


Abstract
We previously described a highly flexible bispecific antibody (bs-mAb) pretargeting procedure using a multivalent, recombinant anti-CEA (carcinoembryonic antigen) × anti-HSG (histamine-succinyl-glycine) fusion protein with peptides radiolabeled with 111In, 90Y, 177Lu, and 99mTc. The objective of this study was to develop a radioiodination procedure primarily to assess PET imaging with 124I. Methods: A new peptide, DOTA-d-Tyr-d-Lys(HSG)-d-Glu-d-Lys(HSG)-NH2 (DOTA is 1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′-tetraacetic acid), was synthesized and conditions were established for radioiodination with yields of ∼70% for 131I and 60% for 124I. Pretargeting with the 131I- and 124I-labeled peptide was tested in nude mice bearing LS174T human colonic tumors that were first given the anti-CEA × anti-HSG bs-mAb. Imaging (including small-animal PET) and necropsy data were collected at several intervals over 24 h. Comparisons were made between animals given 124I-anti-CEA Fab′, 18F-FDG, the same peptide radiolabeled with 111In and pretargeted with the bs-mAb, and the radioiodinated peptide alone. Results: The radioiodinated peptide alone cleared quickly from the blood with no evidence of tumor targeting, but when pretargeted with the bs-mAb, tumor uptake increased 70-fold, with efficient and rapid clearance from normal tissues, allowing clear visualization of tumor within 1–2 h. Tumor uptake measured at necropsy was 3- to 15-fold higher and tumor-to-blood ratios were 10- to 20-fold higher than those for 124I-Fab′ at 1 and 24 h, respectively. Thyroid and stomach uptake was observed with the radioiodinated peptide several hours after injection (animals were not premedicated to reduce uptake in these tissues), but gastric uptake was much more pronounced with 124I-Fab′. Tumor visualization with 18F-FDG at ∼1.5 h was also good but showed substantially more uptake in several normal tissues, making image interpretation in the pretargeted animals less ambiguous than with 18F-FDG. Conclusion: Bispecific antibody pretargeting has a significant advantage for tumor imaging over directly radiolabeled antibodies and could provide additional enhancements for oncologic imaging, particularly for improving targeting specificity as compared with 18F-FDG.
Techniques for pretargeting radionuclides are now widely recognized as methods that improve imaging and therapeutic applications as compared with directly radiolabeled antibodies (1,2). Whereas all pretargeting methods are multistep processes, the advantages they offer in comparison with directly radiolabeled antibodies justify the increased interest engendered. All pretargeting procedures use an antibody that is modified to bind to a specific target antigen and to a secondary compound. Three secondary recognition systems have been described, including streptavidin (StAv)/biotin, antihapten antibody/hapten, and oligonucleotides/complementary oligonucleotides (1,2). In each system, the radionuclide is coupled to a small compound that is identified by the secondary recognition portion of the modified antibody. The small compound clears within minutes from the blood, escaping quickly from the vasculature into the extravascular space, making it readily accessible for binding to the tumor-bound modified antibody, and resulting in greatly improved tumor-to-blood ratios within minutes of its injection (3).
StAv-based pretargeting procedures use radiolabeled biotin that most often has been conjugated to 1,4,7,10-tetraazacyclododecane-N,N′,N′′,N′′′-tetraacetic acid (DOTA) for radiolabeling with 111In, 90Y, or 177Lu, but other compounds could be coupled to biotin to permit a wider range of radionuclide selection. Bispecific antibody (bs-mAb) pretargeting procedures use a hapten that is recognized by the antihapten arm of the bs-mAb. Most commonly, antibodies specific for chelate–metal complexes have been studied as the secondary recognition system in bs-mAb pretargeting procedures (4–7) and, therefore, radionuclide selection was somewhat restricted to radionuclides that were not only tightly bound to the chelate but would also bind fastidiously to the antichelate antibody. Initially, the bs-mAb pretargeting approach simply used a chelate–radiometal complex as the secondary effector (6,8), but it was found that the radionuclide could be bound more stably to the tumor if the secondary effector had 2 hapten components, a process termed an affinity enhancement system (AES) (9). A divalent hapten was prepared synthetically by coupling 2 haptens to a short peptide backbone. For example, Le Doussal et al. (9) coupled two 2,4-dinitrophenyl (DNP) groups to a tyrosine-lysine backbone, with the tyrosine providing a substrate for radioiodination. Later, the antihapten portion of the bs-mAb was directed against indium-loaded diethylenetriaminepentaacetic acid (DTPA) (10). A divalent (In)DTPA-tyrosine-lysine peptide-hapten could be radioiodinated (11) or radiolabeled with 111In. Because several radiometals are not bound stably by DTPA (12,13), this system was applied primarily for imaging with 111In and with 131I for therapeutic studies, but it was also adapted for use with 99mTc/188Re (14,15). Although the peptide core could have also been modified with DOTA for more stable binding of 90Y and 177Lu, the need to retain the DTPA for recognition by the bs-mAb raised concerns that even by preloading the DTPA with indium chloride, some portion of the 90Y could still become bound to the DTPA during the radiolabeling procedure, potentially leading to increased 90Y uptake in the bone.
Recently, we reported a more flexible bs-mAb pretargeting system that is based on an antihapten antibody specific for a synthetic compound, histamine-succinyl-glycine (HSG), which enabled DOTA-bearing peptides to be used with 111In, 90Y, and 177Lu, as well as 99mTc/188Re binding moiety (16). Superior imaging was afforded using a 99mTc-labeled di-HSG peptide with this pretargeting procedure when compared with a 99mTc-Fab′ (3). In this report, we examine the utility of this pretargeting procedure for PET using a new 124I-labeled di-HSG peptide in nude mice bearing carcinoembryonic antigen (CEA)-producing human colon cancer xenografts, with comparisons made to 18F-FDG and an 124I-labeled anti-CEA Fab′.
MATERIALS AND METHODS
Bispecific Antibody
The humanized anti-CEA × anti-HSG recombinant bs-mAb, designated hBS14, which is divalent for CEA and monovalent to HSG, has been described previously (17). The bs-mAb was radioiodinated with Na125I (Perkin Elmer) to a specific activity of 41.4–47.4 MBq/nmol using a chloramine-T method (18). Size-exclusion high-performance liquid chromatography (HPLC) showed the product as a single peak with <5% unbound radioiodine. The immunoreactive fraction to CEA was determined as described previously and was in excess of 90% (17).
Peptide Synthesis
IMP-288 (DOTA-d-Tyr-d-Lys(HSG)-d-Glu-d-Lys(HSG)-NH2; MH+: 1,453) was synthesized on solid phase using the Fmoc strategy. The Sieber amide resin was obtained from Calbiochem-Novabiochem and the Fmoc amino acids were obtained from Advanced ChemTech. The DOTA-tris(t-butyl ester) was obtained from Macrocyclics. The crude peptide was purified by reverse-phase HPLC (RP-HPLC) on a Waters LC 4000 using a radial compression module containing three 40 × 100 mm Nova Pak column segments and a 40 × 10 mm precolumn, using a gradient (100% A, 0.1% ammonium acetate in water to 70:30 A/B, where B is 90% acetonitrile/10% water). IMP-288 was ≥99% pure by HPLC and showed a molecular-ion peak by electrospray mass spectrometry (ESMS) (HT Laboratories, Inc.) consistent with the intended structure.
For radioiodination, IMP-288 was first preloaded by adding a 4.5-fold mole excess of indium chloride in 6 mL of 0.01 mol/L citrate/sodium acetate buffer, pH 4.03, and heating at 50°C for 20 min. This material was purified on a Waters Sunfire Prep C18 5-μm, 19 × 150 mm column. ESMS confirmed a molecular-ion peak consistent with the desired peptide. The In+3-loaded IMP-288 used for radioiodination was given a new designation, IMP-325 (In-DOTA-d-Tyr-d-Lys(HSG)-d-Glu-d-Lys(HSG)-NH2; MH+: 1,565).
