


Abstract
PET with 11C-acetate (11C-ACE) has a high sensitivity for detection of prostate cancer and several other cancers that are poorly detected with 18F-FDG. However, the short half-life (20.4 min) of 11C limits the general availability of 11C-ACE. 18F-Fluoroacetate (18F-FAC) is an analog of acetate with a longer radioactive half-life (18F = 110 min). This study was undertaken to assess the potential usefulness of 18F-FAC as a prostate tumor imaging agent. Methods: We developed an efficient radiosynthesis for 18F-FAC, which has already been adapted to a commercial synthesizer. Biodistribution studies of 18F-FAC were compared with 11C-ACE in normal Sprague–Dawley male rats and CWR22 tumor-bearing nu/nu mice. We also performed a small-animal PET study of 18F-FAC in CWR22 tumor-bearing nu/nu mice and a whole-body PET study in a baboon to examine defluorination. Results: We obtained 18F-FAC in a radiochemical yield of 55% ± 5% (mean ± SD) in ∼35 min and with a radiochemical purity of >99%. Rat biodistribution showed extensive defluorination, which was not observed in the baboon PET, as indicated by the standardized uptake values (SUVs) (SUVs of iliac bones and femurs were 0.26 and 0.3 at 1 h and 0.22 and 0.4 at 2 h, respectively). CWR22 tumor-bearing nu/nu mice showed tumor uptake (mean ± SD) of 0.78 ± 0.06 %ID/g (injected dose per gram of tissue) for 11C-ACE versus 4.01 ± 0.32 %ID/g for 18F-FAC. For most organs—except blood, muscle, and fat—the tumor-to-organ ratios at 30 min after injection were higher with 18F-FAC, whereas the tumor-to-heart and tumor-to-prostate ratios were similar. Conclusion: All of these data indicate that 18F-FAC may be a useful alternative to 11C-ACE tracer for the detection of prostate tumors by PET.
An estimated 1,399,790 new cancer cases were projected for 2006, with about 564,830 Americans expected to die from cancer in that year (1). New cases of prostate cancer for 2006 were estimated to be 234,460 or 16.7% of the total new cancer cases. About 27,350 men were expected to die of prostate cancer, making it the third leading cause of cancer death in men in the United States (1). In recent years, there have been several advances in the detection of prostate cancer by biochemical screening as well as in its treatment (2). Despite these advances and improvements in imaging methods, the ability to delineate prostate cancer at the primary site or to detect recurrent or metastatic tumor foci is still far from satisfactory (3).
Prostate cancer is the most prevalent tumor for which imaging by PET with 18F-FDG has been found to be generally unsatisfactory. The 60%–70% sensitivity of 18F-FDG PET for prostate cancer is not high enough to justify its routine clinical use for staging or restaging of this disease (4–6). The poor performance of 18F-FDG PET is likely related to the low glucose metabolic rate that results from the relatively slow growth of most prostate cancers as well as to other factors, including significant excretion of 18F-FDG into the adjacent urinary bladder, making detection of tumor uptake difficult.
11C-Acetate (11C-ACE) has been used for many years as a tracer for measuring oxidative metabolism in the myocardium (7). After it is taken up by the myocardium, 11C-ACE is converted into acetyl-coenzyme A in the mitochondria, which is followed by rapid metabolism via the citric acid cycle and clearance as CO2. Recently, 11C-ACE has been reported to show high uptake in a variety of tumors, including renal cell carcinomas and hepatomas (8,9). Previous clinical studies with 11C-ACE have reported improved sensitivity, up to 100%, for detection of primary tumors in patients with untreated prostate cancer, by comparison with conventional imaging methods including 18F-FDG PET (10,11), and a 59% sensitivity for detection of recurrent prostate cancer in patients with prostate-specific-antigen relapse (12). Yoshimoto et al. demonstrated that increased 11C-ACE accumulation in tumor cells is caused by enhanced lipid synthesis (13). The mechanism of tumor uptake of 11C-ACE thus appears to be its incorporation into cell membrane lipids; there is little excretion of this agent into the urine and relatively rapid clearance of the tracer from most other tissues because of its oxidative metabolism to 11C-CO2 (13). However, the potential for widespread use of 11C-ACE is limited by the short radioactive half-life (20.4 min) of 11C, which necessitates production with an in-house cyclotron. Even with a high-yield synthesis, only a limited number of patients can be studied from a single-batch preparation. Accordingly, there is considerable interest in identifying positron-emitting radiopharmaceuticals labeled with isotopes with longer half-lives that are suitable for imaging of prostate cancer. One such radiopharmaceutical that has been studied is 18F-fluoroethylcholine, which also appears to be a cell membrane precursor compound (14). Another potential agent is the acetate analog 18F-fluoroacetate (18F-FAC). The purpose of this study was to assess the biodistribution of 18F-FAC in animal models as compared with 11C-ACE, to evaluate its potential as a radiopharmaceutical for imaging prostate cancer. We also report here a very reproducible method of synthesis for 18F-FAC that is faster than the currently available methods and that has already been adapted to a commercial synthesizer (15). Preliminary reports of this work have been presented elsewhere (16,17).
MATERIALS AND METHODS
General
Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich Chemical Co. and used without further purification. H218O was purchased from Rotem Industries. Screw-cap test tubes used for fluoride incorporation were purchased from Fisher Scientific (Pyrex no. 9825). Oasis HLB-6cc cartridges, 500 mg, were purchased from Waters Corp. (part no. 186000115). Vacutainer tubes (5 mL) were obtained from Becton-Dickinson (part no. 366434). The chemical and radiochemical purities of the FAC were assayed by high-pressure liquid chromatography (HPLC) on a Fast Acid Analysis column (part no. 125-0100; Bio-Rad), 100 × 7.8 mm, eluted with 10% acetonitrile in 0.007N H2SO4 solution at a flow rate of 0.6 mL/min and ultraviolet (UV) wavelength at 210 nm in the presence of the unlabeled compound as standard. The radiochemical purity of the product was also checked by radio–thin-layer chromatography (radio-TLC). For the TLC analyses, polygram G/UV254 plastic-backed TLC plates from Whatman were used and eluted with 95% acetonitrile in water as the mobile phase. The TLC plates were analyzed using a Bioscan Inc., System 200 imaging scanner. Residual acetonitrile was determined by gas chromatography with a Varian CP-3800 provided with a flame ionization detector using a DB-Wax column 30 m × 0.53 mm inner diameter at 30°C (5 min) ramped at 90°C/2 min to 250°C and a helium flow rate of 4.5 mL/min. Rodents were obtained from Charles River Laboratories and were housed in a barrier facility with a corncob-bedding that was changed twice a week. Animal handling techniques have been described previously (18).
