


Abstract
Data on expression of the HER2 (erbB-2) receptor in breast carcinoma make it possible to select the most efficient treatment. There are strong indications that HER2 expression possesses prognostic and predictive values in ovarian, prostate, and lung carcinomas as well. Visualization of HER2 expression using radionuclide targeting can provide important diagnostic information. The Affibody ZHER2:342 is a short (∼7 kDa) phage-display–selected protein that binds HER2 with an affinity of 22 pmol/L. The goal of this study was to evaluate whether 111In-labeled HER2:342 can be used for imaging of HER2 overexpression in vivo. Methods: ZHER2:342 was labeled with 111In via isothiocyanate-benzyl-DTPA (DTPA is diethylenetriaminepentaacetic acid) and the conjugate was characterized in vitro and in vivo. Results: 111In-Benzyl-DTPA–ZHER2:342 preserved the capacity to bind living HER2-expressing cells specifically. The affinity of In-benzyl-DTPA–ZHER2:342 to HER2 was 21 pmol/L according to surface plasmon resonance measurements. In nude mice bearing HER2-expressing SKOV-3 xenografts, a tumor uptake of 12% ± 3% injected activity per gram and a tumor-to-blood ratio of about 100 were obtained 4 h after injection. Tumor uptake in vivo was receptor specific, as it could be blocked with an excess of nonlabeled ZHER2:342. HER2-expressing xenografts were clearly imaged 4 h after injection using a γ-camera. Conclusion: 111In-Benzyl-DTPA–ZHER2:342 is a promising candidate for visualization of HER2 expression in carcinomas, using the single-photon detection technique.
There has been an increasing interest in molecular in vivo imaging over the last few years. This approach is based on the paradigm that diseased cells may demonstrate a distinctive pattern of expression or processing of gene products. Accumulation of a tracer in a given tissue due to interaction with a product of an aberrantly expressed gene is defined as molecular targeting (1). When a nuclide, which emits radiation detectable outside the human body, is attached to a tumor-targeting molecule, a site of accumulation of the targeting agent can be identified in vivo by noninvasive procedures. This can provide information on the localization or biologic status of the tumor.
One of the molecular targets, which have been identified for radionuclide imaging, is HER2 (1,2). This transmembrane protein belongs to the human epidermal growth factor receptor (HER) tyrosine kinase receptor family. Increased HER2 activity is associated with increased proliferation and decreased apoptotic capacity. Expression of HER2 in normal tissues is low or it is not expressed there (3). It has also been shown that breast cancers expressing HER2 respond well to anthracycline (e.g., doxorubicin)–based chemotherapy (4), and HER2 expression can be used for the selection of patients for such treatment. The clinical practice guidelines of the American Society of Clinical Oncology recommend evaluating HER2 expression on every primary breast cancer at the time of either diagnosis or recurrence (5). Even today, detection of HER2 expression provides important diagnostic information with regard to selection of treatment strategy. In the future, the diagnostic value of HER2 detection may increase even more, as the data that the expression of HER2 in tumor is associated with short survival of the breast cancer patients (6) and resistance to treatment with tamoxifen (7) or cyclophosphamide–methotrexate–fluorouracil (CMF) therapy (8) await confirmation from high-statistical-power prospective studies. In vivo radionuclide imaging of HER2 expression could help to avoid the risk of false-negative results associated with biopsy. However, it may also provide information that is unavailable by other means, such as anatomically correlated information on HER2 expression in individual metastases, before and after treatment. In this manner, in vivo imaging has the potential of being used to monitor disease progression and guide the management of various therapies targeting HER2—for example, monoclonal antibodies and small inhibitory molecules—in an individualized, patient-specific manner.
Behr et al. (9) have presented an excellent example of the clinical utility of radionuclide imaging of HER2 expression in breast cancer patients. It was shown that the use of 111In-DTPA-trastuzumab (DTPA is diethylenetriaminepentaacetic acid) enabled identification not only of patients responding to trastuzumab treatment (alone or in combination with chemotherapy) but also of patients who may have cardiac toxicity associated with such treatment.
The sensitivity of radionuclide imaging is determined by many factors related to tumor biology and physiology, detection technique, and radioactive tracer properties. The majority of targeting agents, which are used for tumor targeting or are considered for such application, are monoclonal antibodies and their derivatives or peptide ligands to receptors that are overexpressed in tumors. Comparative analysis of the biodistribution of antibodies and their fragments has demonstrated that the smaller the molecular weight, the higher the radiolocalization indices (10–12). Thus, creation of a relatively low-molecular-weight substance, which specifically binds HER2 in vivo, may improve clinical diagnostics. In fact, animal studies with such antibody products as (Fab′)2 or derivatives of single-chain variable fragments (scFv) demonstrated that low-molecular-weight targeting molecules have advantages in comparison with intact immunoglobulin (13,14). Still, creating a HER2-targeting molecule with a size smaller than a scFv is of interest. The use of peptide receptor ligands or their analogs has enabled the production of imaging agents for many types of receptors overexpressed in tumors (15,16). However, at the moment, this method is not possible for HER2, as a natural ligand to this receptor has not been identified. Moreover, it is believed that HER2 signaling occurs by heterodimerization with other receptors of the HER family and that a natural ligand to HER2 may in fact not exist (17).
One way to select a HER2-binding protein smaller than a single-chain fragment is through the use of the variant of phage-display called Affibody technology (18,19), which uses the domain scaffold of the immunoglobulin-binding staphylococcal receptor protein A. The 58-amino-acid-long cysteine-free Affibody protein provides a robust framework, independent of disulfide bonds for its folding. Randomization of 13 solvent-accessible surface residues of the protein A domain was used to create a library containing about 109 members, enabling the isolation of high-affinity ligands for virtually any tumor-associated protein target. The selection of the Affibody ZHER2:4, which binds with high specificity to HER2, was recently reported by Wikman et al. (20). The initial affinity of the ZHER2:4 was 50 nmol/L. The use of affinity maturation enabled selection of an Affibody molecule (Affibody molecule is used in this article instead of Affibody® molecule), ZHER2:342 (Fig. 1), with an affinity of 22 pmol/L (21). Binding affinity in the nanomolar or subnanomolar range is considered an important prerequisite for the use of protein for radionuclide diagnostics (11).

