


Abstract
Pancreatic cancer is a silent disease that most commonly presents in an already metastatic form. Current treatment options provide little survival benefit. Radiolabeled PAM4 IgG, a monoclonal antibody that recognizes a unique epitope associated with a mucin found almost exclusively in pancreatic cancer, has shown encouraging therapeutic effects in animal models and in early clinical testing (90Y-humanized PAM4 IgG, 90Y-clivatuzumab tetraxetan). The studies reported herein examine a new pretargeting procedure for delivering therapeutic radionuclides. Methods: We prepared a humanized, recombinant tri-Fab bispecific monoclonal antibody (bsmAb) (TF10) using specificity for targeting pancreatic cancer of PAM4 and another Fab binding to a hapten (histamine-succinyl-glycine [HSG]) and tested this in a pretargeting setting with a 90Y-1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid-di-HSG-peptide (pretargeted radioimmunotherapy [PT-RAIT]). Nude mice bearing established Capan-1 human pancreatic cancer xenografts were given TF10 and then received the 90Y peptide as a single bolus dose 19 h later, or the therapy cycle was fractionated weekly. Other studies examined different combinations with gemcitabine. Results: PT-RAIT of 18.5 MBq (∼50% of its maximum tolerated dose [MTD]) was as effective as the MTD of 90Y-PAM4 IgG (5.55 MBq). Three monthly doses of 9.25 MBq of PT-RAIT combined with a monthly cycle of gemcitabine (3 weekly, 6-mg doses) significantly enhanced survival, compared with PT-RAIT alone. Adding gemcitabine as a radiosensitizer to 9.25 MBq of PT-RAIT enhanced objective responses. Weekly fractionation of the PT-RAIT, as compared with a single treatment, improved responses. Conclusion: PAM4-based PT-RAIT with 90Y hapten peptide is an effective treatment for pancreatic cancer, with less toxicity than 90Y-PAM4 IgG, in this model. Combinations with gemcitabine and dose fractionation of the PT-RAIT enhanced therapeutic responses.
Each year, nearly 38,000 people in the United States are newly diagnosed with pancreatic cancer, and almost 34,000 die from the disease because currently there are no effective therapies (1). Patients with stage 3 locally advanced disease likely will have local radiation or gemcitabine therapy, but the median overall survival is only 11.0 mo (2). Patients with metastatic disease are currently treated with gemcitabine and erlotinib. Recent attempts to augment this treatment with bevacizumab, although significantly improving the median progression-free survival by 1 mo, have not significantly improved the overall survival (2). Several other clinical trials are being conducted to improve on these results, including one that combines Nab-paclitaxel with gemcitabine that has promising early results (3,4).
We reported the development of a monoclonal antibody, PAM4, highly specific for pancreatic carcinoma, with 85% or more of pancreatic carcinomas expressing the PAM4-reactive mucin (5,6). Recent immunohistology studies have shown that the PAM4-antigen is present in early pancreatic intraepithelial neoplasia lesions. An ELISA for detecting PAM4-mucin in the blood appears to be more specific and sensitive for detecting pancreatic carcinoma than CA19-19 (7). Radioconjugates of PAM4 were effective in targeting human pancreatic carcinoma xenografts; thus, PAM4 radioconjugates may be useful in the detection and treatment of pancreatic cancer. Indeed, animal studies showed 90Y-labeled PAM4 IgG to be effective in controlling the growth of established xenografts (8). Small, repeated doses of gemcitabine added to 90Y-PAM4 therapy improved the response, and even small doses of 90Y-PAM4 IgG given to mice together with an equivalent of the recommended gemcitabine therapy regimen (i.e., 3 weekly human equivalent doses of 1,000 milligrams of gemcitabine per m2 [6 mg/20-g mouse], followed by 1 wk of rest before repetition of the cycle 2 more times) improved therapeutic responses (9). These encouraging data led to a phase I therapy trial with 90Y-1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA)-humanized PAM4 IgG (hPAM4; 90Y-clivatuzumab-tetraxetan; Immunomedics, Inc.) in patients with metastatic pancreatic cancer who failed at least 1 prior chemotherapy regimen. The maximum tolerated dose was 740 MBq (20 mCi)/m2, based on dose-limiting hematologic toxicity; encouragingly, 1 patient at the 555 MBq (15 mCi)/m2 dose and 2 patients at the 740 MBq (20 mCi)/m2 dose had 32%–51% shrinkage of their pancreatic lesions. Another 4 patients at various dose levels had stable disease (10). Currently, a phase Ib/II trial is underway to examine 3 weekly treatments of 90Y-hPAM4 IgG combined with gemcitabine (200 mg/m2 given 2 d after each 90Y dose) in a frontline setting for patients with metastatic pancreatic cancer (11).
We recently reported the development of a novel humanized tri-Fab bispecific antibody, TF10, as an agent for pretargeted radioimmunotherapy (PT-RAIT). The construct consists of 2 PAM4-Fab arms for tumor binding and a third Fab that binds to a unique hapten, histamine-succinyl-glycine (HSG) (12,13). This pretargeting system markedly amplifies the tumor-to-nontumor signal ratios, allowing for the clear detection of tumors within an hour of the radiolabeled hapten-peptide injection (14). Tumor uptake of peptide with this pretargeting system appeared quite promising for therapy when the peptide was labeled with 90Y (15,16). Herein, we report the initial therapeutic evaluation of this new pretargeting system in mice and show how it can be combined with gemcitabine for enhanced antitumor responses.
MATERIALS AND METHODS
Reagents
The preparation of humanized tri-Fab bispecific antibody TF10 and IMP-288 (DOTA-d-Tyr-d-Lys(HSG)-d-Glu-d-Lys(HSG)-NH2) was described previously (12,16,17). DOTA-hPAM4 IgG was provided by Immunomedics, Inc. Gemcitabine HCl (Gemzar) was purchased from Eli Lilly and Co.