Statistical Methods
To compare differences in uptake of 18F-FAC and 11C-ACE, the Student t test was performed. Differences at the 95% confidence level (P < 0.05) were considered significant.
Synthesis of Substrate Ethyl O-Mesylglycolate (2)
To a 250-mL round-bottom flask were added ethyl glycolate (5.0 g, 48 mmol) and methanesulfonyl chloride (4.0 mL, 51.2 mmol) dissolved in methylene chloride (50 mL). The solution was cooled to 0°C, and triethylamine (4 mL, 28.6 mmol) was added dropwise with stirring. After stirring for 1 h at room temperature, the reaction mixture was extracted with ice-cold 1N HCl (50 mL) and then with water (3 × 50 mL). The organic phase was separated and dried over anhydrous sodium sulfate and then concentrated. The crude mixture was then purified by silica gel column chromatography, using petroleum ether and ethyl acetate as eluent, to give ethyl O-mesylglycolate (2) as a colorless oil in a 73% yield.
1H NMR (CDCl3, 300 MHz): δ 1.31 (t, 3H, CH2CH3); 3.21 (s, 3H, OCH3); 4.26 (q, 2H, OCH2-CH3); 4.76 (s, 2H, OCH2CO).
Synthesis of Fluoroacetate (Nonradioactive Standard)
The synthesis of the nonradioactive standard was performed according to published work (19). To a mixture of ethyl fluoroacetate (10.6 g, 0.1 mol) and 20 mL of water containing a few drops of an alcoholic solution of phenolphthalein, finely powdered barium hydroxide octahydrate was added in small portions while the mixture was stirred vigorously. Approximately 16–20 g of barium hydroxide were consumed before the reaction mixture became alkaline. The resulting mixture was filtered, and the filtrate was evaporated to dryness in vacuo. The residue was dissolved in 10 mL of water, and barium fluoroacetate was precipitated by the addition of ethyl alcohol (60 mL). The precipitate (13 g) was added in portions to 28 g of stirred concentrated H2SO4. The mixture was then slowly distilled at reduced pressure (15–20 mm), and, at ∼200°C; approximately 7 g of crude fluoroacetic acid were collected in the receiver. Redistillation of this distillate over the temperature range of 164°C–170°C yielded 4 g of pure fluoroacetic acid, which solidified in the receiver (literature melting point 35.2°C).
1H NMR (CDCl3, 300 MHz): δ 4.91 (s, 1H, F-CH); 5.03 (s, 1H, F-CH); 8.95 (s, 1H, COOH).
Production of 18F-Fluoride
18F-Fluoride was produced at our institution by the 18O(p,n)18F reaction through proton irradiation of 95% enriched 18O-water, using either the JSW BC16/8 cyclotron (The Japan Steel Works Ltd.) or the CS15 cyclotron (The Cyclotron Corp.). The radiosynthesis of 18F-FAC is shown in Scheme 1:
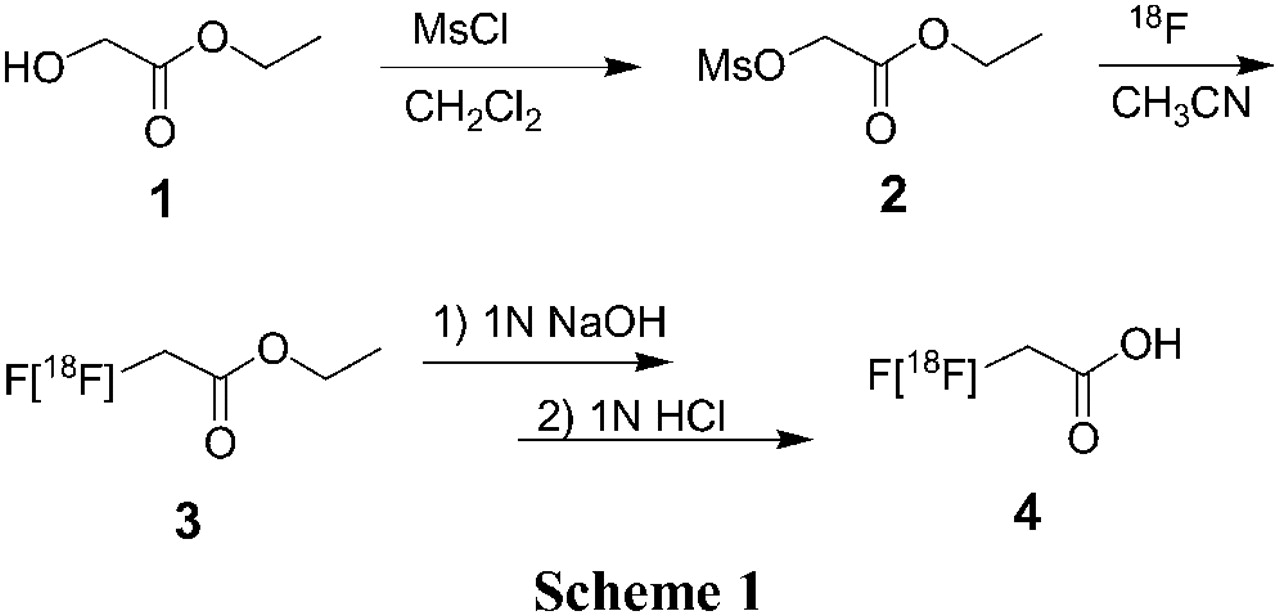
Radiosynthesis of 18F-Fluoroacetic Acid (4)
18F-Fluoride, 3.7–5.55 GBq (100–150 mCi), was added to a Vacutainer tube containing 6.0 mg Kryptofix 2.2.2. and 1.0 mg K2CO3. Water was azeotropically evaporated from this mixture in an oil bath at 110°C using HPLC-grade acetonitrile (3 × 0.5 mL) under a stream of nitrogen. After the final drying sequence, 300 μL of acetonitrile were added, and the redissolved 18F residue was transferred to a 10-mL Pyrex brand tube with a screw cap containing 1 mg of ethyl O-mesylglycolate (2). A 3-mm glass bead was also added to the tube for a more homogeneous energy distribution during the fluoride labeling, and the tube was capped firmly on a specially designed, remotely operated capping station. The contents were vortexed before being heated in an oil bath at 100°C for 5 min.