Amino acid sequence of ZHER2:342. HER2-binding site is formed by helices 1 and 2. Helix 3 provides structural rigidity of Affibody molecules.
The choice of radiolabel for the targeting protein is dependent on the purpose of the procedure, the biokinetics of the tracer, and the imaging device to be used. Apparently, the best way to estimate overexpression of HER2 in vivo would be quantification using PET. Taking into account the time required for tumor accumulation and clearance from nonspecific compartments, we have several options: the use of 18F, the use of generator-produced 68Ga, and the use of cyclotron-produced long-lived positron emitters (22). However, all positron-emitting labels potentially suitable for recombinant proteins have a general problem: A quick introduction in wide clinical practice cannot be expected. The majority of current methods for labeling of proteins and peptides with 18F are time consuming and complicated (23). The use of recently proposed chemoselective conjugation labeling via oxime formation can be a breakthrough in development of rapid and high-yield radiofluorination of short synthetic peptides (24), but its utility for larger recombinant proteins requires careful development and evaluation. Although 68Ga generators, which provide nuclide in chemical form suitable for labeling, are available now from a commercial source (25), they still are not approved for human use on a large scale. Production of long-lived positron emitters is currently limited to a few research centers. For this reason, the in vivo quantification of HER2 expression using PET would, most likely, be confined in the near future to a small number of research-oriented university hospitals. At the same time, data on visualization of HER2 expression using 111In-trastuztamab (9) clearly suggest that even semiquantitative imaging using SPECT can provide valuable diagnostic information. The wide availability of SPECT installations and radionuclides would enable Affibody-mediated imaging of HER2 expression now. For this reason, we directed our efforts toward developing a label for single-photon detection.
Three radionuclides are most commonly used for single-photon imaging—namely, 99mTc, 123I, and 111In (12). From these, 111In was selected for this study because of good imaging properties, facile logistics of delivery, and well-studied labeling chemistry. Significantly, the indium label possesses residualizing properties—that is, it is trapped intracellularly after internalization and proteolytic degradation of targeting protein by tumor cells (26), which might improve tumor accumulation. Derivatives of DTPA are often used as bifunctional chelators for the attachment of 111In to targeting antibodies and peptides. They demonstrate adequate chelate stability when coupled to peptides with fast in vivo kinetics and are easy to handle (27–29). For this reason, the isothiocyanate derivative of benzyl-DTPA was selected for this study.
The overall goal of the present study was to prepare and evaluate the indium-labeled conjugate on the basis of affinity-matured anti-HER2 Affibody molecule ZHER2:342 for the imaging of overexpression of HER2 in malignant tumors.
MATERIALS AND METHODS
The ZHER2:342 was provided for this study by Affibody AB as a 1.5-mg/mL solution in phosphate-buffered saline (PBS). Isothiocyanate-benzyl-DTPA was purchased from Macrocyclics, indium(III) chloride was from Mallinckrodt, and 111In-indium chloride was from Tyco Healthcare Norden AB, Mallinckrodt. High-quality Milli-Q water (resistance higher than 18 MΩ/cm) was used for preparing solutions. Buffers were prepared using common methods from chemicals supplied by Merck. Buffers, which were used for conjugation and labeling, were purified from metal contamination using Chelex 100 resin (Bio-Rad Laboratories). NAP-5 size-exclusion columns were from Amersham Biosciences.
The radioactivity was measured using an automated γ-counter with a 3 in. NaI(Tl) detector (model 1480 Wizard; Wallac Oy). 111In was measured using both photopeaks and the summation peak (energy setting, from 140 to 507 keV). Distribution of radioactivity along the instant thin-layer chromatography (ITLC) strips was measured on a Cyclone Storage Phosphor System (Perkin Elmer) and analyzed using the OptiQuant image analysis software (Perkin Elmer). Cells were counted using an electronic cell counter (Beckman Coulter).
Data on cellular uptake and biodistribution were analyzed by a 2-tailed t test using GraphPad Prism (version 4.00 for Windows; GraphPad Software) to determine any significant differences (P < 0.05).
Conjugation and Labeling Chemistry
Conjugation of isothiocyanate-benzyl-DTPA to ZHER2:342 was performed according to the method described by Mirzadeh et al. (30), using a chelator-to-protein molar ratio of 1:1. Briefly, 300 μL of ZHER2:342 (450 μg) were mixed with 43 μL of freshly prepared solution (1 mg/mL) of isothiocyanate-benzyl-DTPA in 0.07 mol/L sodium borate buffer, pH 9.2. The total volume was adjusted to 500 μL with 0.07 mol/L borate buffer, after which the mixture was vortexed for about 30 s and then incubated overnight at 37°C. After incubation, the reaction mixture was purified on a NAP-5 size-exclusion column, preequilibrated with 0.2 mol/L sodium acetate buffer, pH 5.3, according to the manufacturer's instructions. The eluate was vortexed and aliquoted into portions containing 50 μg of benzyl-DTPA–ZHER2:342 conjugate and stored at −20°C before labeling.
To evaluate the efficiency of isothiocyanate-benzyl-DTPA coupling to ZHER2:342, 2 samples were analyzed by high-performance liquid chromatography and online mass spectrometry (HPLC-MS) using an Agilent 1100 HPLC/MSD. The mass spectrometer was equipped with electrospray ionization and single quadropol. The software used for analysis and evaluation was ChemStation Rev. B.01.03. (Agilent). Thirty microliters of diluted sample (1:1 in solution A, 0.1% trifluoroacetic acid [TFA] in Milli-Q water) were loaded onto a Zorbax 300SB-C8 Narrow Bore (Agilent Technologies) (2.1 × 150; 3.5 μm) RPC-column, equilibrated with solution A with 20% solution B (0.1% TFA in acetonitrile) and eluted at a flow rate of 0.5 mL/min. After 2 min with 20% B solution, the proteins were eluted using a linear gradient from 20% to 70% solution B in 15 min. The mass spectrometer was run according to the manufacturer's recommendations.