Radiolabeling
IMP-288 was radiolabeled with 111InCl3 or 90YCl3 (PerkinElmer), according to previously published methods (16). The product had less than 3% unbound radionuclide that was captured, with excess diethylenetriaminepentaacetic acid added to the product at the completion of the labeling procedure. The final specific activity was 36.8 MBq (0.995 mCi)/nmol for 111In-IMP-288 and 102.7 MBq (2.8 mCi)/nmol for 90Y-IMP-288.
Animal Studies
Capan-1 (American Tissue Culture Collection) human pancreatic tumors were initially established by implanting (subcutaneously, 1 × 107 cells into the right flank) in 4- to 5-wk-old female athymic NCr nu/nu nude mice from National Cancer Institute Animal Production or athymic NCr nu/nu nude mice obtained from Taconic Farms. We subsequently found that tumors grew more consistently and rapidly when tissue culture cells were supplemented with a tumor suspension prepared from a serially propagated, subcutaneously grown tumor. When tumors reached approximately 2 cm3, they were minced in buffer and passed through a cell dissociation sieve, 50-mesh screen (Sigma-Aldrich Inc.), to create a 15%–20% tumor suspension. This suspension was then supplemented so that each mouse received 9 × 106 Capan-1 cells in 0.25 mL of the tumor suspension.
All agents, except gemcitabine, were administered intravenously in 0.2 mL or less. Radiolabeled hPAM4 IgG was adjusted to 50 μg with unlabeled hPAM4 IgG. For pretargeting studies, the doses of TF10 and IMP-288 were adjusted to yield a prescribed molar ratio (i.e., moles of TF10 to IMP-288 administered, typically 12:1). Because IMP-288 was always radiolabeled at a fixed maximum specific activity (see above), the amount of TF10 administered was adjusted according to the amount of IMP-288 used to deliver the prescribed amount of radioactivity. Animals were monitored daily, with body weights measured weekly. Animals showing any signs of morbidity or a loss greater than or equal to 20% in body weight were euthanized and censored as a treatment-related toxicity.
Tumor sizes were measured in 3 dimensions at the onset of the study and then once or twice per week thereafter. A partial response was declared when tumor volume decreased by at least 50% or more from the maximum volume. When tumors were no longer measurable or visible, a complete response was declared. The duration of response was judged from the maximum response to when progression occurred. When the product of these measurements, tumor volume, was greater than or equal to 3.0 cm3, the animal was removed from study. Comparisons were based on a log rank test, using the time required for tumors to reach 3.0 cm3 as a survival surrogate. Analysis was performed using GraphPad Prism software (version 4.0; GraphPad Software).
Hematologic toxicity was assessed in non–tumor-bearing, 8-wk-old female NIH Swiss mice (National Cancer Institute). Animals were bled retroorbitally after local proparacaine anesthesia 1 d in advance and then weekly after treatment (∼50 μL). Blood was added to 1 mL of lysing buffer. After 10 min, the cells were centrifuged and washed in phosphate-buffered saline (PBS) and finally resuspended in 2 mL of PBS–formalin. One hundred microliters were added to 0.9 mL of PBS–formalin and read by flow cytometry (Facscalibur; BD Biosciences) using preset scatter windows. The percentage change in each animal's total white blood cell (WBC) count from its baseline determination was calculated and averaged (n = 5 per treatment). Treatments consisted of an intravenous injection of 5.55 MBq (0.15 mCi) of 90Y-hPAM4 IgG or 18.5 MBq (0.5 mCi) of 90Y-IMP-288 targeted in advance 16 h earlier with TF10 (molar ratio, 12:1).
To assess renal toxicity, groups of 11 NIH Swiss mice were given PT-RAIT alone or in combination with gemcitabine. PT-RAIT groups included 18.5, 25.9, and 33.3 MBq (0.5, 0.7, and 0.9 mCi, respectively) of 90Y-IMP-288 given at week 0. Combination groups included 18.5 and 25.9 MBq (0.5 and 0.7 mCi, respectively) of 90Y-IMP 288 PT-RAIT given at week 0 with gemcitabine (6 mg, intraperitoneally) administered at weeks 0, 1, and 2. Mice were monitored for 36 wk and necropsied, and kidneys were placed in 10% buffered formalin, embedded in paraffin, and sectioned for histopathologic evaluation.
RESULTS
Two groups of animals given 9.25 and 18.5 MBq (0.25 and 0.5 mCi, respectively) of the 90Y-labeled IMP-288 targeted in advance with TF10 were evaluated initially. Another group of animals was given 5.55 MBq (0.15 mCi) of 90Y-DOTA-hPAM4 IgG. All animals tolerated PT-RAIT with no appreciable loss in body weight (≤5%). One animal given 90Y-hPAM4 IgG was removed because of weight loss at 7.4 wk.
Tumors (∼0.40 ± 0.10 cm3 at onset) increased in size after treatment, some more than doubling, before they began to regress 2 wk after treatment (Figs. 1A–1C). Seven of 10 tumors in the 9.25-MBq-treated group progressed within 1–5 wk after reaching their maximum antitumor response, whereas only 2 tumors in the 18.5-MBq group progressed over this same time. Only 1 tumor progressed to 3.0 cm3 by week 20 in the 90Y-hPAM4 IgG group. At 19 wk, 3 of the remaining animals in the 2 PT-RAIT groups that had progressing tumors were retreated. The animals tolerated the second dose without weight loss. Two animals with less than or equal to 0.5-cm3 tumors at the time of retreatment responded at each dose level, whereas 1 approximately 2.5-cm3 tumor continued to progress after a second 18.5-MBq dose. This finding is consistent with earlier results with 131I-PAM4 IgG, in which the survival time was reduced with increasing tumor size (18). Using time to progression to 3.0 cm3 as a surrogate for survival, we determined that the median survival time was 16.3 wk for the 9.25-MBq PT-RAIT group, a significant survival advantage over untreated animals (5.4 wk; P < 0.001). The median survival for 18.5 MBq of PT-RAIT and 5.55 MBq of 90Y-hPAM4 IgG was not reached after 30 wk, when the study was terminated. Several animals had no evidence of residual tumor remaining at the implantation site at the conclusion of the study.
