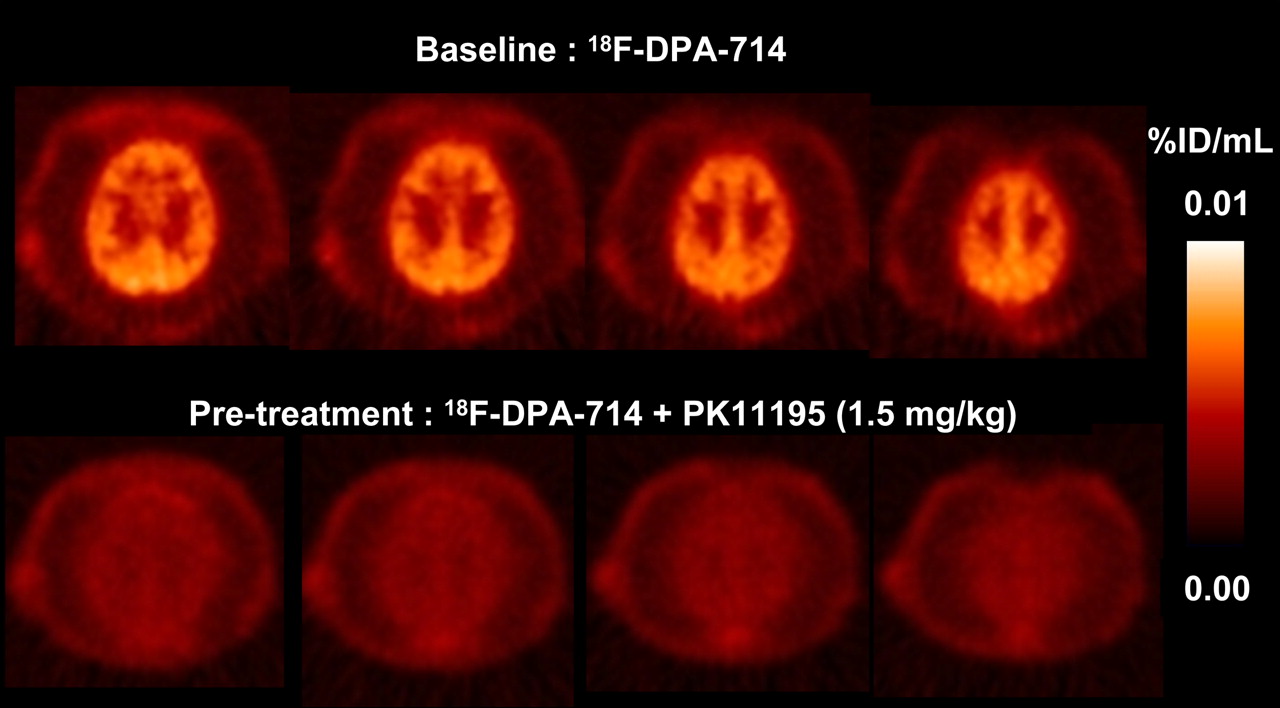
PET studies were conducted with an anesthetized P. hamadryas baboon to evaluate the in vivo regional distribution kinetics of 18F-DPA-714. First, in a baseline study, dynamic PET brain imaging commenced a few minutes before the intravenous administration of 18F-DPA-714 (100 MBq in 2 mL of saline; specific activity, 270 GBq/μmol) and was terminated 60 min after injection. This acquisition was followed by a whole-body acquisition for 2 min at each of 6 bed positions. 18F-DPA-714 was capable of penetrating the blood–brain barrier, with accumulation in the brain (Fig. 5). Second, in a blockade study, pretreatment with the TSPO ligand PK11195 (1.5 mg/kg) was used to determine the specificity of 18F-DPA-714 uptake. PK11195 effectively blocked the uptake of 18F-DPA-714 in the brain (Fig. 5). Finally, a displacement study was conducted to assess the reversibility of radioligand binding. The displacement study involved the administration of nonlabeled DPA-714 (1 mg/kg) 20 min after the 18F-DPA-714 injection.