For labeling, 50 μg conjugate were mixed with a predetermined amount of 111In and incubated at room temperature for 60 min. In some cases—for example, for biodistribution experiments—buffer was exchanged for sterile PBS using a NAP-5 column. For cell studies, the reaction mixture was diluted with PBS.
For routine quality control of the labeling, ITLC SG (silica gel–impregnated glass fiber sheets for ITLC; Gelman Sciences Inc.) eluted with 0.2 mol/L citric acid was used. In this system, radiolabeled Affibody molecules remain at the origin, free indium migrates with the front of solvent, and 111In-isothiocyanate-DTPA complex has a Rf of 0.4.
Biacore Analysis of In-Benzyl-DTPA–ZHER2:342 Affinity
Affinity measurements were performed using BIAcore 3000 (BIAcore AB) with Sensor Chip CM5. To evaluate the effect of both chelator and metal on binding affinity, benzyl-DTPA–ZHER2:342 was conjugated with a nonradioactive indium of natural isotopic composition in acetate buffer, pH 5.5. To ensure complete saturation of chelators, a 10-fold molar excess of indium over benzyl-DTPA–ZHER2:342 was used. HER2 was immobilized using amine chemistry at a low level (∼20 response units) according to the manufacturer's instructions. Conjugate was injected for 600 s at 5 concentrations ranging from 16 pmol/L to 6.6 nmol/L. The results were evaluated with BIAevaluation 4.0 (BIAcore AB) using a 1:1 interaction model.
Cell Binding and Retention Studies
The binding specificity of the obtained conjugates was tested on HER2-expressing SKOV-3 ovarian cancer cells. Labeled conjugate (7 ng) was added to 2 groups of Petri dishes (3 dishes; diameter, 3.5 cm; 2−5 × 105 cells per dish). One group of dishes in each experiment was presaturated with a 1,000-fold excess of nonlabeled ZHER2:342 for 10 min before the labeled ZHER2:342 was added. Cells were incubated with labeled conjugate for 1 h at 37°C and incubation medium was collected. Cell dishes were washed 6 times with cold serum-free medium and treated with 0.5 mL trypsin/ethylenediaminetetraacetic acid (EDTA) solution (0.05% trypsin/0.02% EDTA in buffer; Flow Irvine) for 10 min at 37°C. When cells were detached, 0.5 mL complete medium were added to each dish and the cells were resuspended for radioactivity measurement.
Cellular retention of radioactivity after interrupted incubation was studied in the following way: Culture dishes containing 2–5 × 105 SKOV-3 cells were washed in serum-free medium and then incubated for 2 h with culture medium containing 7 ng 111In-benzyl-DTPA–ZHER2:342. The dishes were then washed 6 times with cold serum-free culture medium, fresh complete medium was added, and the cells were incubated at 37°C. At predetermined time points (0.5, 1, 2, 4, 12, and 29 h after change of medium), incubation medium was collected from 3 culture dishes and cells were detached from culture dishes by trypsin treatment as described. The radioactivity associated with the cells and the culture medium was measured. The fraction of the initial cell-associated radioactivity was analyzed as a function of time.
To facilitate interpretation of the results of the cellular studies, we attempted to determine the internalized fraction by acid wash in a manner similar to a method applied earlier to epidermal growth factor–based conjugates (31). However, validation experiments demonstrated that this method could not completely remove radioactivity from the cell surface after incubation on ice, probably because of the very strong binding of 111In-benzyl-DTPA–ZHER2:342 to HER2 (data not shown). This means that acid wash would overestimate internalized radioactivity. For this reason, a quantitative internalization assay was not performed.
Tumor Uptake and Biodistribution of 111In in SKOV-3 Xenograft-Bearing Nude Mice After Subcutaneous Injection of 111In-Benzyl-DTPA–ZHER2:342
The animal study was approved by the local Ethics Committee for Animal Research. Female outbred BALB/c nu/nu mice (10- to 12-wk old on arrival) were acclimatized for 1 wk at the Rudbeck Laboratory animal facility before subcutaneous injection of ∼5 × 106 SKOV-3 cells in the hind leg 4−8 wk before the experiment. The mice were maintained using a standard diet, bedding, and environment, with free access to food and drinking water.
Twenty-eight BALB/c nu/nu mice with SKOV-3 xenografts were randomly divided into 7 groups of 4 animals each. One group was injected subcutaneously (neck area) with 375 μg of nonlabeled ZHER2:342 in PBS. One hour later, all mice were injected subcutaneously with 111In-benzyl-DTPA–ZHER2:342 at a protein dose of 1 μg (∼100 kBq) in 100 μL PBS. All injections were tolerated well. At 1, 4, 12, 24, 48, and 72 h after injection, 1 group of mice was injected intraperitoneally with a lethal dose of Ketalar/Rompun solution (20 μL of solution per gram of body weight; Ketalar [ketamin], 10 mg/mL [Pfizer]; Rompun [xylazin], 1 mg/mL [Bayer]). The mice were killed by heart puncture with a 1-mL syringe rinsed with diluted heparin (5,000 IU/mL; Leo Pharma). Blood was collected in preweighed vials. Mice in the blocking group were killed 4 h after injection. Heart, lung, liver, spleen, pancreas, kidney, stomach, salivary glands, brain, and tumor as well as samples of muscle, small and large intestines, and bone were excised and collected in weighed plastic bottles. Organs and tissue samples were weighed and their radioactivity was measured using an automatic γ-spectrometer. Uptake was calculated and expressed as the percentage injected activity per gram (% IA/g).