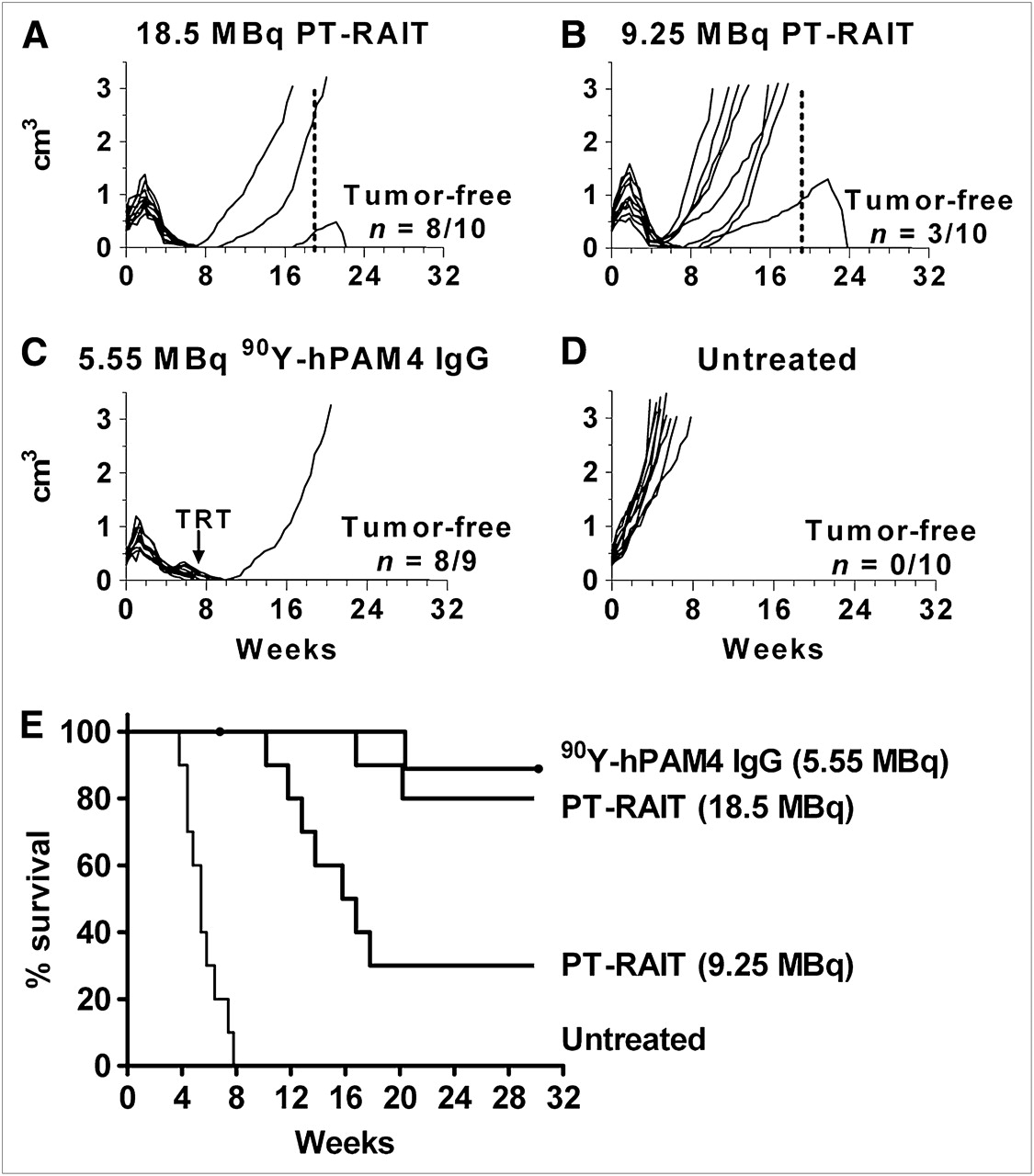
PT-RAIT (TF10–90Y-IMP-288) and RAIT (90Y-hPAM4 IgG) of established Capan-1 human pancreatic tumor xenografts in nude mice (n = 10/group). (A–D) Individual curves for each animal in 4 groups. (E) Representation of survival data based on time required to reach 3.0 cm3. (A) Mice received 481 μg of TF10, followed by 18.5 MBq (0.5 mCi) of 90Y-IMP-288 16 h later. (B) Mice received 241 μg of TF10, followed by 9.25 MBq (0.25 mCi) of 90Y-IMP-288 16 h later. (A and B) Second PT-RAIT treatment was given to 3 animals on week 19 (dotted line). (C) Mice received 5.55 MBq of 90Y-hPAM4 IgG (50 μg). One animal in RAIT group died of treatment-related toxicity (TRT). (D) Mice were not treated.