After the reaction mixture had been cooled to room temperature, 7 mL of water were then added. A solid-phase purification was performed using a Waters (HLB-6cc) Oasis cartridge preconditioned previously with ethanol (5 mL), which was followed by water (8–10 mL). The radioactive sample was applied to the cartridge and rinsed with 4 × 6 mL of additional water to eliminate any unreacted fluoride. The radiolabeled intermediate 3 was then eluted from the solid-phase media with ethanol (7 mL). To the ethanol eluant 200 μL of 1N NaOH were added, and the hydrolysis was completed while the ethanol was being removed under reduced pressure on a rotary evaporator provided with a water bath at 40°C. The residual mixture was then neutralized with 1N HCl (180 μL), diluted with 5 mL of 0.9% saline solution, and passed through a sterile 0.22-μm filter to render an isotonic, sterile, and nonpyrogenic solution ready for injection. The radio-TLC showed the intermediate compound 3 with an Rf value of 0.88, whereas the Rf value of 18F-FAC 4 was 0.16. The analytic HPLC showed only one radioactive peak corresponding to the product 18F-FAC, eluting at ∼4.30 min. The total preparation time was ∼35 min.
Radiosynthesis of 11C-ACE
11C-ACE was synthesized using a Grignard Chemistry module (Siemens CTI). Briefly, after irradiation to produce 11C-CO2, the radioactive gas was bubbled through 50 μL of 3 mol/L methylmagnesium bromide, diluted with 1.0 mL of diethyl ether. Sterile water (0.25 mL) was added and the ether was evaporated at 145°C with helium gas. The intermediate was hydrolyzed by addition of 0.5 mL of 10% phosphoric acid, and the product was distilled at 145°C into USP (United States Pharmacopeia) 0.9% saline solution for injection. After the pH was adjusted to 4.5–8.5 with 2 or 3 drops of 8.4% sodium bicarbonate, the product was passed through a sterile 0.22-μm filter to render an isotonic, sterile, and nonpyrogenic solution ready for injection. Radiochemical and chemical purities were determined by analytic HPLC (Bio-Rad Fast Acid column); the product 11C-ACE eluted at ∼3.6 min. The total preparation time was ∼15 min.
Biodistribution Study in Sprague–Dawley Male Rats
All animal experiments were conducted in compliance with the Guidelines for the Care and Use of Research Animals established by the Animal Studies Committee at our institution. Housing of the animals, the heat lamps used to warm the animals during the procedures, temperature monitoring via a rectal thermometer, the hemodynamic monitoring system, and anesthetic procedures have all been described previously (18). After tracer administration, the animals were allowed to wake up and maintain normal husbandry until euthanasia by cervical dislocation.
Mature male Sprague–Dawley rats (160–200 g) were used in this study and were given standard rat chow and water ad libitum. In all animal studies, all of the radiopharmaceuticals diluted with 0.9% saline were used at >98% radiochemical purity. The animals were anesthetized with 1%–1.5% isoflurane and injected intravenously with 740 kBq (20 μCi) of 18F-FAC via the tail vein. At 20 min and 1, 2, and 4 h after injection (n = 5 per time point), the animals were sacrificed, and blood samples from a heart puncture, tibia and fibula bones, abdomen muscle, and other selected tissues were harvested, blotted dry, and weighed, and their radioactivity was determined in a Beckman Gamma 8000 well scintillation counter as reported previously (20). For comparison, another set of animals was also injected with 925 kBq (25 μCi) of 11C-ACE and sacrificed after 20 and 60 min (n = 5 per time point), and their organs were removed and analyzed as described. The injected dose (ID) was calculated by comparison with dose standards prepared from the injected solution of appropriate counting rates, and the data were expressed as percentage ID per gram of tissue (%ID/g).
Biodistribution Study in CWR22 Tumor-Bearing Male nu/nu Mice
Athymic nu/nu male mice, 4- to 6-wk old, were obtained from Charles River Laboratories. The CWR22 tumor line was a gift from Dr. Thomas G. Pretlow (Case Western Reserve University, Cleveland, OH). The CWR22 tumor was propagated in the animals by the implantation of minced tumor tissue, obtained from a previously established tumor, into the subcutaneous tissue of the flanks of the mice. For the maintenance of high serum androgen levels, mice were implanted subcutaneously with 12.5 mg of 60-d-releasing testosterone pellets from Innovative Research of America. Three weeks after tumor implantation, the mice of average weight 25.9 g (n = 4 per group) were anesthetized with 1%–2% isoflurane and injected with 370–444 kBq (10–12 μCi) of 18F-FAC via the tail vein. After 30 min and 2 h after injection, the animals were sacrificed. For comparison, another set of animals (n = 4 per group) was likewise injected with 740–925 kBq (20–25 μCi) of 11C-ACE and sacrificed after 30 min. Blood and selected tissues obtained as before were weighed, and their radioactivity was determined in a Beckman Gamma 8000 well scintillation counter. Biodistribution data were calculated as previously reported (20) as %ID/g (mean ± SD).
Small-Animal PET
Athymic nu/nu male mice, 4- to 6-wk old (n = 2), with CWR22 tumor implanted as described, were imaged. The mice (average weight, 25.9 g) were anesthetized with 1%–2% isoflurane and injected with an average of 10.8 MBq (294 μCi) of 18F-FAC. Static small-animal PET images (10-min static scan) were acquired with a microPET R4 scanner (Siemens Medical Solutions USA, Inc.) (21) at 30 min and 1 and 2 h after injection, and the tracer accumulation in the tumor was analyzed.
Data Collection and Imaging Reconstruction
The method of data collection for small-animal PET has been described previously (22). The list-mode raw data were first sorted into 3-dimensional sinograms, and static images were reconstructed by the maximum a posteriori (MAP) statistical reconstruction algorithm with a smoothing parameter (β = 0.1). All corrections (normalization, scatter, and attenuation) were performed during the reconstructions using the manufacturer's provided software. The MAP reconstruction method can produce an improved spatial resolution and favorable noise properties in comparison with the conventional filtered backprojection method (23,24). Regions of interest (ROIs) were drawn around each tumor or other organs (heart, spine, back muscle, and background) in the transaxial small-animal PET images that included the entire tumor or organ volume. The analysis was based on results normalized to the ID of radioactivity, and ratios of organs to muscle with background correction were calculated as the average radioactivity concentration in the tumor or other organs divided by the average radioactivity concentration in the muscle according to the following formula: where a.c. is the activity concentration (Bq/mL).
where a.c. is the activity concentration (Bq/mL).