γ-Camera Imaging
Two animals were injected with 3 MBq (5 μg) 111In-benzyl-DTPA–ZHER2:342 into the tail vein. Immediately before imaging (4 h after injection), animals were killed by overdosing with Rompun/Ketalar. Imaging was performed using a Siemens e.CAM γ-camera (Siemens Medical Systems) equipped with a medium-energy, general-purpose collimator at the Department of Nuclear Medicine, Uppsala University Hospital.
Static images (10 min, 550,000 counts), obtained with a zoom factor of 3.2, were digitally stored in a 256 × 256 matrix. The pixel size was 2.4 mm. The scintigraphic results were evaluated visually and analyzed quantitatively using Hermes software (Nuclear Diagnostics). Quantitative analysis was performed with equal, 13-pixel regions of interest (ROIs) drawn over the tumor, contralateral thigh, kidneys, and liver. Tumor-to-nontumor ratios were calculated based on counts in the whole ROI.
RESULTS
Conjugation and Labeling
Application of the method published by Mirzadeh et al. (30) was simple and straightforward, even at a low chelator-to-peptide ratio. Results of HPLC-MS analysis suggest that the majority (66%) of ZHER2:342 Affibody molecules bore a single benzyl-DTPA group, and 14% 2 benzyl-DTPA, whereas 20% remained unconjugated. Incubation of benzyl-DTPA–ZHER2:342 with 111In-indium chloride in 0.2 mol/L sodium acetate buffer, pH 5.3, enabled a labeling efficiency of ∼90% after 30 min and >95% after 60 min. Additional purification of the labeled product using a NAP-5 size-exclusion column increased the radiochemical purity to >98%. The major side product could be identified as a complex of 111In with benzyl-DTPA. A specific radioactivity of 8 GBq/μmol was obtained. To ensure that labeling was mediated by benzyl-DTPA, a control experiment was performed whereby peptide was treated the same way as during the coupling procedure, but neat 0.07 mol/L borate buffer, pH 9.2, was added instead of isothiocyanate-benzyl-DTPA solution. Incubation of treated peptide with indium in acetate buffer did not provide any labeling. This indicates that indium binding was associated with benzyl-DTPA and not with occasional chelating amino-acid sequences of ZHER2:342.
On-Rate, Off-Rate, and Affinity of In-Benzyl-DTPA–ZHER2:342 Binding to Extracellular Domain of HER2
Biacore analysis of the In-benzyl-DTPA–ZHER2:342 interaction with the immobilized extracellular domain of HER2 indicated that manipulations associated with chelator coupling and labeling had no negative effect on the HER2-binding capacity of ZHER2:342. Our data showed that it has both a fast on-rate (4.4 × 106 M−1·s−1) and a slow off-rate (9.5 × 10−5 s−1), resulting in an affinity of 21 pmol/L, which is comparable with an affinity of 22 pmol/L for nonconjugated peptide. The off-rate corresponds to dissociation down to 50% of the initial binding level in ∼3 h.
Cell-Binding and Retention Experiments