The 1 animal in the 90Y-hPAM4 IgG group that experienced excessive toxicity suggested the maximum tolerated dose (MTD) had been exceeded. Weekly monitoring of blood counts confirmed that animals given 5.55 MBq of 90Y-hPAM4 IgG experienced approximately a 90% decrease of their WBC count starting 1 wk after treatment and lasting for 1 more week, before recovering by week 8 (Fig. 2). Animals given 18.5 MBq of 90Y-IMP-288 PT-RAIT experienced a transient 70% decrease in WBC counts that rebounded to a 30% loss in baseline counts by the next week, with full recovery by week 7. Because renal toxicity was shown previously to be dose-limiting for PT-RAIT (15), NIH Swiss mice were given various treatments and monitored for 9 mo. Mice averaging approximately 22 g at the onset of treatment had no appreciable weight loss in any of these groups. One animal given gemcitabine alone began losing weight at week 27 and was found dead at week 29, and another animal given 25.9 MBq (0.7 mCi) of PT RAIT plus gemcitabine developed a distended abdomen and diarrhea at week 34 and was removed from the study. All of the remaining animals were euthanized in week 36. Kidney weights in the PT-RAIT and PT-RAIT–plus–gemcitabine groups at necropsy were similar to the sham-buffer control or gemcitabine-alone groups. Histologic assessment found no evidence of substantial morphologic changes in the kidneys in the PT-RAIT or PT-RAIT–plus–gemcitabine group when compared with the sham-buffer control or gemcitabine-alone groups (Table 1). None of the changes were suggestive of the low incidence of severe renal pathology noted previously (15). The previous study used a first-generation di-HSG-peptide, 90Y-IMP-241. A comparison of renal uptake of 111In-IMP-288 and IMP-241 3 h after injection revealed that 111In-IMP-288, compared with 111In-IMP-241, had 30% lower uptake (1.52 ± 0.47 and 2.29 ± 0.52 percentage injected dose per gram, respectively; n = 8/group, P = 0.008), with identical blood concentrations for both peptides. Thus, although 33.3 MBq (0.9 mCi) of 90Y-IMP-241 caused severe renal toxicity in a few animals, 33.3 MBq (0.9 mCi) of 90Y-IMP-288 was tolerated in all animals. In this regard, the therapy data shown in Figure 1 for 18.5 MBq of PT-RAIT represent responses at a dose that was perhaps only 55% of the MTD, whereas the 90Y-hPAM4 IgG was given at a level slightly above its MTD.

Hematologic toxicity in NIH Swiss mice given single treatment of TF10-pretargeted 90Y-IMP-288 or 90Y-hPAM4 IgG (5.55 MBq, 50 μg). PT-RAIT group received 481 μg of TF10, followed by 18.5 MBq (0.5 mCi) of 90Y-IMP-288 16 h later.
Histologic Assessment of Renal Toxicity in NIH Swiss Mice (22.5 ± 1.7 g) Given PT-RAIT
For combination studies, we elected to use a PT-RAIT dose of just 9.25 MBq, since a majority of the tumors respond but then progress after receiving this dose. The combination of gemcitabine with PT-RAIT was examined in 2 different settings. In the first, a small amount of PT-RAIT was added to a standard course of gemcitabine therapy. Animals were given a gemcitabine regimen that mimicked a standard treatment cycle given to patients (e.g., 3 weekly doses of the human equivalent dose of 1,000 mg/m2 gemcitabine [6 mg/20-g mouse intraperitoneally]; after a 1-wk rest, the cycle was repeated 2 more times). The TF10-pretargeted 90Y-IMP-288 was given 19 h after the first gemcitabine dose of each cycle (i.e., 9.25 MBq of PT-RAIT monthly × 3). In the second setting, various doses and schedules of gemcitabine were added to a single 9.25-MBq dose of TF10-pretargeted 90Y-IMP-288 to determine whether gemcitabine could augment PT-RAIT.
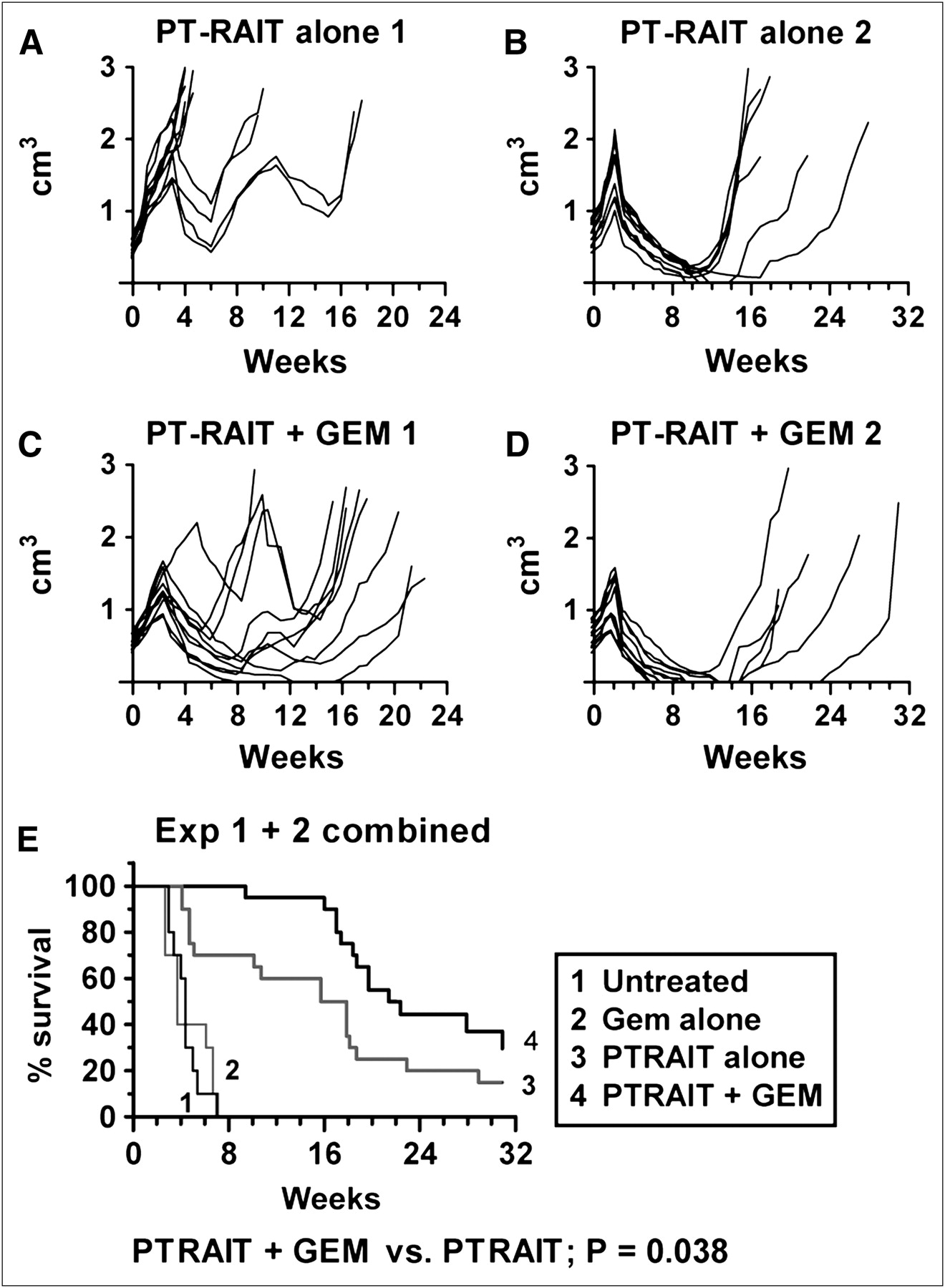