Baboon Study
PET was performed with an ECAT EXACT HR+ tomograph (Siemens CTI Corp.). A 3-min transmission scan was performed using rotating 68Ge/68Ga line sources, before tracer injection and collection of initial emission scans. A male baboon (weight, 23.4 kg) was injected with 370 MBq (10 mCi) of 18F-FAC, and 7-min static emission scans of the whole body were performed at 1 and 2 h after injection. As an index of 18F-FAC uptake, the standardized uptake value (SUV) was calculated for circular ROIs drawn around the brain, liver, kidney, muscle, and bone. To minimize the dependency on the injected activity, the SUV was defined as the average radioactivity concentration in the organ divided by the total injected activity according to the following formula:
RESULTS
Radiochemistry
Our radiosynthetic method produced 18F-FAC with high radiochemical and chemical purities without the need for preparative HPLC. We obtained the final product in a radiochemical yield of 55% ± 5% (mean ± SD) and a radiochemical purity of >99% (n = 22, decay corrected to end of bombardment) in ∼35 min and practically free (<1%) of any unreacted 18F-fluoride. To calculate the specific activity of the final product, we synthesized fluoroacetic acid in the previously described manner (19). The UV trace at 210 nm of the analytic HPLC column showed a mass peak from which the amount corresponding to the mass of fluoroacetic acid present could be determined. Comparison of this peak with the response to a calibrated FAC standard allowed calculation of the mass of 18F-FAC present in the final product. The average specific activity of the final compound ranged from 74 to 129.5 GBq/μmol (2,000–3,500 Ci/mmol) at the end of synthesis (EOS) (n = 5). No radioactive peak corresponding to the unhydrolyzed intermediate (3) or any mass peak corresponding to the starting substrate (2) (which eluted with retention times of 6–7 min) was present. A mass peak corresponding to the saline solvent eluted at the column front and was well separated from the desired radioactive peak. No residual acetonitrile was present, as determined by gas chromatographic analysis. Aliquots of 11C-ACE were obtained from batches destined for clinical use at our institution (25) and ranged from 3.7 to 18.5 GBq (100–500 mCi) in 7.8–8.8 mL of USP saline solution at EOS. The chemical and radiochemical purities were >99%.
Biodistribution Study in Sprague–Dawley Male Rats
Biodistribution data were determined with 11C-ACE (Fig. 1A) at 20 min and 1 h after injection and with 18F-FAC (Fig. 1B) at 20 min and 1, 2, and 4 h after injection. Biodistribution data of 11C-ACE showed relatively rapid clearance (30%–60%) at 1 h, from most of the organs studied. Radioactivity from the pancreas, however, did not clear after 1 h (Fig. 1A, 0.84 ± 0.07 %ID/g [mean ± SD] at 20 min vs. 0.77 ± 0.20 %ID/g at 1 h; P = not significant [NS]). Biodistribution data of 18F-FAC showed that this tracer cleared significantly slowly from those organs investigated, including the pancreas (Fig. 1B, 0.36 ± 0.01 %ID/g at 20 min; 0.32 ± 0.02 %ID/g at 1 h; 0.25 ± 0.01 %ID/g at 2 h; 0.21 ± 0.01 %ID/g at 4 h; P < 0.001). After the injection of 18F-FAC, the radioactivity in bone increased significantly over time (Fig. 1B, 0.42 ± 0.02 %ID/g at 20 min; 0.76 ± 0.03 %ID/g at 1 h; 1.21 ± 0.05 %ID/g at 2 h; 1.87 ± 0.15 %ID/g at 4 h; P < 0.001), likely indicating defluorination. There was no difference in uptake in the normal prostate between 18F-FAC and 11C-ACE at 1 h (Figs. 1A and 1B, 0.28 ± 0.05 %ID/g vs. 0.28 ± 0.04 %ID/g, respectively; P = NS). We observed a slight but significant decrease in 18F-FAC uptake in the normal prostate over time (Fig. 1B, 0.30 ± 0.05 %ID/g at 20 min; 0.28 ± 0.05 %ID/g at 1 h; 0.23 ± 0.01 %ID/g at 2 h; 0.20 ± 0.03 %ID/g at 4 h; P = 0.002).

Rodent biodistribution study. (A) Biodistribution of 11C-ACE in Sprague–Dawley rats (n = 5 per group) at 20 min and 1 h after injection. (B) Biodistribution of 18F-FAC in Sprague–Dawley rats (n = 5 per group) at 20 min and 1, 2, and 4 h after injection. (C) Comparison of biodistribution of 11C-ACE vs. 18F-FAC in CWR22 tumor-bearing male nu/nu mice (n = 4 per group) at 30 min after injection. Changes in biodistribution for 18F-FAC from 30 min to 2 h after injection are also shown.