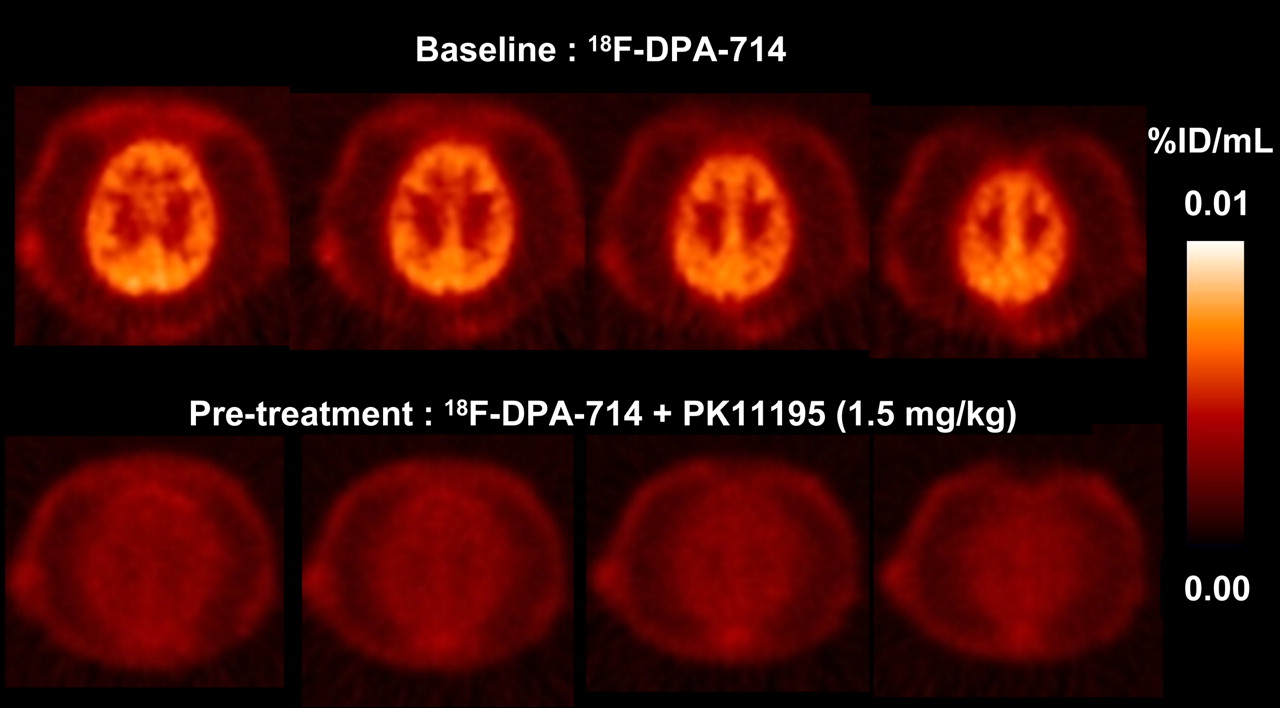
P. hamadryas baboon PET summation images of selected transaxial brain slices over 60 min.
Time–activity curves representing the uptake of 18F-DPA-714 in the whole baboon brain in each of the 3 PET studies are shown in Figure 6. In the baseline study, 18F-DPA-714 uptake reached a maximum within 10 min and remained at approximately the same level for the following 50 min. The results of the displacement study appeared to be analogous to those of the baseline study during the first 20 min; however, after the injection of nonlabeled DPA-714, the curve rose to a sharp peak followed by complete washout of the radioligand. In contrast, pretreatment with the TSPO-specific ligand PK11195 resulted in an initial increase in 18F-DPA-714 uptake followed by a rapid decrease to the washout level, identical to that seen in the displacement study. The whole-body image visibly depicted the high uptake of 18F-DPA-714 in the heart, kidneys, adrenal glands, and salivary glands (Fig. 7A). The injection of nonlabeled DPA-714 20 min after radioligand administration led to the complete displacement of 18F-DPA-714 binding in these peripheral regions (Fig. 7B).