Two studies were performed to examine monthly PT-RAIT dosing added to a standard gemcitabine treatment. In study 1, 6 tumors in the PT-RAIT–alone group progressed rapidly after the first 9.25-MBq treatment, resulting in a median survival of only 4.8 wk (Fig. 3A). In contrast, 9 of 10 tumors in the paired group of animals given PT-RAIT and gemcitabine all responded to the first treatment cycle, with a median survival of 18.1 wk (Fig. 3C), a significant improvement over the PT-RAIT–alone group (P < 0.002). Because many more tumors in the PT-RAIT–alone group progressed more rapidly in this study than observed in Figure 1, a second study was initiated (Figs. 3B and 3D). For this second study, 12.95 MBq were administered as the initial PT-RAIT dose, followed by 2 monthly cycles of 9.25 MBq of the TF10-pretargeted 90Y-IMP-288 alone or together with gemcitabine. This treatment regimen increased the median survival to 20.4 wk in the PT-RAIT–alone group, with 4 animals being tumor-free after a 31-wk monitoring period. The initial spurt of tumor growth in the combination group, compared with animals that received only PT-RAIT, appeared to be stunted. Six animals that received 3 full cycles of gemcitabine with PT-RAIT were tumor-free at this same time, but there was only a trend toward a significant survival advantage as compared with PT-RAIT alone (P = 0.13). When the data from both studies were combined, a modest (16.8 vs. 21.9 wk) but significant survival advantage for the combination group (P < 0.038) was observed (Fig. 3E). Gemcitabine alone had no effect on tumor progression in either study, with an average median survival of just 4.4 wk, compared with 3.7 wk for untreated animals.

PT-RAIT enhancement of standard gemcitabine treatment. Capan-1 pancreatic tumor xenografts in nude mice (10/group) were treated with PT-RAIT (9.25 MBq) alone or in combination with gemcitabine in 2 separate studies. Study 1 was monitored for 22 wk and study 2 for 31 wk. PT-RAIT was given on weeks 0, 4, and 8, and gemcitabine was given once weekly for weeks 0–2, 4–6, and 8–10. Gemcitabine was always given 19 h in advance of 90Y-IMP-288 injection. PT-RAIT dose was 3 × 9.25 MBq in study 1; in study 2, the first PT-RAIT dose was 12.95 MBq, followed by 2 × 9.25 MBq. (A–D) Growth curves for individual animals in PT-RAIT and PT-RAIT–plus–gemcitabine treatment groups. (E) Survival curve representation of combined data from both studies, which is based on time when tumors reached 3.0 cm3. Combined survival curves for gemcitabine alone and untreated are included. (A) On weeks 0, 4, and 8, mice received 241 μg of TF10, followed by 9.25 MBq of 90Y-IMP-288 16 h later. (B) On week 0, mice received 337 μg of TF10, followed by 12.95 MBq (0.35 mCi) of 90Y-IMP-288 16 h later. On weeks 4 and 8, they were given 241 μg of TF10, followed by 9.25 MBq of 90Y-IMP-288 16 h later. (C) A + gemcitabine. Gemcitabine (6 mg) was given weekly for weeks 0–2, 4–6, and 8–10. Gemcitabine was given 19 h in advance of the 90Y-IMP-288 injection. (D) 337 μg of TF10, followed by 12.95 MBq (0.35 mCi) of 90Y-IMP-288 16 h later. Gemcitabine dosing was same as that given in C. GEM = gemcitabine.
The second investigation examined whether gemcitabine could enhance the activity of PT-RAIT. Several different dosing regimens were studied. One tested the best timing for gemcitabine administration by giving 6 mg of gemcitabine at either 19 h before or 22 h after 90Y-IMP-288. Although the literature consistently advises gemcitabine should be given 6–24 h before radiation exposure (19–22), because 90% of the radioactivity in a PT-RAIT setting is removed from the body within 1 d, perhaps a later gemcitabine dose might spare any enhanced sensitization of normal tissues while still offering some additional cytotoxicity to the tumor. In this study, the median survival time for PT-RAIT alone was 6.0 wk, and for the untreated group it was 3.5 wk. Animals receiving gemcitabine 19 h in advance had a significant survival advantage over animals given gemcitabine 22 h after the 90Y-IMP-288 (5.7 vs. 8.2 wk), but it was not significantly better than PT-RAIT alone. Importantly, the single 6-mg gemcitabine dose given 19 h in advance of 90Y-IMP-288 in this study produced the same minor (≤5%) body weight loss as did PT-RAIT alone, and therefore further escalation of the PT-RAIT dose could be tolerated.