Radiolabeling
IMP-288 was radiolabeled with 111InCl3 (IsoTex) using the previously reported procedure (16). A number of conditions for radioiodinating IMP-325 using Na131I were evaluated, but the following procedure provided the most consistent yields. Briefly, 4 μL of a 2 mmol/L solution of IMP-325 in 0.5 mol/L sodium phosphate, pH 4.0, were added to 100 μL of a buffer composed of 1.0 mol/L phosphate, 1.0 mol/L acetic acid, and 0.75 mol/L ammonium hydroxide, pH 2.0. The entire contents were transferred to the manufacturer's vial containing 370 MBq of Na131I (MDS Nordion or Perkin Elmer). Initially, the peptide and radioiodine solution were transferred to an IODO-GEN–coated vial (Pierce); however, we found that the iodination reaction proceeded more reliably when IODO-GEN was added as a microparticulate. For this procedure, 100 μg of IODO-GEN dissolved in 20 μL of dimethylformamide (DMF) were added to the vial containing the peptide-iodide solution and mixed briefly on a vortex mixer. After 15–20 min, the contents of the reaction vial, with a 150-μL wash, was added to 400 μL of a freshly prepared quenching buffer (0.1 mol/L cysteine in 0.5 mol/L sodium phosphate, pH 7.4) that was suspended on the top of a Waters Oasis HLB column (1 mL, 30 mg). Washing the column with 2.0 mL of the cysteine buffer followed by 2.0 mL of water removed the unbound radioiodide. The labeled peptide was eluted with 400 μL of 1:1 ethanol/water. Radiolabeling yields averaged between 70% and 80%, with specific activities ranging from 25.9 to 44.4 GBq (0.7–1.2 Ci)/μmol (all specific activity estimates assume that 100% of the peptide was recovered after purification). By RP-HPLC, the labeled peptide showed 2 peaks, likely representing mono- and diiodinated species. The purified products typically had <4% unbound radioiodine.
A similar microparticulate IODO-GEN procedure was used for 124I radiolabeling of the peptide; however, 6 nmol of a sodium iodide solution were added to the radiolabeling mixture to improve yields. Na124I was purchased from Eastern Isotopes. After 30 min at room temperature, the reaction was quenched and separated in an identical manner as stated above. Radiolabeling yields with 124I were commonly 50%–60%, with an estimated specific activity of ∼29.6 GBq/μmol. By RP-HPLC, the diiodinated peptide represented ∼40% of the product with ≤1.0% unbound 124I.
The purified 124I-IMP-325 used for animal studies was diluted in phosphate-buffered saline containing 1 mg/mL ascorbic acid to 74 MBq/mL. The product was stored overnight at 4°C and then shipped the next morning for injection in animals. RP-HPLC analysis of the 124I-peptide stored in this manner showed an increase from 0.8% to 5.4% unbound 124I after 17 h, which increased to ∼10% after 30 h. However, a 1:100 diluted sample used to analyze the initial postlabeling run was reexamined after it was stored at 25°C overnight in the HPLC autoinjector, and it showed the amount of unbound 124I was unchanged from the previous day. Thus, the higher concentration of the product that was administered to the animals resulted in some radiolysis of the 124I-peptide.
124I-Labeled humanized anti-CEA (hMN-14, labetuzumab; Immunomedics (16)) Fab′ was prepared using a similar procedure as described for radioiodinating the bs-mAb but, as with the 124I-peptide radiolabeling, 6 nmol of a sodium iodide solution were added to increase incorporation. The final product was isolated in phosphate-buffered saline, pH 7.4, containing 1% human serum albumin, and adjusted to a concentration of 74 MBq/mL. It was stored at ∼4°C overnight and then shipped by same-day courier to Memorial Sloan-Kettering Cancer Center (MSKCC) and Center for Molecular Medicine and Immunology (CMMI) for animal injections. Size-exclusion HPLC showed a single peak with <1% unbound radioiodine.
In Vivo Pretargeting
All animal studies were performed in compliance with the Guide for the Care and Use of Laboratory Animals in accordance with National Institutes of Health policies. Female nude mice of 8–10 wk of age (athymic NCr-nu/nu, National Cancer Institute colony; Taconic Laboratories) were implanted subcutaneously with 1 × 107 LS174T human colon cancer cells (American Tissue Culture Collection) that were freshly harvested from culture and washed in phosphate-buffered saline, pH 7.4, before implantation (0.5 mL). Seven to 11 d after implantation, studies were initiated.
After an initial study that confirmed optimal pretargeting with the 131I-IMP-325 could be achieved using the same conditions as reported previously for other labeled di-HSG peptides (3,17) (not shown), 13 animals were given 40 μg (0.5 nmol) of the hBS14 bs-mAb, containing a trace amount of 125I-hBS14 (74 kBq), followed 24 h later with ∼1.07 MBq (0.05 nmol) of the radiolabeled peptide. External scintigraphy and tissue counting were used to determine uptake and distribution at 1, 4, and 24 h. Appropriate windows were set in the γ-scintillation counter to discriminate each of the radioactive elements, correcting the 125I channel for downscatter of 131I. Two animals receiving the 131I-peptide alone were imaged at 2 and 4 h and then necropsied. In addition, 8 animals were given 40 μg of the hBS14 bs-mAb followed 24 h later with 0.05 nmol (1.07 MBq) of 111In-IMP-288. Five animals were imaged at 1 h and then necropsied, with the other 3 animals imaged and necropsied at 24 h. None of the animals was given a radioiodine-blocking regimen deliberately, so that dehalogenation could be assessed in the images (e.g., thyroid and stomach uptake). Imaging was performed for 20 min using an ADAC Solus imaging system with a high-energy collimator for 131I and a medium-energy collimator for 111In, with animals placed in prone position on the face of the collimator. No background correction was applied, but the intensity was varied by adjusting the color scale so that the pixels in the region with the most activity first became saturated, with the exception of the 1-h images, where the high activity in the urinary bladder was ignored. The adjusted nuclear images were then flipped to match photographs of the animals lying on the collimator.
The next series of studies compared the biodistribution and targeting of 124I-IMP-325 to 18F-FDG- or 124I-labeled hMN-14 anti-CEA Fab′. Two sets of animals were examined: one at MSKCC (microPET and necropsy at conclusion of imaging) and one at CMMI (necropsied at 3 planned intervals). Imaging studies were performed at MSKCC using a Focus 120 microPET unit (Concorde Microsystems, Inc.). A minimum of ∼20 million events was acquired over ∼5 min using an energy window of 350–750 keV, a coincidence timing window τ of 6 ns, and a span of 3. These list-mode data were sorted into 2-dimensional histograms by Fourier rebinning, and transverse images were reconstructed by filtered backprojection and by ordered-subsets expectation maximization (OSEM) iterative reconstruction into 128 × 128 × 94 (0.86 × 0.86 × 0.76 mm) for the Focus 120. The microPET imaging studies were quantitative—that is, the voxel values in the reconstructed images were in terms of percentage injected dose per gram (%ID/g). Data were corrected for the duration of the image acquisition, dead time, randoms, branching ratio, and physical decay of the radionuclide using hardware and software (ASIPro) provided by the manufacturer (Concorde Microsystems). The fully corrected counting rate densities (in cps/voxel) were then converted to activity concentrations (MBq/mL) by applying a measured system calibration factor (MBq/mL/cps/voxel) applicable to mice.
At MSKCC, 3 LS174T-bearing nude mice were given an intravenous injection of the hBS14 bs-mAb (160 μg; 2 nmol). Twenty-four hours later, an intravenous injection of 124I-IMP-325 (0.2 nmol; 7.4 MBq in 0.1 mL) was given (n = 3). No radioiodine-blocking regimen was administered. All 3 animals were anesthetized and imaged at ∼1–2, 4–6, and 24–27 h after the radiolabeled peptide injection and necropsied after the final imaging session. Three other groups of animals were examined. One group (n = 3) was fasted overnight and given an intravenous injection of 18F-FDG (30 MBq per mouse; Eastern Isotopes) and, after imaging at ∼1.5 h, were necropsied for tissue distribution determinations. Other animals received ∼7.4 MBq (0.2 nmol) of 124I-IMP-325 alone (no pretargeting; n = 3) or 124I-hMN-14 Fab′ (7.4 MBq, 12 μg; n = 3), followed by sequential imaging at the same intervals as the pretargeted animals, and then necropsied at the conclusion of the last imaging study.
All images were adjusted without background correction. The maximum intensity for the images of the pretargeted animals and the animals given 18F-FDG was adjusted so that the highest-activity voxels of the tumor corresponded to the highest-intensity gray scale. Whenever possible, the maximum intensity of the sequential images of the individual animals was adjusted to the identical intensity used for the animal's 1-h image, so that quantitative comparisons could be made of the tumor and tissue distribution over the specified time interval for that animal.
At CMMI, groups of 5 LS174T-bearing animals were necropsied at 1, 3, and 24 h after the radioactivity injection. The animals received identical doses for the pretargeted peptide, peptide alone, and 124I-hMN-14 Fab′. An additional group was given 7.4 MBq of 124I-hMN-14 Fab′ that was supplemented with unlabeled hMN-14 Fab′ to increase the total protein dose to 100 μg.