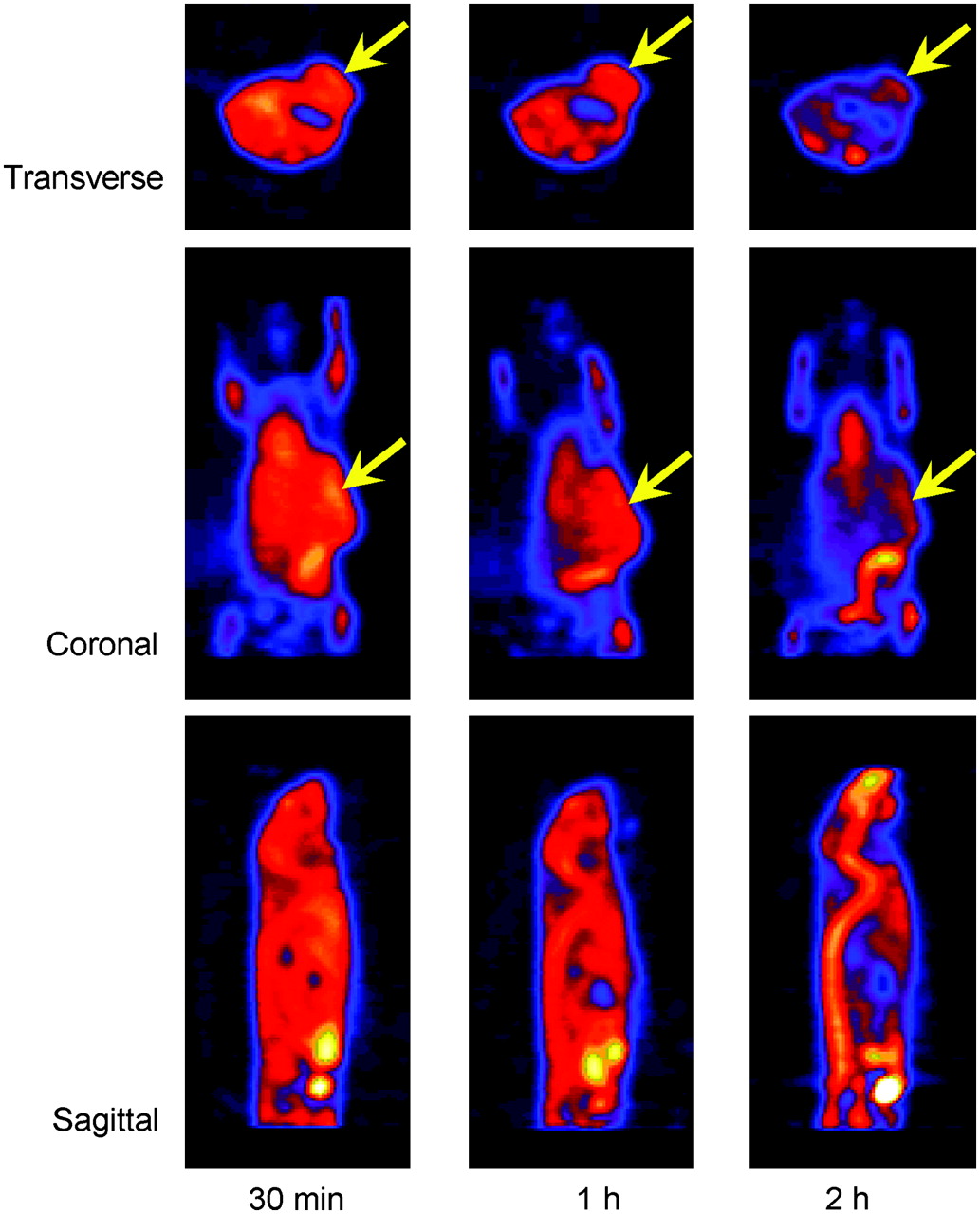
Small-animal PET images of mouse show 18F-FAC uptake in CWR22 tumors implanted in left flank: 18F-FAC uptake in mouse at 30 min and 1 h and 2 h after injection of about 10.7 MBq (290 μCi). Transverse, coronal, and sagittal images are shown. Images were reconstructed by the MAP method (β = 0.1). Arrows indicate localization of tumors.
Biodistribution Study in CWR22 Tumor-bearing Male nu/nu Mice
Analysis of the biodistribution data was performed for 11C-ACE at 30 min after injection and with 18F-FAC at 30 min and 2 h after injection (Fig. 1C). As in the case of the rat study, extensive defluorination was also observed in these mice, and the bone uptake increased significantly from 2.59 ± 0.36 %ID/g at 30 min to 5.23 ± 1.64 %ID/g at 2 h (P = 0.023). The uptake in normal prostate at 30 min was significantly higher with 18F-FAC compared with 11C-ACE (2.60 ± 0.50 %ID/g vs. 0.60 ± 0.17 %ID/g, respectively; P < 0.001). Indeed, except for the spleen at 3.63 ± 0.0.32 %ID/g for 11C-ACE versus 2.79 ± 0.21 %ID/g for 18F-FAC (P < 0.004), significantly higher uptake of 18F-FAC was found in all organs studied, including the tumor, which was ∼5 times higher at 30 min (0.78 ± 0.06 %ID/g for 11C-ACE vs. 4.01 ± 0.32 %ID/g for 18F-FAC; P < 0.001). Table 1 compares the tumor-to-organ ratios for both tracers at 30 min and at 2 h for 18F-FAC. We observed that, with the exception of the tumor-to-blood ratio (1.60 ± 0.27 for 11C-ACE vs. 0.93 ± 0.17 for 18F-FAC; P = 0.004), the tumor-to-muscle ratio (2.32 ± 0.32 for 11C-ACE vs. 1.43 ± 0.14 for 18F-FAC; P = 0.006), and the tumor-to-fat ratio (5.45 ± 2.68 for 11C-ACE vs. 3.45 ± 1.04 for 18F-FAC; P = NS), the 18F-FAC ratios were consistently higher at 30 min after injection than those for 11C-ACE at 30 min (P < 0.005). All ratios were even higher with 18F-FAC after 2 h (P < 0.005). The tumor-to-heart and tumor-to-prostate ratios were similar between the 2 radiotracers at 30 min (P < 0.005), and the 18F-FAC ratios did not change after 2 h (P < 0.005). Because of the short half-life of 11C, no comparison was possible between the 2 tracers at this later time. The extensive defluorination observed is responsible for the lower tumor-to-bone ratio obtained for 18F-FAC at 2 h versus that at 30 min (Table 1).
Comparison of 11C-ACE and 18F-FAC Tumor-To-Organ Ratios After Biodistribution Study in CWR22 Tumor-Bearing Male nu/nu Mice
Organ-to-Muscle Ratios (with Background Correction) After Injection of 18F-FAC to Male nu/nu Mice (n = 2) Implanted with CWR22 Minced Tumor Tissue in Left Flank
Small-Animal PET Study
The small-animal PET images (static collection, 10 min) at 30 min and 1 and 2 h after injection of 18F-FAC showed clear delineation of CWR22 tumor in the left flank of mice. Several organs (such as heart and vertebrae) showed high uptake of 18F-FAC (Fig. 2). Data for muscle were obtained from the forelimb. The ratios of tumor to heart, vertebrae, and muscle with background correction are shown in Table 2. The ratios of tumor to muscle of 2 mice at 2 h after injection were 1.44 and 1.40, respectively. The coronal view in Figure 2 also shows activity present in what appears to be the bowel.