In an effort to improve responses, 6 additional dosing schemes that fractionated gemcitabine into 2–3 doses over a period of 1–2 d were examined (Table 2). None of these schemes proved to be more effective than a single 6-mg dose given 19 h before 90Y-IMP-288, without causing substantial weight loss. Overall, the addition of gemcitabine to PT-RAIT treatment decreased the intensity of the initial growth spurt 1–3 wk after PT-RAIT and increased the number of partial-response/complete-response results. However, tumor progression occurred at approximately the same time. Figure 4 shows an example of this in animals treated with PT-RAIT alone or PT-RAIT plus gemcitabine (dosing regimen 4 in Table 2), in which there is an increase in the objective response rate but the duration of response was not changed appreciably. In this treatment regimen, animals lost an average of 20% of their weight within 1 wk but recovered fully by week 3 (PT-RAIT alone averaged only ≤5% loss), which could allow an additional treatment cycle to be added. However, the amount of weight loss would not permit escalation of the PT-RAIT dose, and because only approximately 30% of the MTD was given, a better response could be achieved by simply administering higher doses of PT-RAIT without gemcitabine. In contrast, a single 6-mg gemcitabine dose given before PT-RAIT yielded no additional toxicity, indicating that further escalation in the PT-RAIT dose is possible, and therefore this dosing regimen could improve the response.
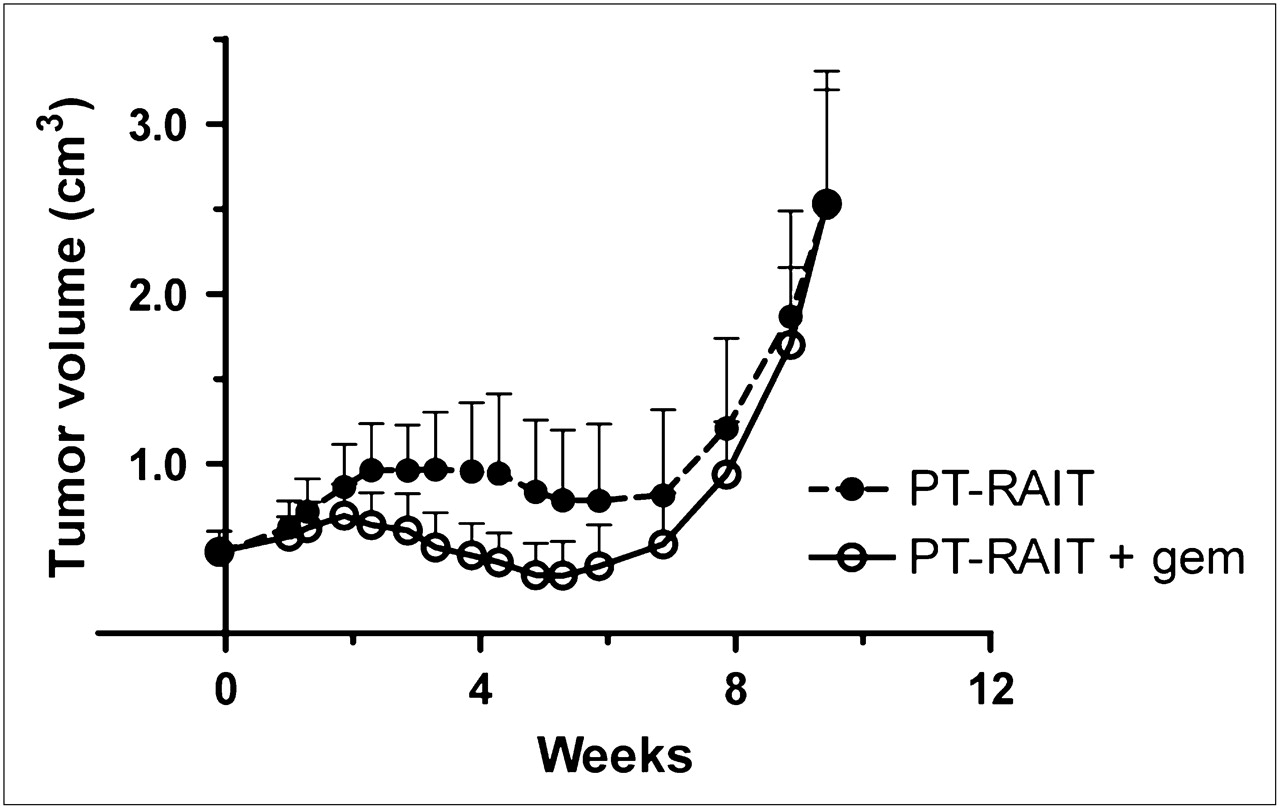
Capan-1 pancreatic tumor xenografts in nude mice (n = 11; mean ± SE) treated with PT-RAIT alone (9.25 MBq) or combined with gemcitabine (2 × 4 mg) given at 19 and 1 h in advance, respectively, of PT-RAIT dose. GEM = gemcitabine.
Effect of Nontherapeutic Gemcitabine Dosing on Single Dose of PT-RAIT
Dose fractionation has been reported as another potential way to improve responses (23–27). Animals were given a single 22.2-MBq injection of TF10-pretargeted 90Y-IMP-288, or this total dose was split into 2 or 3 equal fractions and given 1 wk apart (Fig. 5). The best responses were associated with a fractionated dosing regimen, with 2 × 11.1 MBq of 90Y-IMP-288 spaced 1 wk apart having the greatest survival advantage (2 doses vs. 1 dose; P = 0.013; 1 vs. 3 or 2 vs. 3 doses were not significantly different at a P value of 0.21). Weight loss peaked at approximately 10% 1 wk after the single dose. The maximum weight loss was similar in the 2 fractionated dose groups, occurring 1 wk after completion of the full 22.2-MBq treatment. Thus, fractionation did not reduce toxicity based on weight loss but did provide more robust and complete responses than a single treatment.