RESULTS
Radioiodination of IMP-325
A number of iodination reactions of the peptide IMP-325 were performed with 125I, 131I, and 124I. The initial reactions were performed using IODO-GEN–coated vials under conditions typically used for the radioiodination of peptides (19). Most notably, this procedure uses a pH of 7.5 for the radioiodination reaction. However, using this method, the radiolabeled peptide contained several different products by HPLC, most likely representing the incorporation of radioiodine on the imidazole ring of the HSG hapten, as well as on the tyrosine (20). By lowering the pH of the reaction mixture, the imidazoles would be protonated, making them less reactive with electrophilic iodide. Additional test labels performed at progressively lower pH confirmed that the products had fewer iodinated species, until at a pH of 4.0 or lower, only 2 primary peaks were seen by RP-HPLC. These 2 peaks were assumed to be mono- and diiodotyrosine peptides based on previously published experience (21). The formation of the second (longer retention time) peak could also be minimized as the pH was lowered and even eliminated if a high molar ratio of peptide to iodide was used.
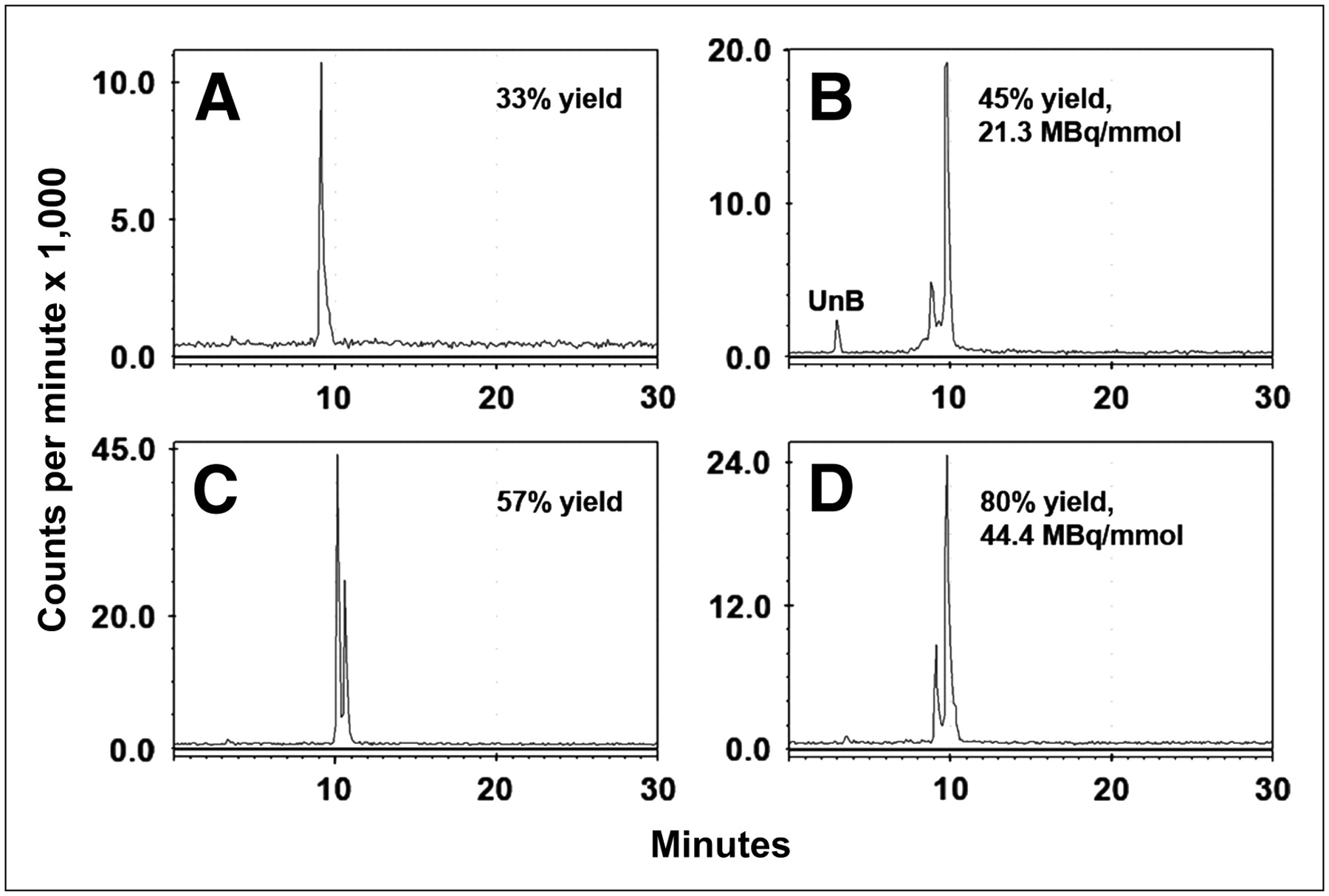
We initially performed the radioiodination in IODO-GEN–coated vials but found the yield to be highly variable, especially with high-specific activity iodide. The addition of cold iodide improved the yield for the 124I-labeled peptide and increased the proportion of the second peak (diiodotyrosine) (Figs. 1A and 1B). The reaction consistency was also improved by dissolving the IODO-GEN in DMF and then precipitating it in the iodine vial in the presence of the peptide (Figs. 1C and 1D (22)). The preparation of 131I-IMP 325 was also sensitive to the ratio of iodide to moles of peptide, but when the reaction was performed as described in the Materials and Methods, the radiolabeling yields (70%–80%) and specific activity (25.9–44.4 GBq/μmol) were consistent. The ratio of monoiodopeptide to diiodopeptide varied from batch to batch of 131I, ranging from 70:30 (monoiodopeptide/diiodopeptide) to 10:90, but within the same batch, the ratios were consistent from run to run.
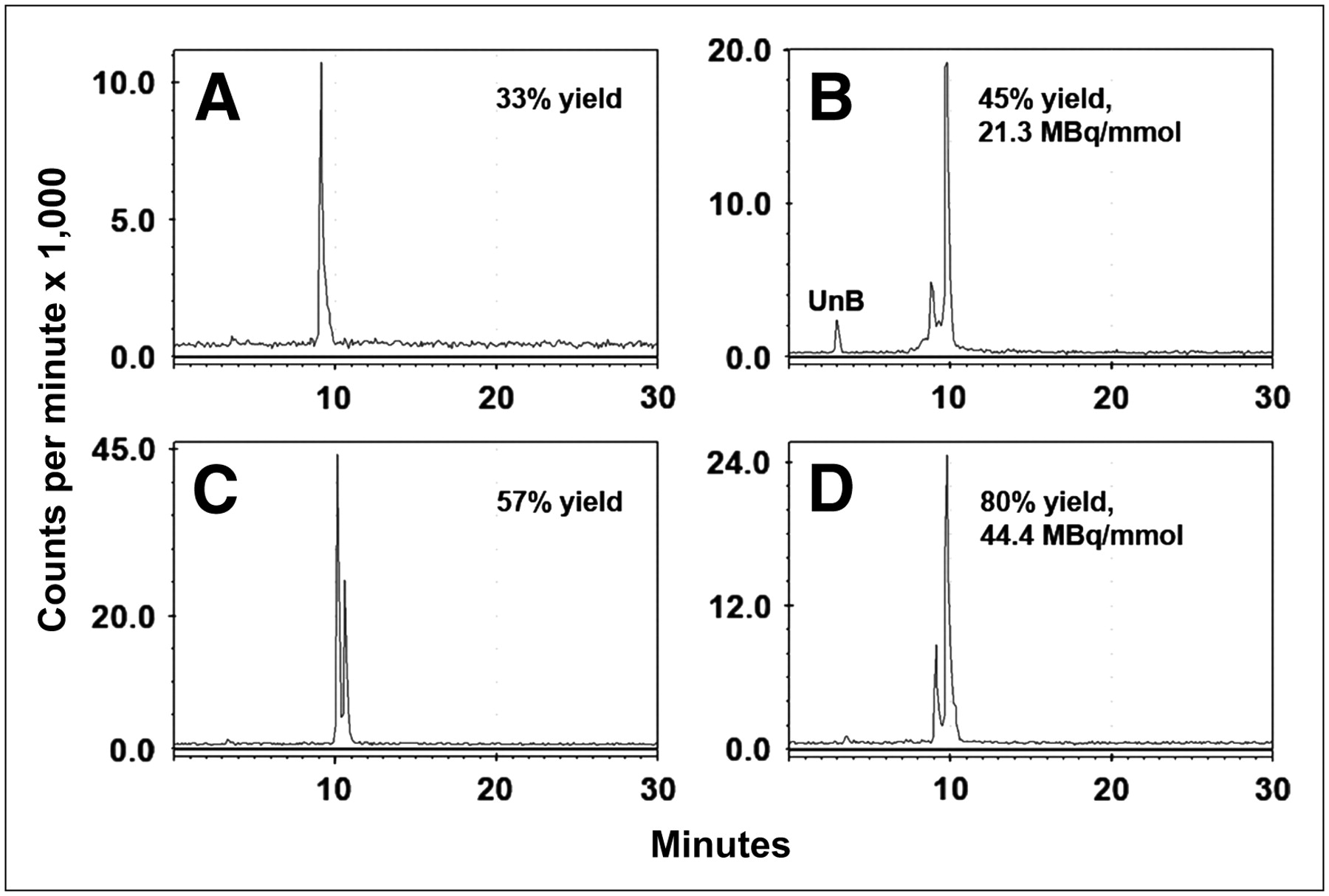
RP-HPLC illustrates final radiolabeled products obtained under different radioiodination conditions. (A and C) 124I-Labeled products obtained using Na124I with (C) or without (A) addition of carrier-added iodide. Addition of carrier improved yields, with formation of both mono- and diiododinated forms. (B and D) 131I-Labeled products prepared using IODO-GEN–coated vials vs. microparticulate IODO-GEN. UnB = unbound radioiodine.