Baboon PET Study
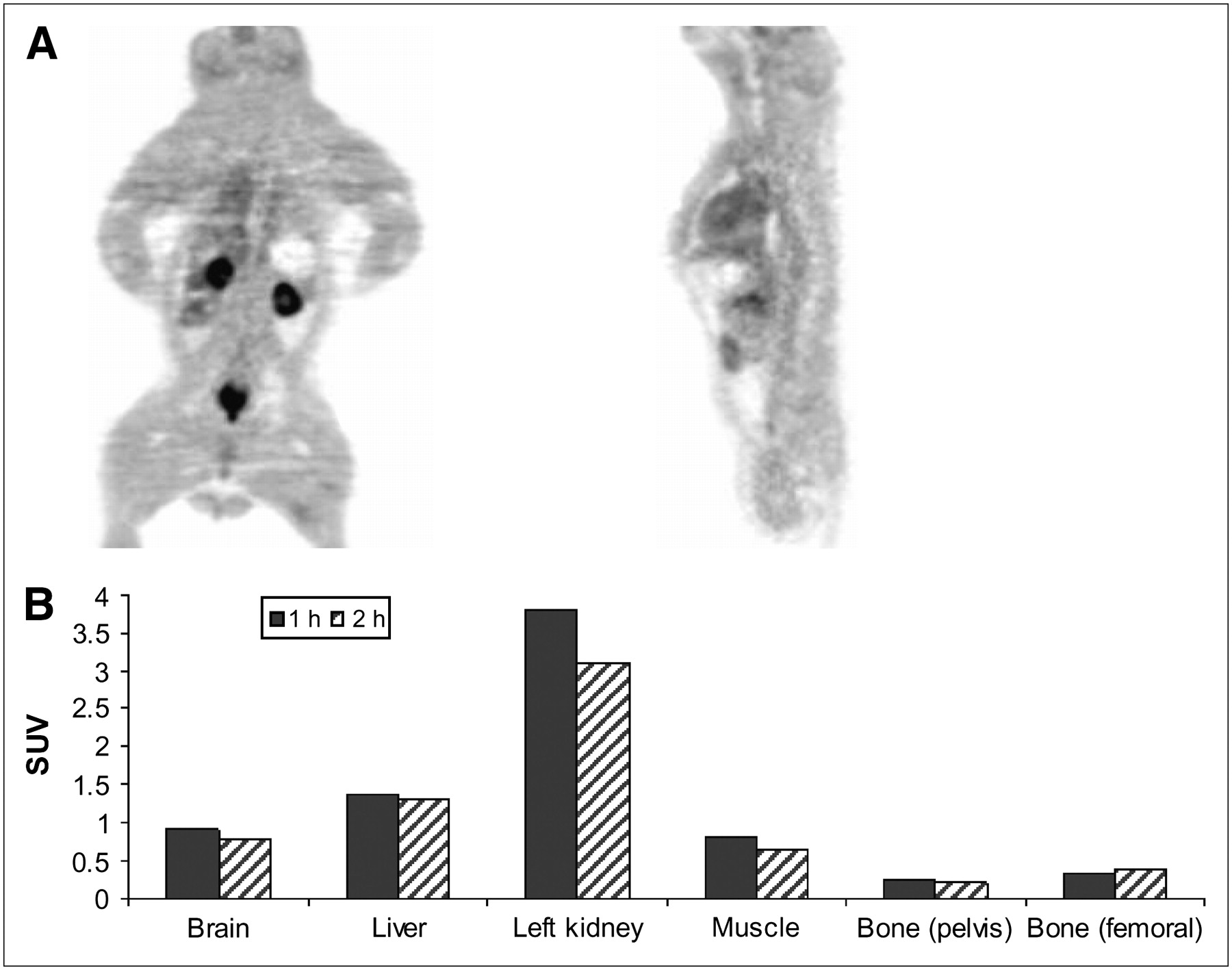
18F-FAC SUV uptake in baboon bone was not significantly different at 1 and 2 h after injection (Figs. 3A and 3B). The SUV of the iliac bone was 0.26 at 1 h and 0.22 at 2 h after injection; that of the femur was also low, 0.3 at 1 h and 0.4 at 2 h, indicating very little defluorination. The largest accumulation of radioactivity at 1 h was found in the kidneys (SUV = 3.7), which was followed by the liver (SUV = 1.3). These values were reduced slightly to 3.1 and 1.2, respectively, at 2 h. There was some radioactivity in the bladder, which indicated the excretion of metabolic by-products from the kidneys. Brain and muscle SUV uptakes were also low, below 1.0.
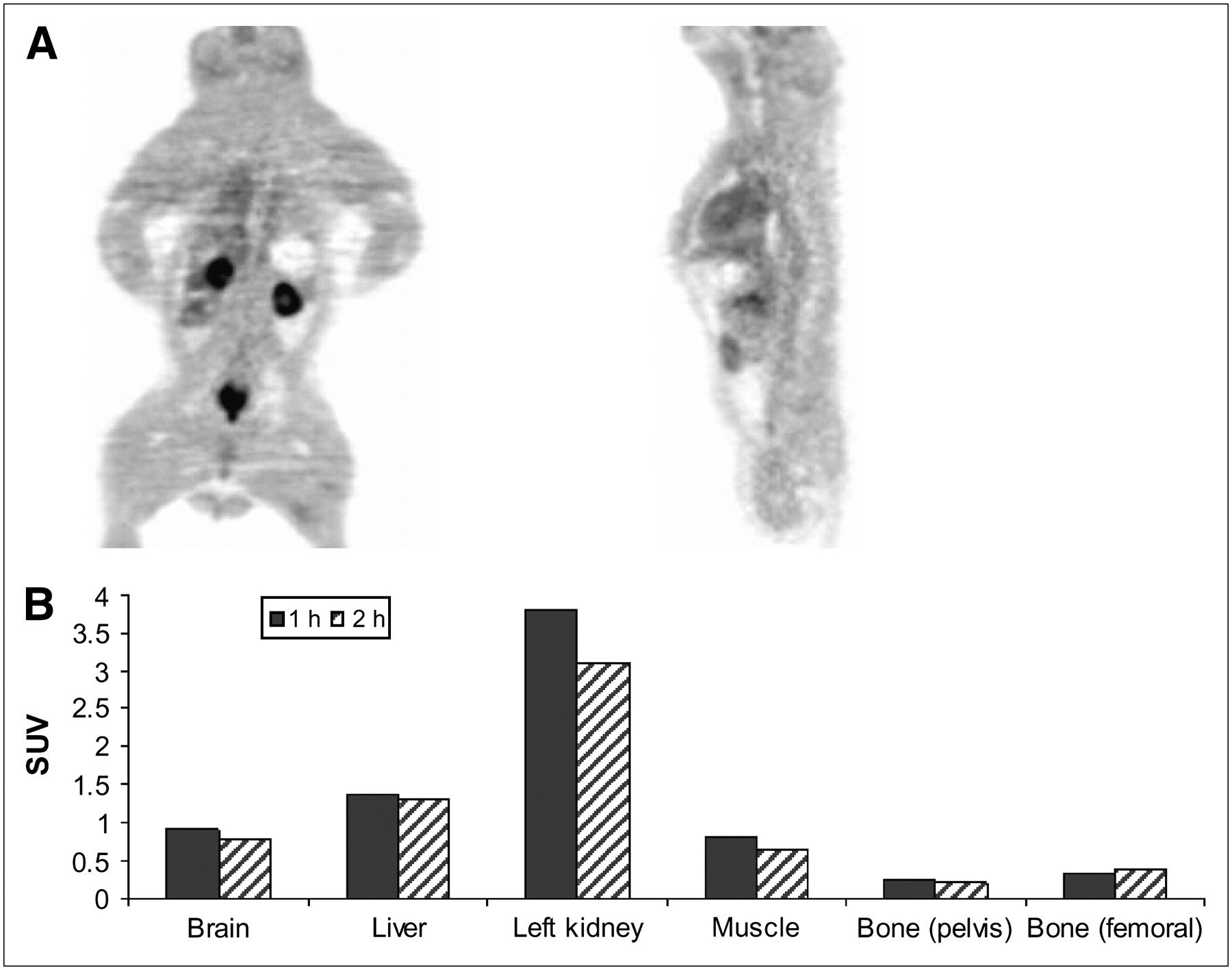
(A) PET of baboon (coronal, right; transaxial, left) taken at 1 h after administration of 370 MBq (10 mCi) of 18F-FAC. PET was performed with ECAT EXACT HR+ tomograph (Siemens CTI Corp.). (B) ROIs were placed on iliac bone and femur and SUVs were calculated. Biodistribution data of rodents showed that 18F-FAC uptake into bone increased over time (Fig. 2), whereas in baboon 18F-FAC uptake in bone was low.