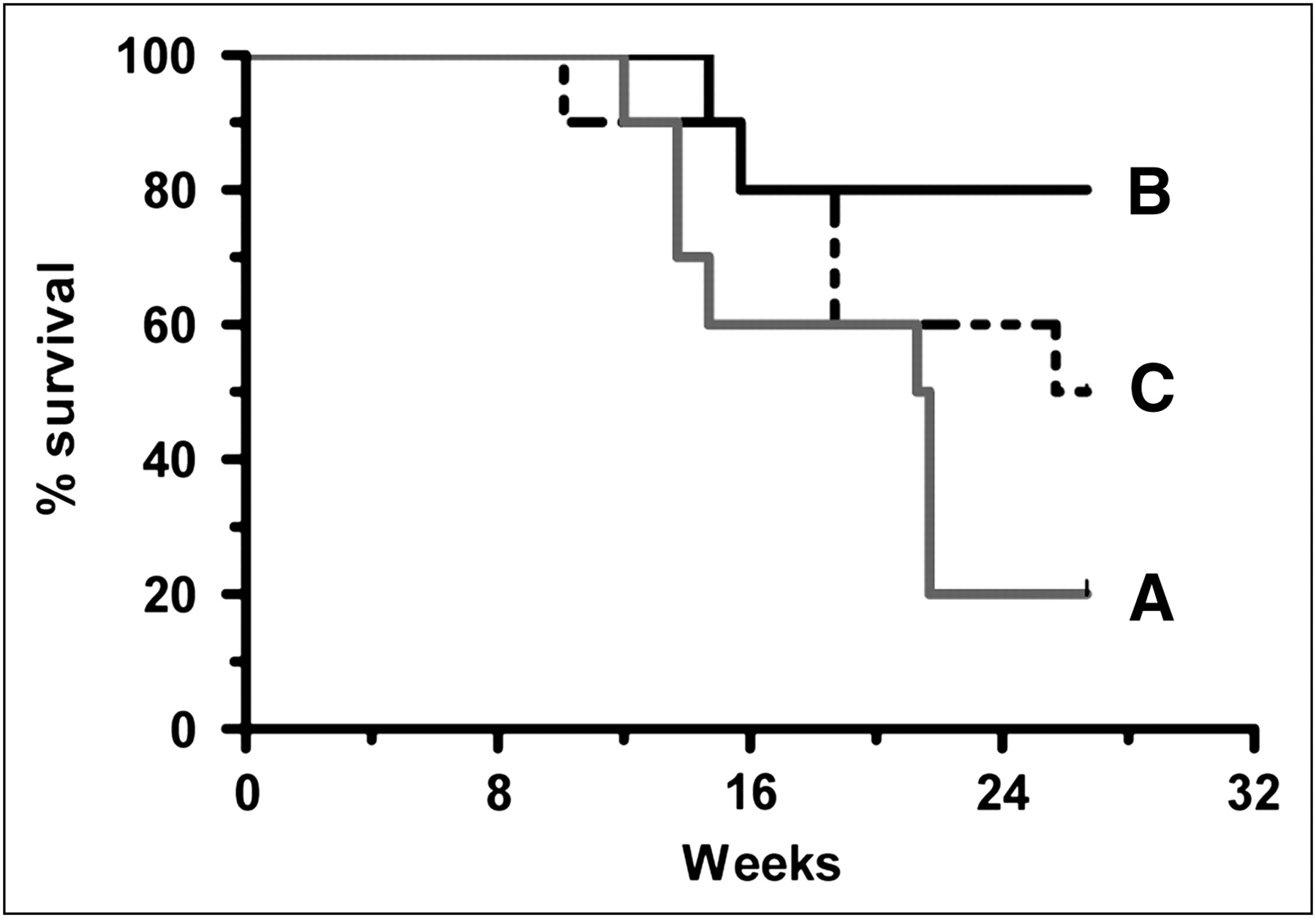
Single dose vs. fractionated PT-RAIT. Capan-1 pancreatic tumor xenografts in nude mice (10/group) were treated with PT-RAIT as single dose of 22.2 MBq (A) on week 0 or fractionated into following 2 or 3 equal fractions: 2 × 11.1 MBq (B) given on weeks 0 and 1 or 3 × 7.4 MBq (C) given on weeks 0, 1, and 2. Study was monitored for 26.7 wk. Survival curve is based on time when tumors reached 3.0 cm3. At end of study, there were tumor-free mice in each group (A, n = 1; B, n = 7; and C, n = 3). Median survival was 21.5 wk (A) and 26.2 wk (C). For group B, median survival was not reached at end of study. Single 22.2-MBq dose vs. 2 doses of 11.1 MBq, P = 0.0125.
DISCUSSION
Pancreatic cancer is an exceptionally difficult disease to treat, but preclinical studies indicated 90Y-labeled PAM4 IgG might be an effective therapeutic, and trials are now underway to test this therapy in patients (11). Because our group has been involved in the development of pretargeting methods for various cancers and found them to be therapeutically superior to directly targeted antibody (15,16), we developed a humanized recombinant bsMAb based on PAM4 for pretargeting pancreatic cancer. Initial studies showed that human pancreatic cancer xenografts could be visualized easily within a few hours of the TF10-pretargeted 111In-IMP-288 injection, with tumor-to-blood ratios exceeding 2,000:1 and biodistribution studies suggesting that this pretargeting procedure could be at least as effective as, but with less hematologic toxicity than, directly radiolabeled PAM4 IgG (12). By reducing hematologic toxicity, targeting in advance also has a better opportunity for being combined with chemotherapy. Gemcitabine is known for its ability to radiosensitize tumors, and it is approved for use for pancreatic cancer, making it a logical choice to be included in a multimodality treatment of pancreatic cancer with PT-RAIT.