In Vivo Pretargeting
Initial studies confirmed that optimal pretargeting with 131I-IMP-325 could be achieved by first administering the hBS14 bs-mAb, followed 24 h later with the radiolabeled peptide (not shown). A 10-fold mole excess of the hBS14 was given in relation to the moles of radiolabeled peptide injected. The targeting properties were first examined using varying doses of the hBS14 bs-mAb, with a constant amount of 131I-peptide given 24 h later.
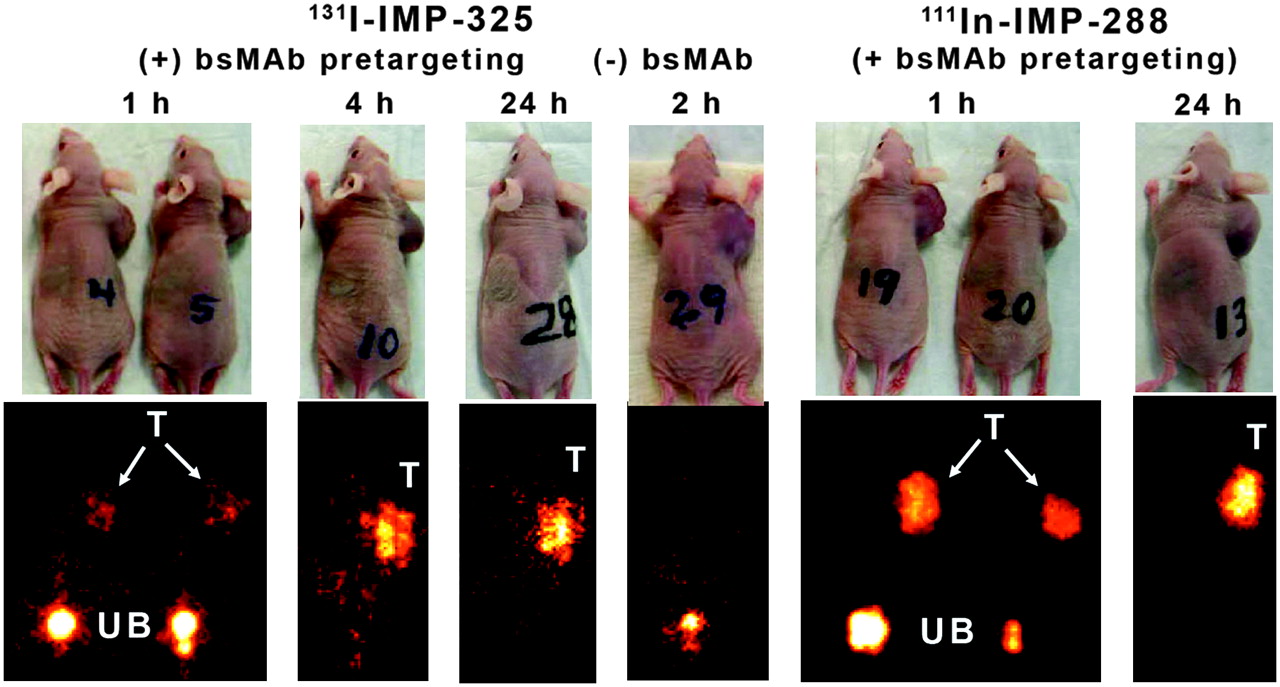
Whole-body imaging studies were performed with 131I-IMP-325 in pretargeted animals at 1, 4, and 24 h after the peptide injection (Fig. 2). At 1 h, most of the 131I radioactivity was already filtered from the kidneys and in the urinary bladder. Tumor uptake was evident, but image intensity could not be increased further without significant septal penetration emanating from the high 131I activity levels in the urinary bladder. Images taken at 2 h from animals that had only received 131I-IMP-325 (without pretargeting) indicated no selective 131I-peptide uptake in the tumor. The 1-h images of the animals pretargeted with 111In-IMP-288 could be adjusted to highlight tumor uptake without artifacts.
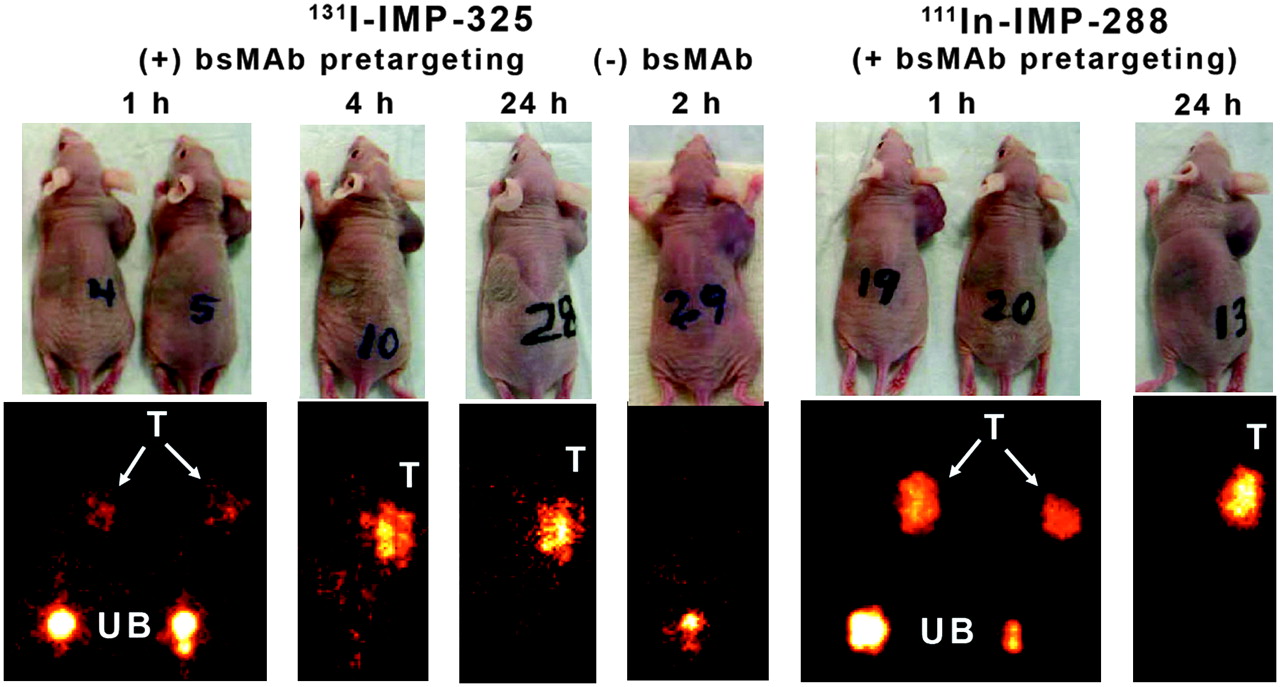
Whole-body γ-images of 131I-IMP-325 in animals given hBS14 bs-mAb 24 h earlier (+ bs-mAb) or animals given only peptide alone (− bs-mAb). Images on the right show pretargeting under the same conditions using 111In-IMP-288. T = tumor; UB = urinary bladder. Arrows show tumor location on photographs of animals.
Necropsy data in Table 1 are given as a %ID per organ to reflect the amount of activity in each tissue as seen by external scintigraphy, but the tissue weights (normal tissue weights taken from 1-h necropsy data) are provided for conversion to %ID/g. The tumors had substantially higher amounts of the pretargeted 131I- or 111In-peptide than any of the normal tissues at 1 h, with >30 times the amount of activity in the tumor than in the liver or spleen and ≥15 times more activity in the tumor than in the blood and lungs. As expected, the kidney and stomach had the highest amount of 131I-activity of the normal tissues, as the peptide is cleared from the body by urinary excretion, with uptake in the stomach indicative of the metabolic processing of radioiodine (no premedication to reduce gastric or thyroid uptake), albeit a portion of this uptake could be attributed to the small amount (∼4%) of unbound radioiodine coinjected with the 131I-IMP-325. Neither of these organs was visualized on the 1-h image, because when expressed as %ID per organ, tumor uptake was ≥15 times higher than in kidneys or stomach. Except for an ∼10-fold increase in the amount of activity in the stomach at 1 h for the 131I-pretargeted peptide compared with the pretargeted 111In-peptide, there were no appreciable differences in the tumor and organ uptake. By 4 h, the radioactivity in the urinary bladder had been voided and, therefore, the 131I-images could be adjusted to illustrate the enhanced tumor uptake, whereas uptake in all normal tissues had decreased. At 4 h, the pretargeted peptide was 70-fold higher in the tumor than with the peptide alone. At 24 h, only the activity in the tumor was visualized, with tumor-to-blood ratios of >200:1.