To demonstrate that the binding was receptor specific, a large excess of nonlabeled ZHER2:342 was added to cells in the control experiments to saturate binding sites on HER2. This caused almost complete blocking of binding, with the reduction of cell-associated radioactivity from 46,000 ± 4,000 to 207 ± 3 cpm per dish (P < 0.005). The results of the binding specificity experiments demonstrated that the binding of 111In-benzyl-DTPA–ZHER2:342 could be prevented by receptor saturation.
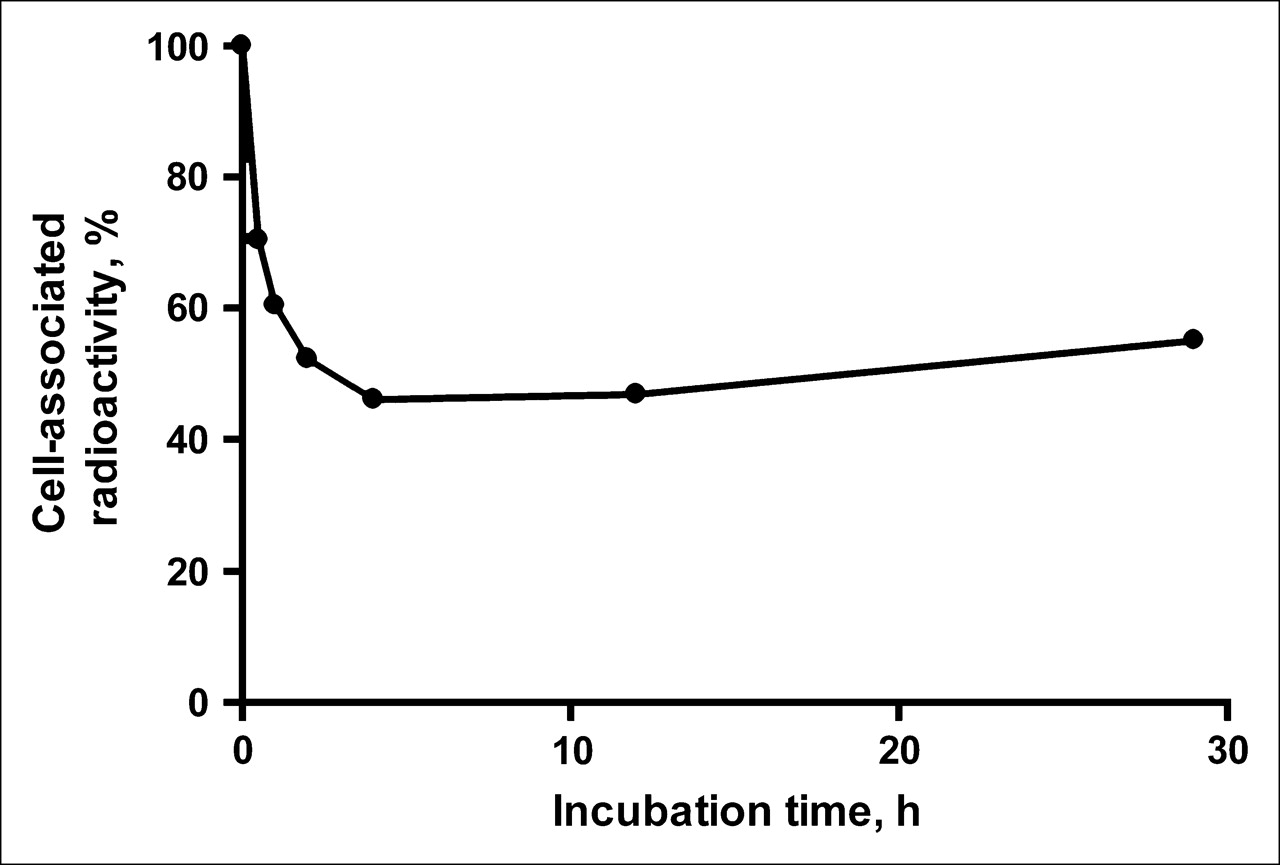
The retention pattern of 111In radioactivity after interrupted incubation of 111In-benzyl-DTPA–ZHER2:342 with SKOV-3 is shown in Figure 2. Two segments characterize the curve. An initial drop of radioactivity during the first 4 h after interrupted incubation was followed by a relatively constant amount of cell-associated 111In with a tendency to slow increase. This curve shape might be explained in the following way: Internalization of bound 111In-benzyl-DTPA–ZHER2:342 seems to be relatively slow and, after a change of culture medium, a substantial part of the conjugate was dissociated in a nondegraded form. Note that the dissociation rate corresponds well with the off-rate obtained by BIAcore analysis. At the same time, a part of the conjugate was internalized, and this made cell association of radioactivity practically irreversible because of the residualizing properties of the 111In label. Reassociation of the conjugate from the medium, with subsequent internalization, led to a slow increase of the cell-associated radioactivity.

Cell-associated 111In radioactivity as function of time after interrupted incubation of SKOV-3 cells with 111In-benzyl-DTPA–ZHER2:342. Cell-associated radioactivity at time zero after interrupted incubation was considered as 100%. Data are presented as mean ± SD (n = 3). Error bars are not evident because they are smaller than • symbols.
Biodistribution
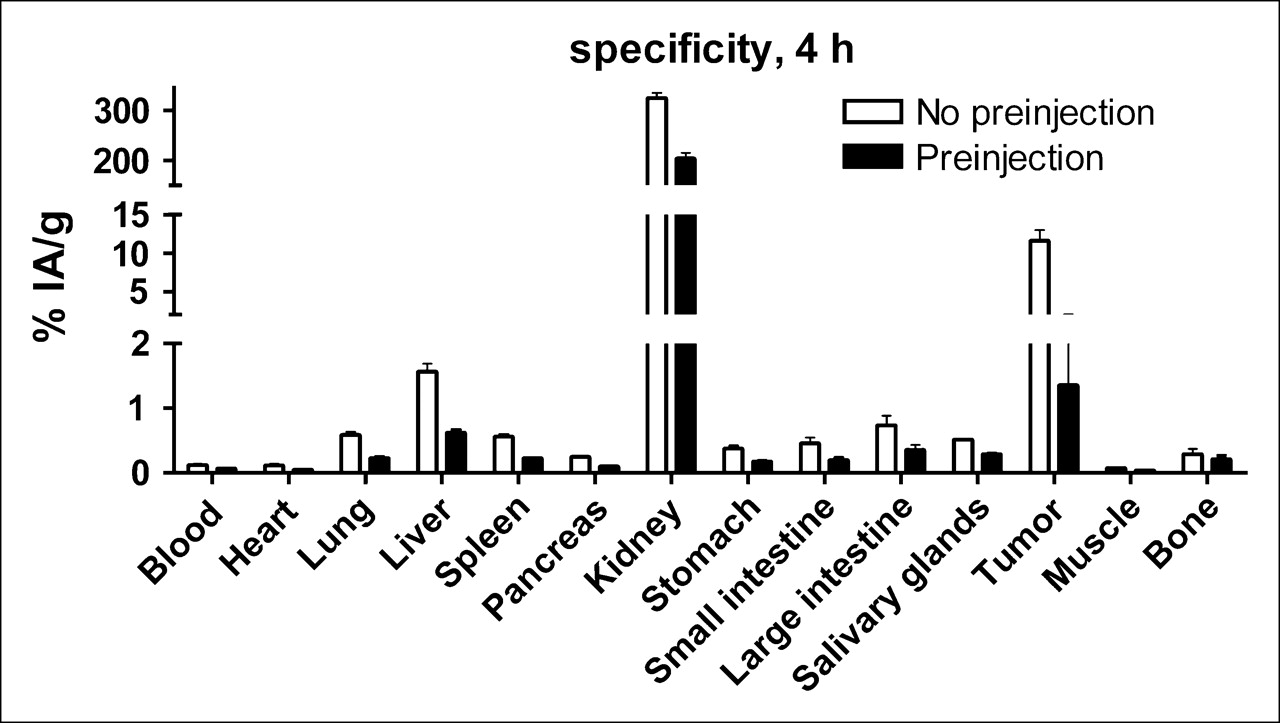
Biodistribution data are shown in Table 1. The distribution of 111In-benzyl-DTPA–ZHER2:342 was characterized by quick blood clearance, with 2.1% ± 0.3% IA/g remaining 1 h after injection and 0.12% ± 0.03% IA/g 4 h after injection. For the elimination phase, the half-life can be estimated to be 12.6 h. A high renal uptake indicates that the substance was cleared almost exclusively through the kidneys. Four hours after injection, when almost all radioactivity was cleared from the rest of the body, kidneys reabsorbed 94% ± 3% of injected radioactivity, whereas liver contained 1.8% ± 0.1%. Besides kidney, the only site of high uptake was tumor. Remarkably, a high tumor uptake was detected at 1 h after injection. At this time point, the radioactivity concentration in the tumor significantly exceeded the concentration in all organs and tissues, including blood. The tumor uptake remained at the same level until the 12-h time point. At 24 h after injection, this value decreased to 8.6% IA/g and, at 72 h after injection, a further reduction