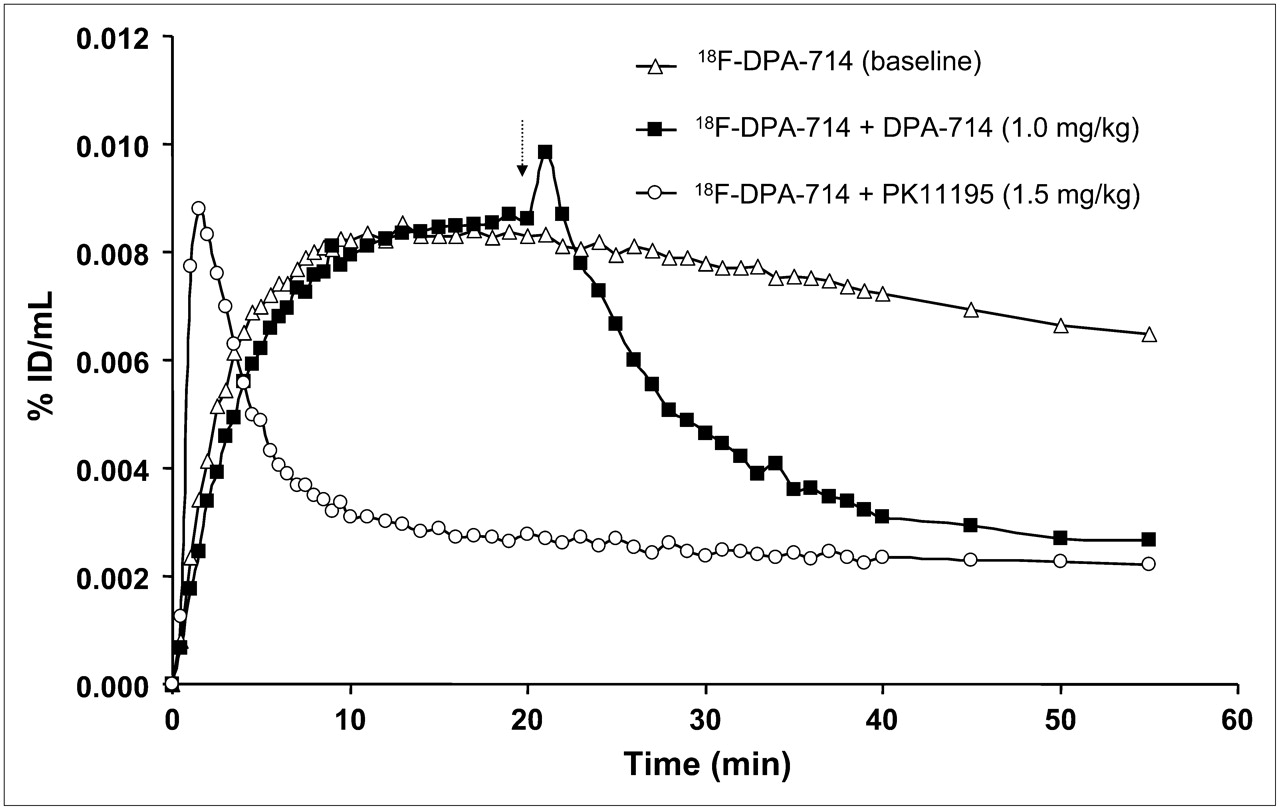
Time–activity curves depicting uptake of 18F-DPA-714 in whole P. hamadryas baboon brain during 60-min PET scan time for all 3 studies: baseline 18F-DPA-714, block 18F-DPA-714 plus PK11195 (1.5 mg/kg), and displacement 18F-DPA-714 plus DPA-714 (1 mg/kg). Arrow indicates time of administration of displacing agent.