Tissue Uptake and Targeting Ratios for Pretargeted or Nonpretargeted 131I-IMP-325 and Pretargeted 111In-IMP-288
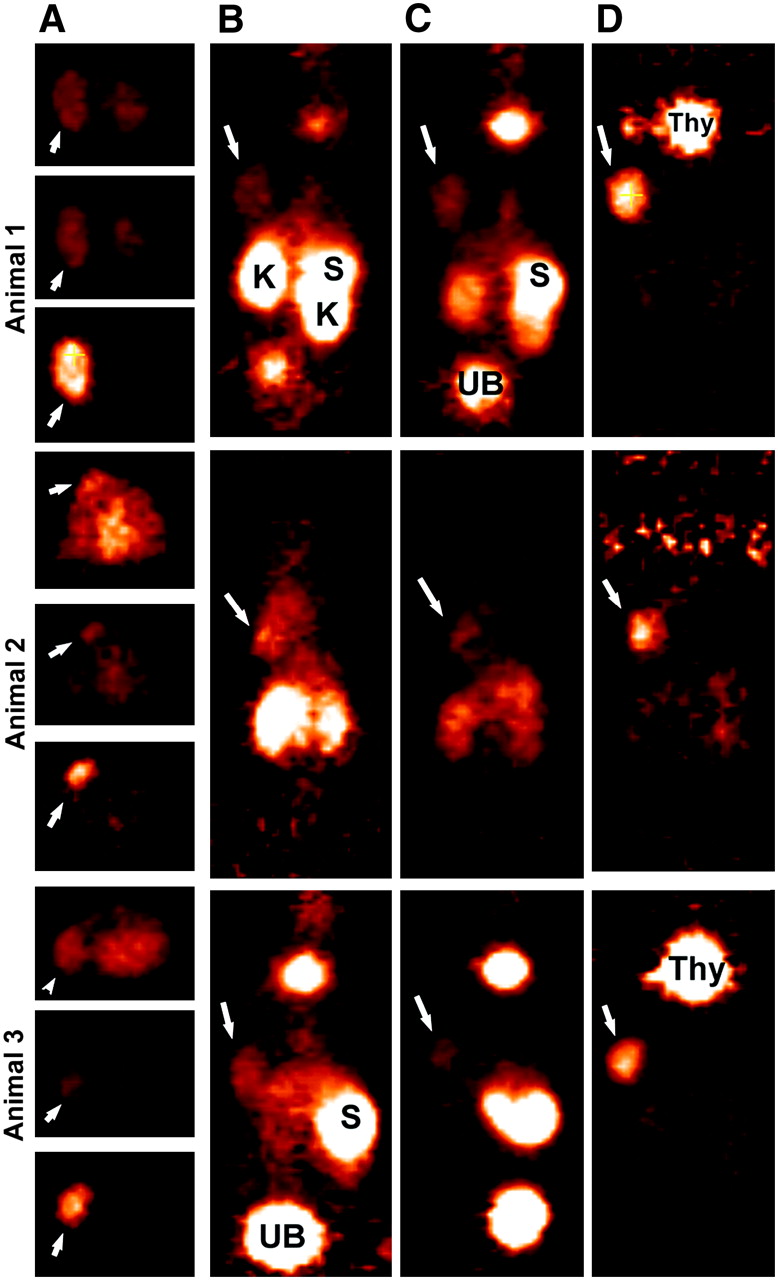
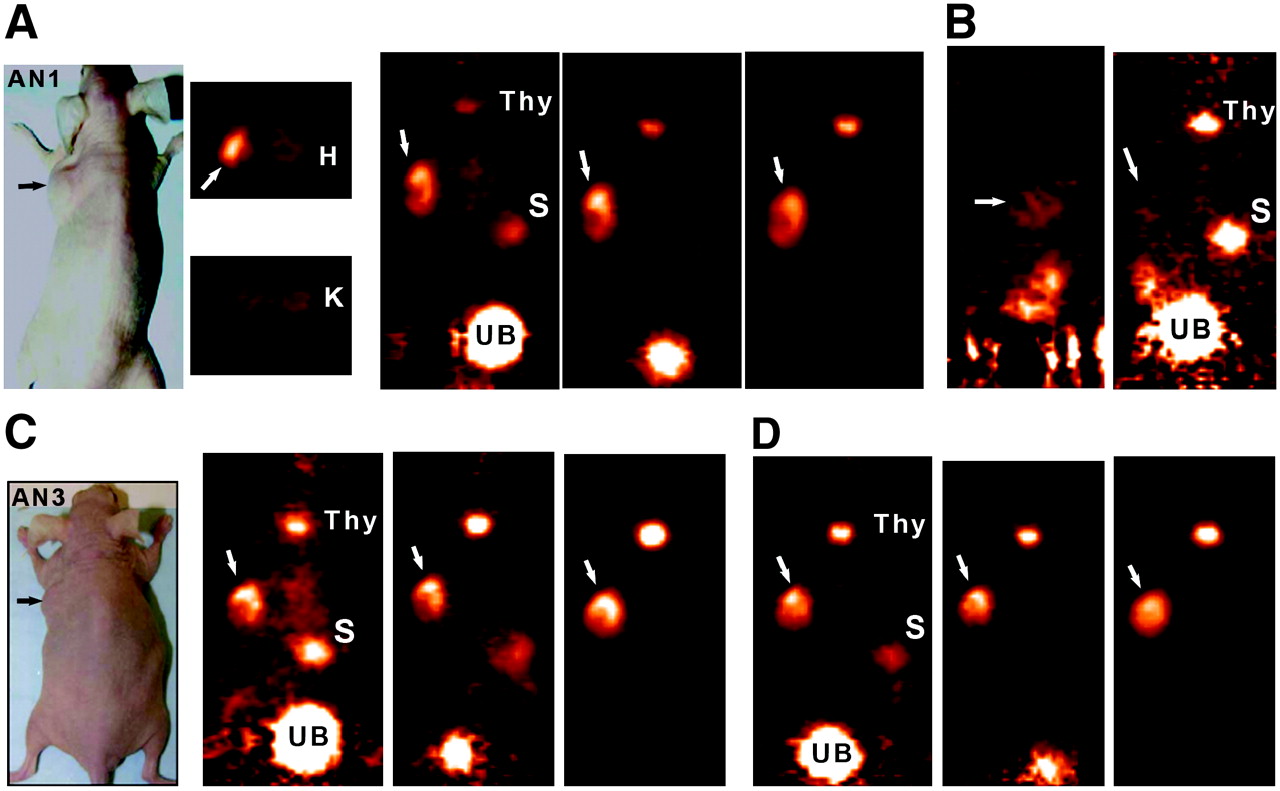
With evidence that the peptide could be radioiodinated and perform well in a pretargeting setting, an evaluation of the 124I-labeled peptide for PET was undertaken. Figures 3–⇓5 compare the images obtained in animals receiving the various agents. Thyroid and stomach activity was more prominent in the pretargeted 124I-peptide images (e.g., Fig. 2 vs. Fig. 3), presumably because of the higher level of unbound 124I but also the tumors in these animals were smaller (∼0.2–0.5 g). The smaller tumors were all clearly seen on the 1-h images without substantial radioactivity observed in any other organ (except for the nonvoided activity in the urinary bladder). A transverse slice through the region of kidneys and tumor is shown for animal 1 and illustrates the relative ease with which the tumor can be visualized against the major organ involved in the excretion of the radiolabeled peptide. Within each of the pretargeted 124I-peptide animals, the image intensity was normalized to the level used at 1 h and, therefore, the coronal slices shown in Figures 3A, 3C, and 3D reflect the quantitative changes occurring from ∼2 to 24 h. Tumor intensity appears unchanged over 24 h, whereas the background in the normal tissues continued to decrease. The region of interest (ROI) for the tumor and left kidney taken from transverse slices over a 24-h period confirmed this and indicated that tumor-to-kidney ratios were initially about 2:1, increasing by nearly 2-fold at 24 h (Table 2). Necropsy data from a separate group of animals indicated the same trend of relatively constant tumor activity with decreasing activities in normal tissues (Table 3). The necropsy results indicated that tumor-to-blood ratios were 9.1 ± 2.6 at 1 h, which doubled at 3 h, and then increased to >200:1 by 24 h.