of tumor uptake to 5% ± 1% IA/g occurred. To investigate whether this tumor uptake is receptor mediated, 4 animals were each preinjected with 375 μg of nonlabeled Affibody to saturate HER2 receptors on tumor cells. This receptor blockage led to an 8.5-fold decrease in tumor uptake, from 12% ± 3% IA/g to 1.3% ± 0.7% IA/g (P < 0.001) (Fig. 3). The blockage resulted in a significant (P < 0.05) reduction of uptake in liver, heart, lung, spleen, pancreas, stomach, and salivary gland. According to the Human Protein Atlas (http://www.proteinatlas.org/index.php), an expression of HER2, though not high, has been found in all of these organs in humans. It is quite possible that the murine counterpart is also expressed in the same tissues. However, the degree of this reduction was smaller, 1.5–2 times, which might indicate that only part of the uptake in these organs was receptor mediated. A significant reduction in uptake was observed in the kidneys as well. However, it is difficult to determine whether this was a blocking of binding from blood or a blocking of negatively charged patches in the proximal tubulae, where peptides are nonspecifically taken up from the primary urine after glomerular filtration. The radioactivity uptake in the brain was negligibly small, which is expected for hydrophilic peptides. Despite a possible receptor-mediated uptake in several tissues, this uptake was low, which resulted in a remarkably high tumor-to-nontumor radioactivity accumulation ratio (Fig. 4). Excluding kidneys, this ratio exceeded 5 for all measured organs and tissues already at 1 h after injection. Because of a quick blood clearance, the tumor-to-blood ratio was 5.7 at 1 h after injection, approached a value of 100 at 4 h after injection, and was close to 200 by 12 and 24 h after injection.

Specificity of 111In-benzyl-DTPA–ZHER2:342 uptake in vivo. One group of animals was preinjected with 375 μg ZHER2:342 to saturate HER2 receptors 1 h before injection of radiolabeled conjugate. All animals were injected with 1 μg 111In-benzyl-DTPA–ZHER2:342. Data are mean ± SD (n = 4).

Tumor-to-organ ratio of 111In-benzyl-DTPA–ZHER2:342 in tumor-bearing nude mice. Ratios are calculated from animals of Table 1. Data are mean ± SD (n = 4).
Biodistribution of 111In-Benzyl-DTPA–ZHER2:342 in Mice Bearing SKOV-3 Xenografts
γ-Camera Imaging
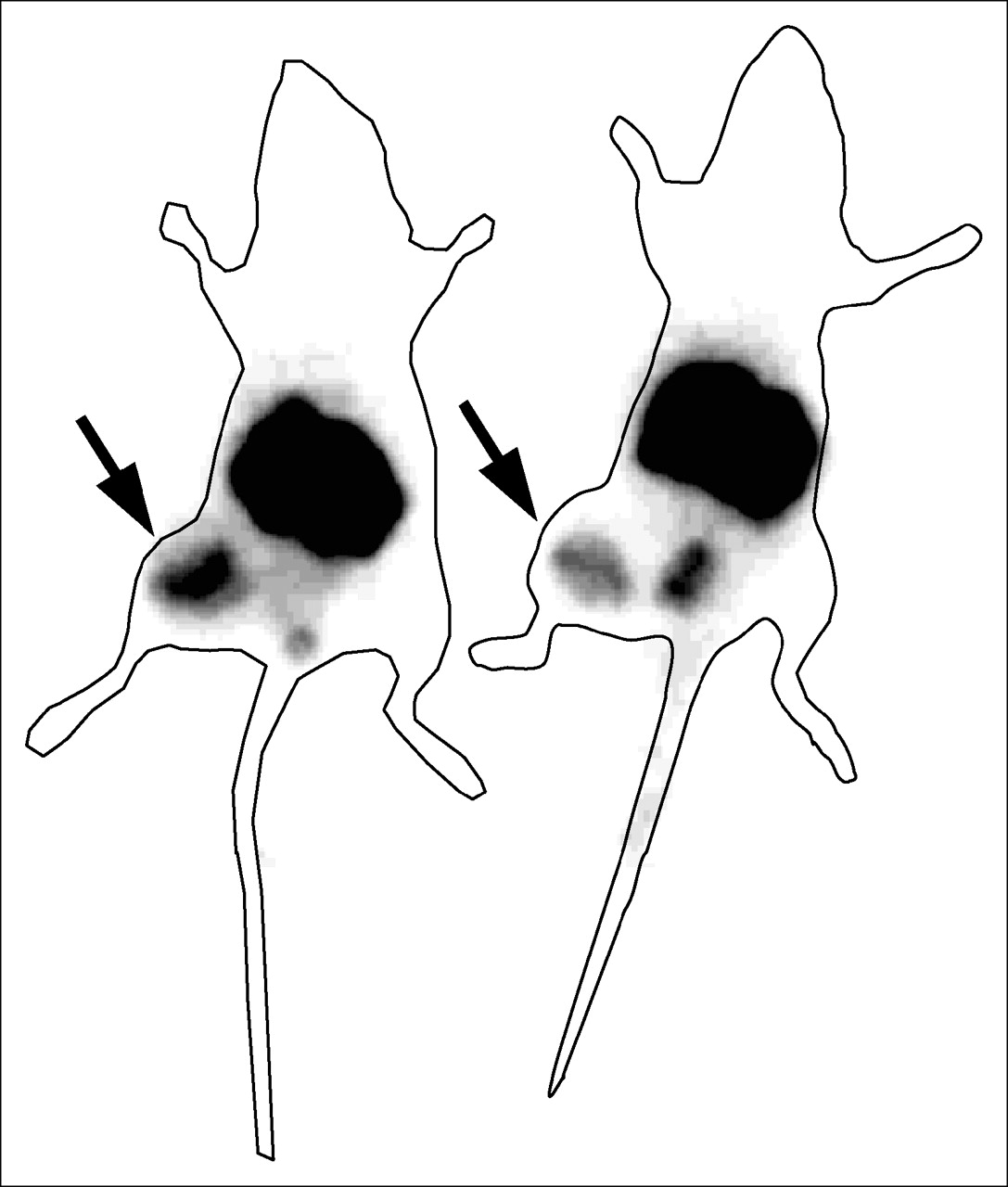
Images acquired 4 h after the administration of the 111In-benzyl-DTPA–ZHER2:342 to immunodeficient mice bearing subcutaneous SKOV-3 tumors revealed a high tumor localization of the radioactivity (Fig. 5). As predicted from the biodistribution studies, the renal route of elimination of the conjugates also led to substantial kidney retention. Accumulation of radioactivity in the urinary bladder was also visualized in all animals. Tumor-to-nontumor ratios were (average ± maximum error) 18 ± 2 for contralateral thigh, 0.11 ± 0.01 for kidneys, and 9 ± 1 for liver.