DISCUSSION
Fluoroacetate is a toxic compound that occurs naturally in the South African poison plant Dichapetalum chymosum as well as in other Dichapetalum species. Similar to acetate, fluoroacetate has been found to be a substrate for acetyl coenzyme A synthase, but with a lower specificity (Km [Michaelis–Menten constant] = 3.12 × 10−2 for FAC vs. 5.13 × 10−4 for ACE) and a 10% Vmax (maximum velocity) versus that of ACE (100%) (26). Fluoroacetate toxicity is due to its conversion (in vivo) to fluorocitric acid, which inhibits aconitase and leads to the inhibition of the tricarboxylic acid (TCA) cycle (27). In brain tissue, fluoroacetate is taken up by glial cells and causes inhibition of the glial TCA cycle (28). In this study we used trace quantities of fluoroacetate labeled with 18F (18F-FAC), an analog of 11C-ACE with a longer radioactive half-life (110 min). We investigated the behavior of 18F-FAC in vivo as a possible PET tracer for tumor detection. It has been estimated that the human median lethal dose (LD50) for FAC is approximately 2–10 mg/kg (29). If we consider the specific activity of the 18F-FAC that was produced in this study (2,000–3,500 Ci/mmol, 74–129.5 GBq/μmol at EOS), then the clinical dose of nonradioactive FAC for a PET study, assumed to require approximately 296–370 MBq (8–10 mCi), is at most 4.5 ng/kg (assuming 70-kg body weight). This is 10−6 times the human LD50. This amount is, therefore, well below levels that could cause any pharmacologic effect.
We used the same ethyl O-mesylglycolate precursor as reported by Jeong et al. (30), but we modified the synthesis by decreasing the amounts of starting substrate and acetonitrile, by the treatment of starting 18F-fluoride with Kryptofix 2.2.2./K2CO3, as well as by modifications in time, temperature, and purification and hydrolysis steps used for the synthesis. The solid-phase purification (Oasis HLB) efficiently removed both the residual organic solvent and any unreacted fluoride (as assayed by gas chromatography of the final product and TLC analyses, respectively), and the product was easily eluted with ethanol. We performed the hydrolysis step cleanly in <6 min, on a rotary evaporator provided with a warm water bath at 40°C temperature, while evaporating under reduced pressure the 7 mL of ethanol used to elute the product. In the original radiosynthetic method (30), the incorporation of 18F into the precursor was followed by the removal of all acetonitrile by heating at 80°C with nitrogen purge. When we performed similar experiments, we observed almost a total loss of radioactivity, as the intermediate 18F-ethyl fluoroacetate (Scheme 1, compound 3) has a boiling point around 121°C. Therefore, we opted to decrease the amount of acetonitrile during the first incorporation, which was followed by a solid-phase purification of the intermediate.
The solid phase chosen was an Oasis HLB cartridge, previously evaluated and found to be more efficient than conventional C-18 Sep-Paks (31). After the sample had been neutralized and reconstituted with isotonic saline, we obtained the final product with purity of >99% in ∼35-min synthesis time (from end of bombardment) and free of any unreacted fluoride. Jeong et al. reported a purity of ∼95% in 70–90 min, with some contaminating fluoride still present (30). The present synthetic method has been successfully adapted to a commercial synthesizer for a full automation of the synthesis and minimum radioactivity exposure to the operator (15).
We decided to perform a normal rat biodistribution study first, followed by a tumor-implanted athymic nu/nu mouse study, to compare the in vivo tissue uptake and metabolism of both tracers under normal physiologic conditions, and under a genetically challenged state. The rat biodistribution study showed that there was a rapid uptake of 11C-ACE from most organs, with rapid clearance except from the pancreas (Fig. 1A). 18F-FAC also showed a rapid uptake with slower clearance than that of 11C-ACE. It has been reported that this rapid uptake and clearance of 11C-ACE are due, respectively, to blood flow and the oxidative metabolism associated with the organ tissue (22). In contrast, because of its lack of oxidative metabolism when it enters into the tissues, 18F-FAC shows a relatively slower clearance than that of 11C-ACE.
Our rat biodistribution data also showed that 11C-ACE does not clear readily from the pancreas (Fig. 1A). According to the study by Shreve and Gross, the retention of 11C-ACE in the pancreatic tissue is due to the incorporation of 11C-ACE into the citric acid cycle, from which it is routed to various amino acids (32). From these intermediates, the 11C-ACE is incorporated into zymogens in acinar tissues that comprise >85% of the human pancreas (33). Because of its ability to inhibit the TCA cycle, 18F-FAC is not incorporated into zymogens, and this may lead to the lower retention of 18F-FAC that was observed in pancreatic tissue (Fig. 1B). The rat biodistribution study also showed that bone activity after injection of 18F-FAC increased significantly over time (Fig. 1B, 0.42 ± 0.02 %ID/g at 20 min, 0.76 ± 0.03 %ID/g at 1 h, 1.21 ± 0.05 %ID/g at 2 h, 1.87 ± 0.15 %ID/g at 4 h; P < 0.001), suggesting defluorination.