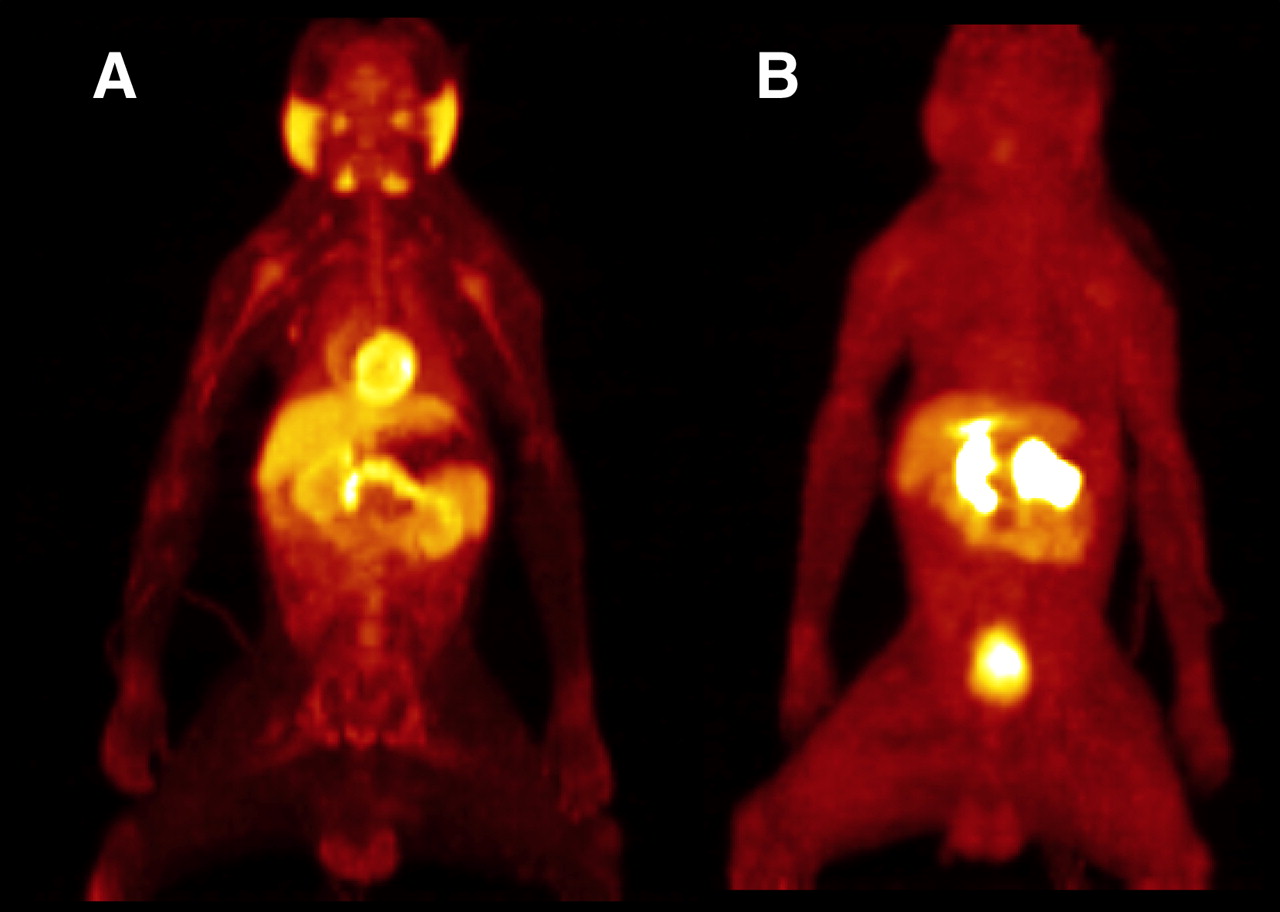
PET images of whole P. hamadryas baboon body 60 min after intravenous injection of 18F-DPA-714 (baseline) (A) and 18F-DPA-714 plus DPA-714 (1.0 mg/kg) (B).