Imaging of HER2 expression in SKOV-3 xenografts in BALB/c nu/nu mice using 111In-benzyl-DTPA–ZHER2:342. Planar γ-camera images were collected 4 h after administration of 111In-benzyl-DTPA–ZHER2:342. Tumors (arrows) were clearly visualized.
DISCUSSION
A number of earlier publications on tumor targeting (10–12) indicate that slow tumor penetration and binding and, simultaneously, slow clearance from blood and normal tissues are the major problem for the diagnostic use of monoclonal antibodies. Smaller targeting molecules—for example, antibody fragments and scFv—provide better radiolocalization indices. The use of the phage-display methodology has an apparent potential for the development of further improved targeting peptides (32). A number of studies have demonstrated the feasibility of phage-display−based selection of short peptides that bind specifically to tumor-associated targets, including HER2 (33–36). However, despite an extensive literature search, we failed to find any research publication describing in vivo tumor-antigen visualization using phage-display−selected short peptides. An analysis of the literature data and our own experience suggested that the major problems in finding good targeting peptides have been insufficient affinity, usually in the micromolar range (32), and loss of binding capacity of the peptides after radiolabeling (34). Both low affinity and loss of binding capacity might be a result of distortion of the free peptide conformation in comparison with its conformation on filamentous phage. We considered that the presence of the robust α-helical backbone of Affibody molecules would improve the tumor-targeting properties and, thus, enable its clinical application. It could be hypothesized that an affinity improvement in this case should outweigh some loss of penetration capacity due to an increase of molecular weight relative to shorter peptides. The challenge would be to select an Affibody molecule that possessed sufficient affinity for successful development of a HER2-targeting agent while retaining binding capacity after chelator coupling and labeling at the same time. Fortunately, the robustness of the Affibody molecule scaffold enabled it to withstand the relatively harsh conditions, which were indicated by Mirzadeh et al. (30) as being crucial for coupling of benzyl-isothiocycanate chelators to antibodies—namely, increased pH, with most efficient coupling at about pH 9.0, and increased temperature. Under these conditions, a coupling reaction proceeded well at a low chelator-to-protein ratio. Biacore experiments demonstrated that the affinity of In-benzyl-DTPA–ZHER2:342 was not compromised in comparison with free peptide. Although Biacore can provide unique information concerning ligand–receptor interactions, this system cannot mimic all interactions of a labeled peptide with living cells. For this reason, confirmation of specific binding to living HER2-expressing SKOV-3 cells was considered to be important information about the binding capacity of 111In-benzyl-DTPA–ZHER2:342.
The high affinity of 111In-benzyl-DTPA–ZHER2:342 provided efficient tumor targeting, enabling a peak accumulation of 14% ± 4% IA/g in murine xenografts. Importantly, this accumulation could be reduced by preinjection of a large excess of nonlabeled ZHER2:342, which demonstrated a saturable binding and indicated a receptor-specific nature of tumor uptake. At the same time, clearance of radioactivity from blood and other organs (except kidneys) was fast, which provided a high tumor-to-nontumor ratio at 4 h after injection. It is of interest to compare 111In-benzyl-DTPA–ZHER2:342 with other 111In-labeled anti-HER2 conjugates, which have been reported recently and where the publications provide biodistribution data for 4 h after injection—that is, 111In-DTPA-CHX-A″-C6.5K-A diabody, scFv dimer (14), and 111In-DOTA-(Fab′)2-trastuzumab (Herceptin) (13). Because this is not an experimental head-to-head comparison, the result should be interpreted with caution. However, the literature comparison shows that tumor accumulation is approximately the same for all conjugates, but the clearance of radioactivity from blood is much quicker for 111In-benzyl-DTPA–ZHER2:342. The tumor-to-blood ratio was about 100 for the Affibody molecule whereas it was <2 for diabody and did not reach unity for (Fab′)2 fragment. If this biodistribution pattern were translated to humans, it would enable imaging of HER2 expression during the same day as the injection, providing obvious economic and logistic advantages compared with radiolabeled conjugates based on larger proteins.