Focus 120 microPET images of 124I-IMP-325 pretargeted with hBS14 (A, C, and D) or alone (not pretargeted; B). (A) From left to right, photograph of AN1, 2 transverse slices (0.82 mm) at level of tumor (arrows) and kidneys (K), taken ∼2 h after injection, and coronal slices obtained at ∼2, 6, and 24 h after injection. (B) From left to right, sagittal and coronal views obtained 1.9 h after injection. (C) From left to right, photograph of AN3 and coronal slices obtained at ∼2, 6, and 24 h after injection. (D) From left to right, coronal slices obtained at ∼2, 6, and 24 h after injection. All pretargeted images are adjusted to same intensity as 2-h image for each animal to illustrate retention and clearance of 124I-IMP-325 over time. At 24-h necropsy, tumors were 0.448, 0.285, and 0.483 g in pretargeted animals and 0.472 g in animal given 124I-IMP-325 alone. H = heart; S = stomach; Thy = thyroid; UB = urinary bladder.
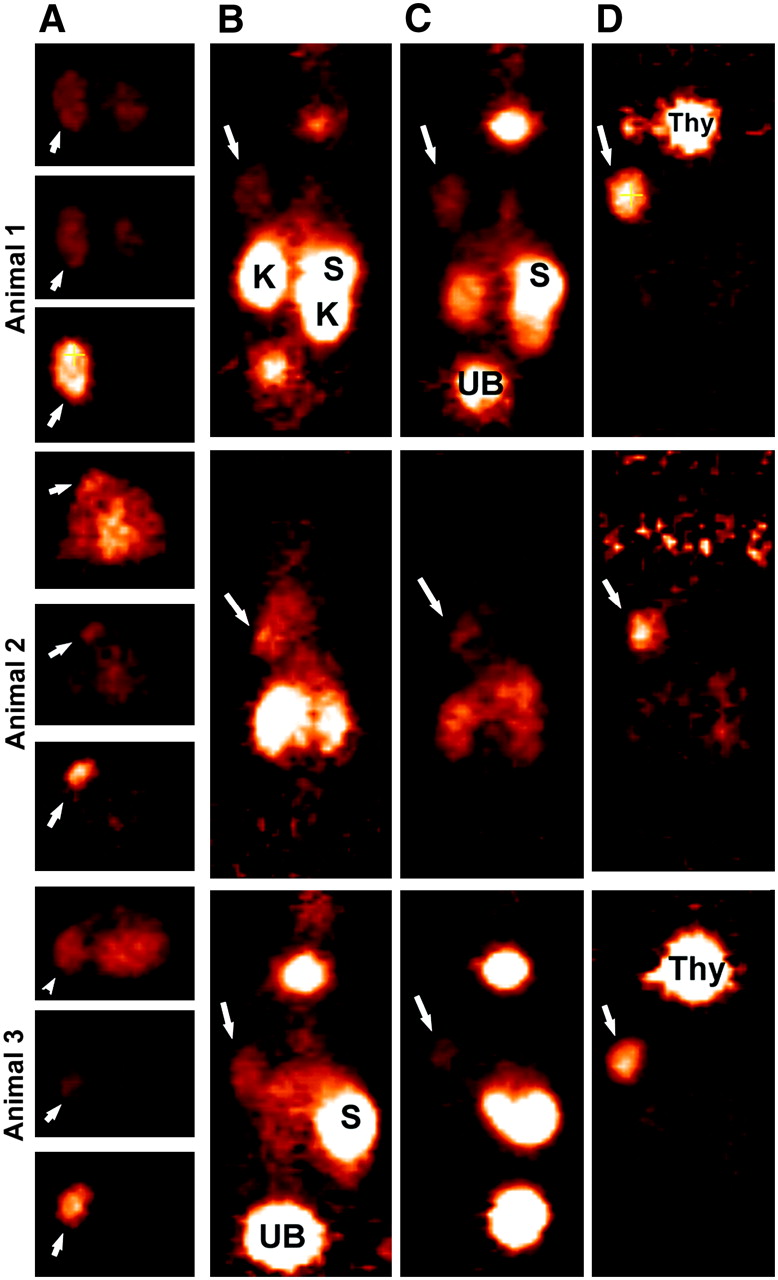
Focus 120 microPET images of 3 animals injected with 124I-hMN-14 Fab′. (A) Transverse slices (0.82-mm thick) in same plane to highlight tumor (arrows) localization at ∼2, 6, and 24 h, from top to bottom, respectively, for each animal. Six-hour images are taken at same intensity as 2-h images, but intensity was increased substantially for 24-h images. Coronal sections taken from 2-, 6-, and 24-h intervals (B, C, and D, respectively) illustrate elevated uptake of radiolabeled Fab′ initially in kidneys (K) and stomach (S). Uptake in these tissues was cleared over time, but there was persistent thyroid (Thy) uptake. Tumor weights at necropsy (24 h) were 0.596, 0.173, and 0.280 g, respectively.

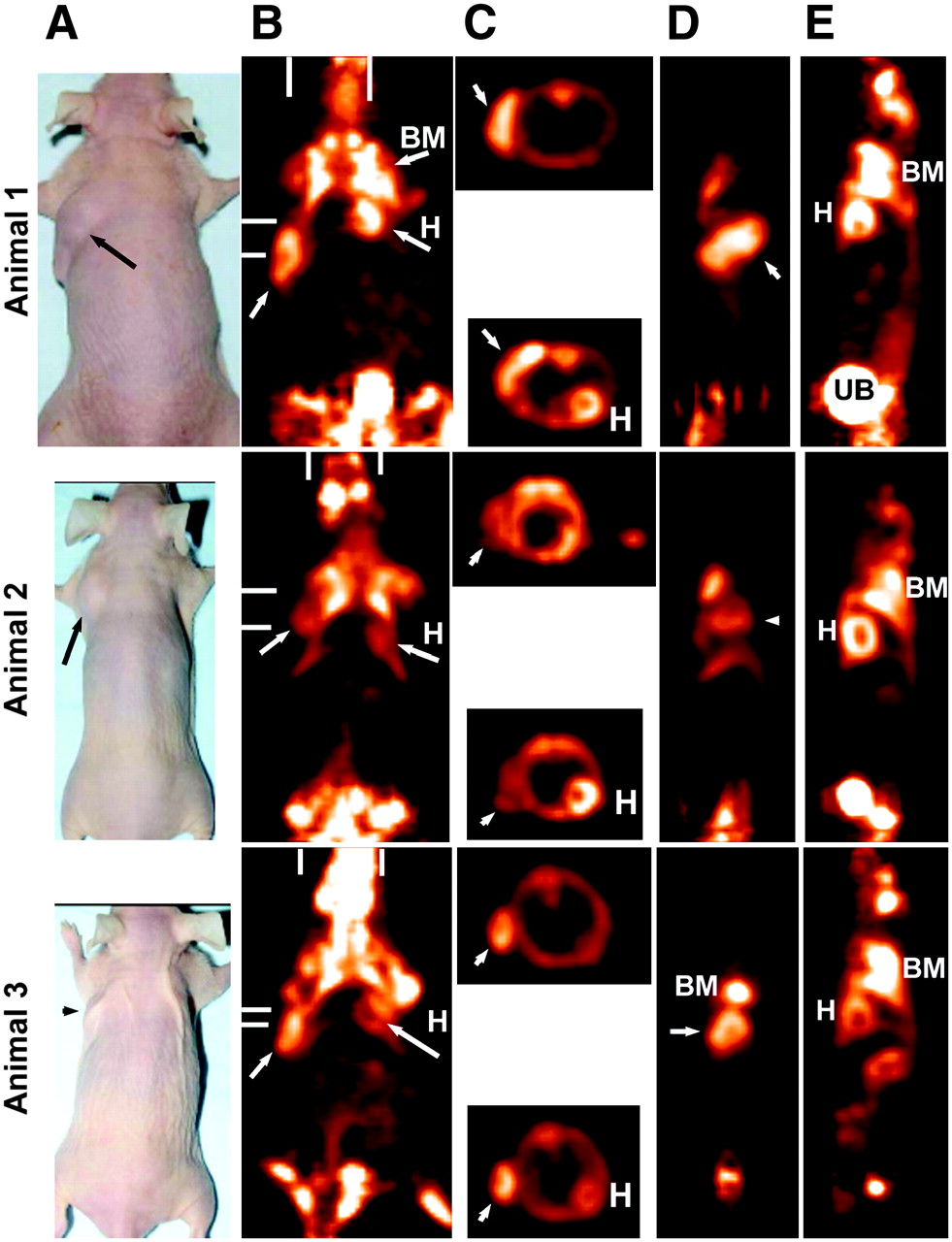
Focus 120 microPET images of animals bearing LS174T human colorectal cancer xenografts 1.7 h after 18F-FDG. All images were taken at same maximum intensity and background set to 0.0. (A) Photographs of 3 tumor-bearing animals. (B) Coronal slice (0.82-mm thick) taken to highlight uptake in tumor (arrows), as well as in bone marrow (BM) of shoulder and in hind quarters. Heart (H) and brain can also be seen in some images. Bars represent planes used for transverse and sagittal sections. (C) Transverse sections that highlight tumor or heart wall uptake. Sagittal sections illustrate the side view of tumor (D) and a plane that highlights heart, bone marrow, and urinary bladder (UB) activity (E). Tumor weights were 0.425, 0.321, and 0.153 g, respectively.