The biodistribution study in athymic nu/nu male mice implanted with the CWR22 tumors is shown in Figure 1C. These immunodeficient mice lack a thymus and are unable to produce T-cells, thus facilitating tumor implantation. However, the metabolism and excretion pathways in these mice are very different from those in normal rats, as indicated by comparison of the tracer uptake in Figures 1A–1C. Normal rats may have the ability to metabolize monofluoroacetate to nontoxic metabolites that are more easily excreted. These athymic animals generally showed a higher 11C-ACE and 18F-FAC uptake in all tissues studied (compare Fig. 1C with Figs. 1A and 1B). The tumor-to-blood, tumor-to-muscle, and tumor-to-fat ratios were higher for 11C-ACE, whereas the tumor-to-heart and tumor-to-prostate ratios were similar for the 2 tracers. The rest of the tumor-to-organ ratios (lung, liver, spleen, kidney, brain, and bone) were higher for 18F-FAC (Table 1). These differences could again be attributed to the lack of oxidative metabolism of 18F-FAC, promoting a higher radioactivity accumulation in most tissues.
Small-animal PET images with 18F-FAC in tumor-bearing mice (Table 2) showed clear delineation of CWR22 tumors implanted in the flank, with the highest contrast relative to muscle or background at 2 h after injection. The tumor-to-muscle ratio was taken because it was observed that muscle uptake does not change much over time; thus, this provides a reference organ for comparison with tumor uptake. It also provides indirectly normalization for the injected activity in the interanimal comparison. These results indicate that 18F-FAC is retained longer in tumor tissue than in other organs, suggesting that it is a useful tracer for PET tumor imaging. The coronal and sagittal images (Fig. 2) also show the liver and kidney as the major metabolic organs. We have not yet determined the precise mechanism for the incorporation of 18F-FAC into tumors. Some preliminary experiments with 18F-FAC uptake in isolated tumor cells (data not presented) have indicated that, contrary to 11C-ACE (13), a low amount of radioactivity (<1%) was present in the isolated cell membrane lipid fraction, and the rest was recovered in the aqueous layer. These preliminary results are in agreement with the low 18F-FAC fat uptake values of 0.09 ± 0.03 %ID/g at 20 min in mature male Sprague–Dawley rats, decreasing to 0.05 ± 0.01 %ID/g at 4 h (data not shown). However, more nonspecific binding, as expressed by the amount of radioactivity found in fat tissue, was observed for these genetically manipulated nu/nu mice implanted with CWR22 tumor line (Table 1 and Fig. 1C), showing 1.25 ± 0.41 %ID/g for 18F-FAC versus 0.19 ± 0.41 %ID/g for 11C-ACE.
18F-FAC imaging of a baboon was performed to examine the amount of defluorination by comparing the radioactivity uptake in bone with that found in rats and mice. On the basis of PET images, the iliac bone and femoral uptake after administration of 18F-FAC was low in the baboon. This suggests that there is less defluorination of 18F-FAC in baboons than in rodents. We have encountered similar species differences in studies with other fluorinated radiopharmaceuticals (34) and have found that their behavior in humans is closer to that found in baboons or other nonhuman primates (35). This preliminary report suggests that high uptake in bone may not be problematic in human studies with 18F-FAC PET. Similar to the small-animal PET study (Fig. 2), the baboon PET study also showed the liver and kidney as the main clearance organs, with some radioactivity found in the bladder (Fig. 3). There was no attempt to catheterize the baboon to collect and analyze the urine, as our major goal in this study was simply to assess defluorination from the 18F-fluorine bone uptake at 2 “snapshot” time points of 1 and 2 h. Bone uptake has been traditionally used by investigators as a measure of defluorination (36–38).
The bladder activity in a clinical setting may result in a higher background during scanning, which could be decreased by having the patient void before imaging, as in 18F-FDG imaging protocols. However, in a preliminary short communication on the imaging of prostate cancer metastases in patients with 18F-FAC (39), the authors found a very clear delineation of prostate cancer metastases that compared favorably with liver accumulation, as measured by SUVs; they also stated that “the mode of excretion was predominantly via the bowel, with low activity in the urine.” These results are contrary to the bladder activity that we observed in the baboon. Interestingly, we did not observe bowel activity in the baboon PET study, but we did observe bowel accumulation in the small-animal PET study (Fig. 2, coronal view). We did not determine whether this activity was in the bowel wall or the luminal contents. On the basis of the short communication of Matthies et al. (39) and our current in vivo results, it appears there is a need for further clinical evaluation of 18F-FAC as a prostate cancer imaging agent and for a precise delineation of its mode of action at the cellular level.
CONCLUSION
The conclusion of this preliminary study is that 18F-FAC is a promising alternative to 11C-ACE for positron tomographic imaging of prostate cancer, and possibly of other neoplasms with relatively low glucose use.
Acknowledgments
This study was supported by a grant from the U.S. Department of Energy (DE-FG02-84ER-60218). Small-animal PET was supported by an NIH/NCI SAIRP grant (R24 CA83060) and by the Small Animal Imaging Core of the Alvin J. Siteman Cancer Center, which is supported by an NCI Cancer Center Support grant (P30 CA91842). The authors thank Lori A. Strong, Jerrel R. Rutlin, Nicole M. Fettig, John A. Engelbach, Lynne A. Jones, and Terry L. Sharp for their technical support with the biodistribution and small-animal PET studies and Sally W. Schwarz for radiopharmaceutical production. We also thank Drs. Jason S. Lewis and Joseph B. Dence for helpful discussions as well as critique of the manuscript.
References
- Received for publication August 9, 2006.
- Accepted for publication November 20, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- [18F]Fluorocholine and [18F]Fluoroacetate PET as Imaging Biomarkers to Assess Phosphatidylcholine and Mitochondrial Metabolism in Preclinical Models of TSC and LAM
- Bacterial infection imaging with [18F]fluoropropyl-trimethoprim
- Preclinical Evaluation of 3-18F-Fluoro-2,2-Dimethylpropionic Acid as an Imaging Agent for Tumor Detection
- Metabolism and Quantification of [18F]DPA-714, a New TSPO Positron Emission Tomography Radioligand
- Prostate Cancer: PET with 18F-FDG, 18F- or 11C-Acetate, and 18F- or 11C-Choline
- 1-11C-Acetate Versus 18F-FDG PET in Detection of Meningioma and Monitoring the Effect of {gamma}-Knife Radiosurgery
- 2-18F-Fluoropropionic Acid as a PET Imaging Agent for Prostate Cancer
- PET of Glial Metabolism Using 2-18F-Fluoroacetate
- 2-18F-Fluoroacetate: A Useful Tool for Assessing Gliosis in the Central Nervous System?