DISCUSSION
Over the last decade, TSPO ligand development has yielded a multitude of new high-affinity compounds (34); however, the range of PET ligands for the TSPO remains limited. 11C-PK11195 is still the most widely used PET tracer for TSPO studies, despite its deficiencies (19). Our goal has been to develop a TSPO-specific ligand with in vivo properties that are an improvement over or that are different from those of 11C-PK11195. In 2005, we reported the synthesis of the first TSPO-specific pyrazolopyrimidine radioligand, 11C-DPA-713 (25). Our current aim was to synthesize an 18F-labeled derivative of DPA-713.
In vitro binding experiments with the fluorine analog DPA-714 demonstrated that substituting the methoxy moiety for the fluoroethoxy moiety had no adverse effect on the TSPO-binding affinity or lipophilicity (Table 1). Steroid assay results indicated that DPA-714 increased pregnenolone production with much greater potency (80% above the baseline levels) than PK11195 and DPA-713 (50% and 1% above the baseline levels, respectively) (Fig. 2). This finding meant that although TSPO affinity was maintained after the exchange of methoxy for fluoroethoxy, the functional activity was effectively reversed. These results suggest a potential therapeutic role for DPA-714 in anxiety-based conditions. Further experiments are currently under way to evaluate this unexpected change in functional activity.
Rats with QA lesions showed high concentrations of 18F-DPA-714 in TSPO-rich peripheral tissues, including the heart and adrenal glands (Fig. 3). Higher levels of 18F-DPA-714 in the ipsilateral striatum and cortex than in the contralateral striatum and cortex (Fig. 4) were consistent with the known ability of QA to increase TSPO expression (35). This increased uptake in lesion-containing brain areas indicated TSPO-specific binding because pretreatment of rats with PK11195, DPA-713, or DPA-714 before radioligand injection inhibited this uptake. 18F-DPA-714 uptake was also inhibited in most peripheral organs in rats in each pretreatment group, demonstrating TSPO-specific binding outside the brain. Interestingly, PK11195 failed to inhibit the binding of 18F-DPA-714 in the adrenal glands, whereas a partial blockade was seen with both DPA-713 and DPA-714 (Fig. 3). These results may provide evidence for the existence of TSPO subtypes or splice variants, and this possibility is currently being evaluated in further studies. Overall, 18F-DPA-714 was observed to cross the rodent blood–brain barrier and specifically bind to the TSPO in areas of neuroinflammation.
The biodistribution and specificity of 18F-DPA-714 were further evaluated in a normal male P. hamadryas baboon in 3 separate PET experiments. The results of the baseline study indicated rapid blood–brain barrier penetration and good uptake of the radioligand in the baboon brain (Fig. 5). Pretreatment with PK11195 resulted in the complete inhibition of 18F-DPA-714 uptake in the brain, suggesting that the uptake observed represented specific binding to TSPO. The whole-body image (Fig. 7A) depicted marked radioligand uptake in TSPO-rich regions similar to that seen in rodent studies. Figure 7B demonstrated the ease with which nonlabeled DPA-714 displaced 18F-DPA-714 in the heart and kidneys, denoting TSPO-specific binding outside the brain. 18F-DPA-714 has yet to be evaluated concurrently with other TSPO-specific radioligands, such as 11C-DAA1106 and 18F-FEDAA1106; however, the shapes of their respective time–activity curves appear to be remarkably similar to that of 18F-DPA-714 (23,36). All 3 time–activity curves display rapid brain uptake, good retention, and effective displacement after the injection of the nonlabeled ligand. The only difference is that 18F-DPA-714 takes slightly longer to reach maximum brain uptake and hence may possess slower kinetics than 11C-DAA1106 or 18F-FEDAA1106 in nonhuman primates. Future studies should involve the evaluation of all 3 radioligands in separate PET experiments with the same subject to enable meaningful comparisons.