ROIs Derived from microPET Images of Animals Given Pretargeted 124I-IMP-325 or 124I-Humanized Anti-CEA Fab′
Comparative Biodistribution (%ID/g and Tumor-to-Nontumor Ratios) of Pretargeted 124I-Peptide Compared with 124I-Anti-CEA Fab′ and 18F-FDG Based on Necropsy Data
The images of the animals given the 124I-peptide alone required considerable intensity enhancement to assess the peptide's distribution even at ∼2 h, as the peptide alone cleared so rapidly from the tissues. Even a tumor of ∼0.5 g could barely be identified on the 1.9-h microPET images and, except for prevoided activity in the bladder and kidneys, there was relatively little activity remaining in the body (Fig. 3B). Over time, the activity levels decreased so dramatically that it was difficult to visually distinguish focal organ or tumor uptake from general body background activity (not shown).
In contrast to the excellent tumor localization in the pretargeted 124I-IMP-325 animals, tumors were less distinct at 2 and 6 h in the animals given 124I-anti-CEA Fab′ because of an appreciably higher level of activity in the kidneys, stomach, and thyroid (Fig. 4). The images and the ROI data (Table 2) show a rapid clearance of radioiodine from the kidneys, but according to the ROI and necropsy data (Table 3), tumor-to-kidney ratios were <1.0 through 6 h. The imaging and necropsy data indicated that the activity in stomach of animals given 124I-Fab′ was 5–8 times higher than in the pretargeted animals at 1 and 3 h, but by 24 h this activity had cleared to just 0.5 ± 0.2 %ID/g, providing a favorable tumor-to-stomach ratio. The 2- and 6-h images were adjusted to the same intensity to illustrate the quantitative changes occurring in the tissue distribution of the radioactivity at these time intervals. Even though the intensity setting of the 24-h images of the animals given 124I-Fab′ had to be increased considerably and, therefore, cannot be quantitatively compared with the earlier images, the tumors (even one that was 0.173 g) were clearly visible without appreciable activity in the normal tissues. Necropsy data indicated that tumor uptake had decreased by as much as 8-fold from the level measured at 1 and 3 h. Importantly, tumor-to-kidney ratios were higher for the pretargeted peptide than with 124I-Fab′ even through 24 h (e.g., P = 0.037 at 24 h using a 2-tailed t test assuming equal variance; Excel 2003). A separate group of animals given a higher protein dose of the Fab′ (100 μg) had similar tumor and tissue uptake values as the lower protein dose (12 μg), indicating that the higher protein dose did not affect tumor localization.
Tumors were relatively well visualized with 18F-FDG at ∼1.7 h after its injection (Fig. 5), but there was substantially more activity in the bone marrow, heart wall, and brain than in the pretargeted animals. The elevated uptake in these tissues is consistent with the known distribution of 18F-FDG, but because these tissues were not included in the necropsy panel, the relative uptake in the bone marrow and heart was assessed by ROI analysis. As shown in Table 4, uptake in the heart wall was about the same or nearly 2-fold higher than the tumor. Bone marrow uptake was 1.2- to 1.7-fold higher than that of the tumor. When compared with the necropsy data from the pretargeted animals at 1 h, the results suggest a higher tumor uptake for the pretargeted animals, but with higher tumor-to-blood ratios for 18F-FDG. The pretargeted animals had improved tumor-to-nontumor ratios for all other tissues, except the kidney.
ROIs Derived from microPET Images for Animals Given 18F-FDG
DISCUSSION
With the excellent sensitivity and spatial resolution—as well as quantification by PET and more recently the fusion of PET/CT images to improve anatomic detail—18F-FDG PET and PET/CT are gaining an increasing role in cancer detection as well as in monitoring the response to treatment (23,24). Nonetheless, the tumor “nonspecificity” of 18F-FDG, which is concentrated avidly in metabolically active and proliferating tissues generally, invites the development of more tumor-specific PET agents. The molecular specificity of antibodies and immune constructs continues to spur interest in their development as highly tumor-specific diagnostic and therapeutic radiopharmaceuticals. Advances in molecular engineering have humanized antibodies to reduce immunogenicity that would allow repeated use, and there are new constructs with more favorable binding and pharmacokinetic properties than traditionally prepared antibody fragments (25,26).
Pretargeting represents another adaptation of antibody targeting that consistently has improved the delivery of radionuclides for imaging and therapeutic applications by greatly improving tumor-to-nontumor ratios—particularly, tumor-to-blood ratios—generated within hours (1,2). Remarkably too, in many instances, pretargeting methods have preserved tumor uptake of the radionuclide at levels comparable with those of directly radiolabeled IgG, whereas tumor uptake of directly radiolabeled antibody fragments is considerably lower, as shown here and elsewhere (1,3). By increasing the amount of radioactivity delivered selectively to the tumor, the resulting stronger signal will enhance detection capabilities for pretargeting methods compared with directly radiolabeled antibody fragments. Monovalent fragments are also quickly washed out of the tumor, whereas pretargeted radioactivity is sustained at a higher level for a longer period of time. With positron-emitters that have a long half-life, such as 124I, delayed images (e.g., 1 d after the 124I-peptide) would continue to have a strong signal emanating from the tumor, with negligible activity in the normal tissues. Positron-emitters with a longer physical half-life would add to radiation exposure but, in a pretargeting setting, the vast majority of the 124I-labeled peptide is eliminated from the body within a few hours and, therefore, the radiation dose to normal tissues would be minimized. Although 124I might not have the same spatial resolution as 18F because of its higher median positron energy (819 vs. 250 keV), when used in pretargeting, 124I should still be a safe and highly sensitive imaging radionuclide. This same pretargeting signal enhancement is seen with single-photon imaging using 99mTc (3), making this a less-expensive alternative to PET, but the enhanced spatial resolution afforded by PET, even with 124I, would likely offer benefits over SPECT in some applications.
Divalent HSG compounds that could be radioiodinated, but contained no additional functionality, were described earlier by Janevik-Ivanovska et al. (27). We were able to establish conditions to preferentially radioiodinate the single tyrosine residue in the HSG peptide used in the current studies, but this peptide also contains DOTA, a chelate that we have shown is capable of binding 111In, 90Y, 177Lu, and 67Ga (3,16,28). Before radioiodination, 111In+3 was added to the peptide to complex DOTA to reduce the potential that other metals may complex with DOTA, thereby ensuring a more homogeneous product. Our investigation revealed that the iodination was optimal when the pH was kept <4.0 to avoid multiple products that occur by iodinating the imidazole ring at higher pH. Under the conditions used, a mixture of di- and monoiodinated peptide was observed, with the mixture ratio varying from one iodide lot to the next, but was consistent for multiple procedures using the same lot of iodide. Specific activities of 29.6–44.4 GBq/μmol were possible using the current supplies of Na131I, with yields of ∼70%. Although Na124I is available in a carrier-free state, a trace amount of iodide was added to improve radiolabeling yields, with yields of 60% and specific activities of 29.6 GBq/μmol possible. The slightly lower radiolabeling yields with 124I may be explained in part by impurities developed in the 124I solution (29). Unlike the previous methods with radiometals, we were unable to develop conditions that would result in a >97% incorporation, which would have eliminated the need for the subsequent purification of the radiolabeled peptide. However, as with the radioiodination of antibodies that require a purification step, the purification of the radioiodinated peptide is a simple 1-step column method. Future studies will require more attention to reduce radiolytic effects if the product requires overnight storage.