An influence of affinity maturation on tumor-targeting properties of anti-HER2 Affibody was evaluated earlier using 125I-para-iodobenzoate as a label for ZHER2:342 (21). In vivo experiments in that study have been performed using the same SKOV-3 cell line and the same BALB/c nu/nu murine strain as in the current study. Comparison of biodistribution of 111In- and 125I-labeled ZHER2:342 could give a good illustration of the difference between nonresidualizing iodine and residualizing indium-benzyl-DTPA labels for ZHER2:342. At 1 h after injection (iodine, 8.2% ± 2.1% IA/g; indium, 12% ± 3% IA/g) and 4 h after injection (iodine, 9.5% ± 2.1% IA/g; indium, 12% ± 3% IA/g), the difference in tumor uptake was within error of measurement, though with a tendency to a higher accumulation of indium. Twenty-four hours after injection, tumor uptake for the indium label was 2 times higher (8.6% ± 0.9% IA/g) than that for iodine (4.1% ± 0.5% IA/g). The blood clearance was slower in the case of radioiodine, possibly because of release of radiocatabolites from excretory organs into the bloodstream. As a result, the 111In label provided a better tumor-to-blood ratio than radioiodine. Comparison of uptake values indicate that there was internalization of ZHER2:342 in tumor xenografts, though this process was relatively slow. An apparent advantage of the iodine label is quick release of radioactivity from kidneys, with 9.5% ± 0.8% IA/g remaining 4 h after injection. In the case of 111In- benzyl-DTPA, almost all injected radioactivity (94%) was reabsorbed in kidneys already at 4 h after injection. An apparent advantage of the use of 111In in comparison with 123I is a much more facile logistic, including the possibility of kit formulation.
γ-Camera imaging experiments demonstrated visualization of HER2-expressing xenografts and confirmed good imaging properties of 111In-benzyl-DTPA–ZHER2:342. Tumors were clearly visualized despite close vicinity to the kidneys. High uptake and retention in kidneys is a general problem for radiometal-labeled peptides and proteins with a size below ∼60 kDa (37). In the case of radionuclide therapy, it is an apparent obstacle. However, this does not seem to be a problem for SPECT of HER2 expression in breast carcinomas, where both primary tumor and most possible metastatic sites are anatomically well separated from the kidneys.
The result of the animal study gives a good reason to believe that 111In-benzyl-DTPA–ZHER2:342 can be used for detection of HER2 expression in malignant tumors in clinical practice. For the moment, such diagnostics would be the most useful for patients who have breast cancer. Treatment with trastuzumab (Herceptin), usually in combination with chemotherapeutics, increases patient survival significantly. However, only patients with HER2 overexpression (15%−25%) may benefit from such treatment. The use of 111In-benzyl-DTPA–ZHER2:342 can help to select eligible patients by assessing HER2 expression also in lesions that are not reachable by biopsy. Furthermore, it is possible that, similarly to 111In-trasuzumab (9), 111In-benzyl-DTPA–ZHER2:342 can help to identify patients at risk of cardiotoxicity due to Herceptin treatment. Further enhancement of the clinical utility of radiolabeled anti-HER2 Affibody is expected when the predictive value of HER2 overexpression is confirmed for selection of patients to tamoxifen or CMF therapy. The clinical potential of radionuclide imaging of HER2 overexpression is not limited only to breast cancer. Predictive or prognostic values of HER2 overexpression in ovarian (38), prostate (39), and lung (40) carcinomas have been reported. Confirmation of this information in prospective studies may further increase the use of 111In-benzyl-DTPA–ZHER2:342.
CONCLUSION
The use of isothiocyanate-benzyl-DTPA enabled labeling of ZHER2:342 HER2 binder with 111In with a nearly quantitative yield. 111In-Benzyl-DTPA–ZHER2:342 preserves its capacity to bind selectively to living HER2-expressing carcinoma cells. Biacore experiments demonstrated that conjugation did not adversely affect the high affinity of ZHER2:342 binding to the extracellular domain of HER2. 111In-Benzyl-DTPA–ZHER2:342 targeted HER2-expressing xenografts with high specificity, which enabled high-contrast imaging of HER2 expression in tumor xenografts in vivo.
Acknowledgments
This study was supported by grants from the Swedish Cancer Society and the Swedish Governmental Agency for Innovation Systems (VINNOVA). We thank Veronika Eriksson and the staff of the animal facility at Rudbeck Laboratory for technical assistance and Dr. Lars Abrahmsén (Affibody AB, Bromma, Sweden) for comments on the manuscript.
References
- Received for publication December 4, 2005.
- Accepted for publication February 11, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Positron Emission Tomography Imaging with 18F-Labeled ZHER2:2891 Affibody for Detection of HER2 Expression and Pharmacodynamic Response to HER2-Modulating Therapies
- Evaluation of 99mTc-peptide-ZHER2:342 Affibody(R) molecule for in vivo molecular imaging
- PET of EGFR Expression with an 18F-Labeled Affibody Molecule
- HER2-Affitoxin: A Potent Therapeutic Agent for the Treatment of HER2-Overexpressing Tumors
- Targeting of HER2-Expressing Tumors Using 111In-ABY-025, a Second-Generation Affibody Molecule with a Fundamentally Reengineered Scaffold
- Efficient Tumor Targeting with High-Affinity Designed Ankyrin Repeat Proteins: Effects of Affinity and Molecular Size
- A 2-Helix Small Protein Labeled with 68Ga for PET Imaging of HER2 Expression
- Molecular Imaging of EGFR: It's Time to Go Beyond Receptor Expression
- Changes in HER2 Expression in Breast Cancer Xenografts After Therapy Can Be Quantified Using PET and 18F-Labeled Affibody Molecules
- Targeting of HER2-Expressing Tumors with a Site-Specifically 99mTc-Labeled Recombinant Affibody Molecule, ZHER2:2395, with C-Terminally Engineered Cysteine
- Design, Synthesis, Radiolabeling and In Vitro and In Vivo Characterization of Tumor-antigen- and Antibody-derived Peptides for the Detection of Breast Cancer
- Affibody Molecules for Epidermal Growth Factor Receptor Targeting In Vivo: Aspects of Dimerization and Labeling Chemistry
- Tumor Receptor Imaging
- Small-Animal PET Imaging of Human Epidermal Growth Factor Receptor Type 2 Expression with Site-Specific 18F-Labeled Protein Scaffold Molecules
- Radionuclide Therapy of HER2-Positive Microxenografts Using a 177Lu-Labeled HER2-Specific Affibody Molecule