CONCLUSION
Taken together, these results indicate that 18F-DPA-714 is a high-affinity radioligand specific for the TSPO. Its suitable in vivo properties and ability to bind to the TSPO with high specificity, along with its significant effect on steroid synthesis, make this ligand an attractive tool for evaluating the TSPO in neuroinflammatory processes.
Acknowledgments
This study was funded in part by the EC-FP6 project DiMI, LSHB-CT-2005-512146.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication August 7, 2007.
- Accepted for publication January 16, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Mutation in the rat interleukin 34 gene impacts macrophage development, homeostasis and inflammation in the brain and periphery
- uPAR Immuno-PET in Pancreatic Cancer, Aging, and Chemotherapy-Induced Senescence
- [18F]DPA-714 PET Imaging in the Presurgical Evaluation of Patients With Drug-Resistant Focal Epilepsy
- PET imaging of fibroblast activation protein alpha (FAP) detects incipient cardiotoxicity due to anthracycline chemotherapy
- Short-Term Colony-Stimulating Factor 1 Receptor Inhibition-Induced Repopulation After Stroke Assessed by Longitudinal 18F-DPA-714 PET Imaging
- A Longitudinal PET/MRI Study of Colony-Stimulating Factor 1 Receptor-Mediated Microglia Depletion in Experimental Stroke
- Structural and Clinical Correlates of a Periventricular Gradient of Neuroinflammation in Multiple Sclerosis
- Individual Mapping of Innate Immune Cell Activation Is a Candidate Marker of Patient-Specific Trajectories of Worsening Disability in Multiple Sclerosis
- Neuroinflammatory Changes in Relation to Cerebrospinal Fluid Viral Load in Simian Immunodeficiency Virus Encephalitis
- 11C-DPA-713 Versus 18F-GE-180: A Preclinical Comparison of Translocator Protein 18 kDa PET Tracers to Visualize Acute and Chronic Neuroinflammation in a Mouse Model of Ischemic Stroke
- Combined PET Imaging of the Inflammatory Tumor Microenvironment Identifies Margins of Unique Radiotracer Uptake
- In Vivo Quantification of Cerebral Translocator Protein Binding in Humans Using 6-Chloro-2-(4'-123I-Iodophenyl)-3-(N,N-Diethyl)-Imidazo[1,2-a]Pyridine-3-Acetamide SPECT
- The Translocator Protein Radioligand 18F-DPA-714 Monitors Antitumor Effect of Erufosine in a Rat 9L Intracranial Glioma Model
- Metabolism and Quantification of [18F]DPA-714, a New TSPO Positron Emission Tomography Radioligand
- Reactive Astrocytes Overexpress TSPO and Are Detected by TSPO Positron Emission Tomography Imaging
- Imaging Microglial/Macrophage Activation in Spinal Cords of Experimental Autoimmune Encephalomyelitis Rats by Positron Emission Tomography Using the Mitochondrial 18 kDa Translocator Protein Radioligand [18F]DPA-714
- Radiation Dosimetry and Biodistribution of the TSPO Ligand 11C-DPA-713 in Humans
- Quantitative, Preclinical PET of Translocator Protein Expression in Glioma Using 18F-N-Fluoroacetyl-N-(2,5-Dimethoxybenzyl)-2-Phenoxyaniline
- 18F-FEAC and 18F-FEDAC: PET of the Monkey Brain and Imaging of Translocator Protein (18 kDa) in the Infarcted Rat Brain
- Radioisotopic Imaging of Neuroinflammation
- Quantification of Translocator Protein (18 kDa) in the Human Brain with PET and a Novel Radioligand, 18F-PBR06
- Comparative Evaluation of the Translocator Protein Radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a Rat Model of Acute Neuroinflammation