Others have reported excellent imaging in the LS174T xenograft model using a variety of engineered 124I-anti-CEA divalent antibody fragments (e.g., minibody and single-chain Fv-Fc) within 18 h, with tumor-to-nontumor ratios of >10:1, including the kidneys (25,26). In contrast, a 64Cu-labeled anti-CEA minibody had substantial retention in the liver partially due to the inherent instability of copper bound by DOTA, whereas smaller fragments radiolabeled with metals will have high renal uptake (30,31). These examples highlight a limitation for any directly radiolabeled construct—namely, highly elevated hepatic or renal uptake when radiometals are used, but much more acceptable ratios when using radioiodine. In our study, tumor-to-kidney ratios with 124I-Fab′ at 24 h were about 6-fold higher than with 99mTc-Fab′ examined previously (3:1 vs. 0.5:1, respectively) (3). Tumor-to-kidney ratios for the 124I-labeled pretargeted peptide achieved this level within just 1 h, and tumor-to-organ ratios for the pretargeted peptide exceeded by as much as 20-fold that of 124I-Fab′, with tumor uptake at 24 h being 10-fold higher with the pretargeted peptide. Thus, pretargeting continued to show an advantage even over radioiodinated Fab′. Lewis et al. (32) reported excellent tumor localization using a StAv pretargeting approach with 64Cu-labeled DOTA-biotin, also showing positive tumor-to-nontumor ratios within 30 min, demonstrating that radionuclides with a short physical half-life would be suitable for pretargeted PET.
Whether the tumors were ∼1.0 g or about 0.1 g, uptake of the pretargeted radioiodinated peptide averaged about 15 %ID/g at 1 h. In contrast, tumor uptake of 18F-FDG at 2 h averaged about 5 %ID/g, with substantially higher uptake in the bone marrow, brain, and heart wall. LS174T is a rapidly growing tumor and, therefore, it should have a high incorporation of 18F-FDG but, as a mucinous colorectal cancer cell line, it might have reduced 18F-FDG uptake. LS174T also highly expresses CEA and should have highly favorable targeting using an anti-CEA antibody. An examination of additional tumor cell lines that vary in antigen expression, growth rate, physiology, and other pertinent parameters would aid in elucidating the broader capabilities or limitations of pretargeted imaging compared with 18F-FDG but, ultimately, a clinical assessment is required.
Although in certain situations the sensitivity and specificity of 18F-FDG for imaging several types of tumors, including colorectal cancer, are excellent (≥90% (33)), there are circumstances in which the specificity of the antibody-directed targeting agent could be more revealing (23,24). An example might be the ability to distinguish whether abdominal uptake of 18F-FDG represents recurrent tumor or a local inflammatory process, in which the expression of a tumor-associated antigen, such as CEA, could provide greater specificity. Kok et al. (34) compared the uptake of 18F-FDG in nude rats bearing an LS174T human colorectal tumor in addition to an Escherichia coli−induced focal infection and found similar uptake and lesion-to-blood ratios over time, indicating that even dual time-point analysis could not reliably discriminate tumor from inflammation. Although the heightened blood flow in inflammatory lesions might lead to an early elevation in the amount of bs-mAb or the radiolabeled peptide, because the bs-mAb lacks Fc receptors and there is no known cross-reactivity of the di-HSG peptides with human blood cells, there should be little, if any, retention of the radiolabeled peptide. Other examples in which ImmunoPET may have an advantage over FDG include prostate cancer and localization of bone or bone marrow metastases. In prostate cancer, FDG PET has not been successfully applied, particularly for local disease, where excreted activity in the urinary bladder masks any uptake in this region. As with any molecular targeting strategy, the image quality will largely depend on the specificity of the targeting agent—in this case, the tumor-binding portion of the bs-mAb. If the target is coexpressed and accessible on normal tissues, then the radiolabeled peptide likely will also have selective uptake in these tissues, much like the enhanced targeting of the intestines seen with the pretargeting system that used an anti-EGP-2 (EpCAM) antibody that had cross-reactivity with the normal intestine (35). Because the bs-mAb must remain accessible at the target so that it can bind to the subsequently administered radiolabeled peptide, targets that would be internalized after bs-mAb binding, or shed and carried away from the initial binding site, would not be expected to be suitable for pretargeting applications. With recombinant engineering, bs-mAb can be made quite small (50–80 kDa) (17,36), allowing the possibility of better penetration against physiologic barriers than larger proteins, and yet continue to provide multivalent binding for enhanced avidity.
Finally, it is important to emphasize that the chemical properties of the peptide can be adjusted to reduce nonspecific uptake in normal organs and allow for rapid urinary excretion with minimal renal retention. This peptide and others that we have described have rather low renal retention, regardless of whether radioiodine or a radiometal, such as 111In, is used. Thus, target-to-liver and target-to-kidney ratios are high enough to allow for easy discrimination against these normal tissues. Because the structure of a receptor ligand must be rigidly followed to allow its binding to the receptor, it is far more likely that these directly radiolabeled ligands could encounter elevated uptake and retention in the liver and kidneys as a normal consequence of their clearance from the body, but there will likely be circumstances in which a ligand could be modified to have more favorable biodistribution without altering its binding specificity and affinity.
CONCLUSION
The current results illustrate the flexibility and capability of the HSG-based pretargeting system by demonstrating the ability to localize a radioiodinated peptide using a recombinant anti-CEA bs-mAb. Rapid tumor uptake with accelerated clearance from normal tissues provided a significant advantage for detecting tumors using the pretargeting method in comparison with 124I-anti-CEA Fab′ or 18F-FDG. Bispecific antibody pretargeting methods could provide an additional dimension for oncologic imaging but may also be applied to other diseases with appropriately selected antibodies.
Acknowledgments
This study was supported in part by grant NJDHSS 06-1853-FS-N0 from the New Jersey Department of Health and Human Resources (CMMI), U.S. National Institutes of Health grants R24 CA83084, R01 CA84596, P50 CA 96439-06, and P30 CA08748, and U.S. Department of Energy grant DE-FG03-95ER62039 (MSKCC). We thank Valerie Longo, Lenka Muskova, Louis Osorio, German R. Tejada, and Dion Yeldell for technical assistance. Drs. McBride, Rossi, Losman, Chang, and Goldenberg are employed by or have financial interests in Immunomedics, Inc., or IBC Pharmaceuticals, Inc. The other authors declare no financial interests.
Footnotes
-
COPYRIGHT © 2006 by the Society of Nuclear Medicine, Inc.
References
- Received for publication February 2, 2006.
- Accepted for publication July 17, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Pretargeting: A Path Forward for Radioimmunotherapy
- {alpha}- Versus {beta}-Emitting Radionuclides for Pretargeted Radioimmunotherapy of Carcinoembryonic Antigen-Expressing Human Colon Cancer Xenografts
- Immuno-PET Using Anticarcinoembryonic Antigen Bispecific Antibody and 68Ga-Labeled Peptide in Metastatic Medullary Thyroid Carcinoma: Clinical Optimization of the Pretargeting Parameters in a First-in-Human Trial
- Diels-Alder Reaction for Tumor Pretargeting: In Vivo Chemistry Can Boost Tumor Radiation Dose Compared with Directly Labeled Antibody
- Quantitative Immuno-SPECT Monitoring of Pretargeted Radioimmunotherapy with a Bispecific Antibody in an Intraperitoneal Nude Mouse Model of Human Colon Cancer
- Phase II Trial of Anticarcinoembryonic Antigen Pretargeted Radioimmunotherapy in Progressive Metastatic Medullary Thyroid Carcinoma: Biomarker Response and Survival Improvement
- Immuno-PET of Cancer: A Revival of Antibody Imaging
- Optimization of Hapten-Peptide Labeling for Pretargeted ImmunoPET of Bispecific Antibody Using Generator-Produced 68Ga
- Pretargeted 177Lu Radioimmunotherapy of Carcinoembryonic Antigen-Expressing Human Colonic Tumors in Mice
- Pretargeted Immuno-Positron Emission Tomography Imaging of Carcinoembryonic Antigen-Expressing Tumors with a Bispecific Antibody and a 68Ga- and 18F-Labeled Hapten Peptide in Mice with Human Tumor Xenografts
- Pretargeted Radioimmunotherapy of Pancreatic Cancer Xenografts: TF10-90Y-IMP-288 Alone and Combined with Gemcitabine
- CD20-targeted tetrameric interferon-{alpha}, a novel and potent immunocytokine for the therapy of B-cell lymphomas
- A Novel Method of 18F Radiolabeling for PET
- Improved Therapeutic Results by Pretargeted Radioimmunotherapy of Non-Hodgkin's Lymphoma with a New Recombinant, Trivalent, Anti-CD20, Bispecific Antibody
- A Novel Bispecific, Trivalent Antibody Construct for Targeting Pancreatic Carcinoma
- Multifunctional Antibodies by the Dock-and-Lock Method for Improved Cancer Imaging and Therapy by Pretargeting
- Bispecific Antibody Pretargeting of Radionuclides for Immuno-Single-Photon Emission Computed Tomography and Immuno-Positron Emission Tomography Molecular Imaging: An Update