The primary goal of these studies was to assess how to best administer a PT-RAIT dosing regimen to optimize response, with gemcitabine as part of this regimen. A single dose of PT-RAIT alone was quite effective in treating established Capan-1 tumors, and because we showed earlier (12) that directly radiolabeled hPAM4 IgG has an exceptionally high tumor uptake with rapid blood clearance in this model, this result was not surprising. However, 90Y-hPAM4 IgG responses were associated with more severe and protracted hematologic toxicity than for PT-RAIT that achieved similar responses at doses less than the MTD. Because the primary toxicity of gemcitabine is hematologic, its combination with PT-RAIT should be better tolerated than a directly radiolabeled IgG. Nevertheless, we found previously that gemcitabine could be combined with 90Y-PAM4 IgG, either to radiosensitize tumors to improve the response of the radioimmunoconjugate or when small amounts of 90Y-PAM4 IgG were added to a full gemcitabine treatment regimen (9,28). These observations led to a phase I clinical trial that determined the MTD of 90Y-hPAM4 IgG to be 740 MBq/m2 (20 mCi/m2) in patients with metastatic pancreatic cancer who had failed at least 1 prior therapy (10). A phase Ib trial to determine the MTD of 3 weekly injections of 90Y-hPAM4 IgG being given with a dose of gemcitabine (200 mg/m2) each week as a frontline treatment for metastatic pancreatic cancer is showing promising responses (11).
Two different gemcitabine–plus–PT-RAIT combination treatment strategies were examined. One explored the possibility of adding PT-RAIT to a full gemcitabine treatment. Animals were given gemcitabine in 3 weekly, 6-mg injections, a dose based on Food and Drug Administration conversion tables to be a human equivalent dose of 24.4 mg/kg or approximately 1,000 mg/m2 (29). A single dose of PT-RAIT at approximately 30% of the estimated MTD was administered 19 h after the first gemcitabine dose. This treatment cycle was repeated every 4 wk for a total of 3 treatment cycles. With gemcitabine alone, the tumors progressed while still undergoing treatment, with no survival advantage over the untreated animals. Animals tolerated 3 PT-RAIT treatment cycles given every 4 wk (total PT-RAIT dose was 27.27 MBq), with minimal loss in body weight and superior tumor control over gemcitabine alone. However, the gemcitabine–plus–PT-RAIT group had a significant survival advantage over the PT-RAIT–alone group. Thus, adding PT-RAIT to a standard gemcitabine treatment could improve this treatment modality, but with PT-RAIT having superior antitumor effects, it would be more logical clinically to consider PT-RAIT as the primary therapeutic.
Graves et al. reported previously in the LS174T colorectal cancer model, using an anti-TAG 72-streptavidin fusion protein/90Y-biotin approach, that gemcitabine could enhance the antitumor response with less toxicity than when combined with a 90Y-labeled fusion protein (21). In our pretargeting model, PT-RAIT in combination with gemcitabine (4 mg/dose) given at −19 and −1 h was too toxic to allow further increases in the PT-RAIT dose. Instead, it appeared that a single 6-mg dose of gemcitabine given 19 h in advance of 90Y-IMP-288 produced the best antitumor response without additional toxicity. This amount of gemcitabine, which is approximately equal to a human dose of 1,000 mg/m2, is considerably higher than that used in patients for radiosensitization purposes (30). Additionally, this combination appeared to allow for a higher objective response rate without extending survival. Thus, it will be important to continue to investigate other agents that might be combined with PT-RAIT plus gemcitabine in hopes of developing induction and consolidation strategies that promote significant and more durable responses with manageable toxicities.
Another important finding was that fractionation of PT-RAIT is superior to a single dose. Although not statistically different, a trend supporting the use of 2 fractions given 1 wk apart, compared with dividing the dose further into 3 smaller fractions, was observed (Fig. 5). However, these results might support the position of Violet et al., who found that hyperfractionation of an 131I-anti-CEA antibody is less effective therapeutically (31).
We did not determine an MTD for PT-RAIT but did show that 33.3 MBq (0.9 mCi) of the TF10-pretargeted 90Y-IMP-288 was well tolerated over a period of 9 mo, with no evidence of morphologic changes in the kidney, suggesting that higher doses might be tolerated. At this same dose level, earlier studies with a different bsmAb and 90Y-labeled hapten-peptide, IMP-241, found 2 of 10 animals with evidence of end-stage renal disease (15). We have since developed a different hapten-peptide, IMP-288, which has similar blood clearance but approximately 30% lower renal uptake than IMP-241, which likely explains the higher tolerance with 90Y-IMP-288. Animals coinjected with 25.9 MBq of TF10-pretargeted 90Y-IMP-288 and 6 mg of gemcitabine had no significant loss in weight or histologic evidence of renal toxicity, indicating that the addition of gemcitabine, which could have sensitized normal tissues to radiation, would be tolerated at highly effective PT-RAIT doses. Thus, these studies support the safety of this combination. Clearly, however, clinical studies will need to be guided by specific renal dosimetry to account for highly selective uptake of the peptide in the cortex of the kidneys, as suggested by others (32), along with careful correlative monitoring of indicators of renal toxicity, such as creatinine and creatinine clearance.
CONCLUSION
In this model, PAM4-based bsmAb targeting in advance with a 90Y-labeled hapten-peptide, with and without the addition of gemcitabine, is a promising therapeutic for pancreatic cancer that can provide responses at least equally as effective, but with less toxicity than, directly radiolabeled 90Y-PAM4 IgG.
Acknowledgments
We thank Ali Mostafa, Jayson Jebsen, Tom Jackson, Christine Johnson, and Dion Yeldell for technical assistance. This work was supported in part by NIH grant CA115755.
Footnotes
-
COPYRIGHT © 2009 by the Society of Nuclear Medicine, Inc.
References
- Received for publication June 26, 2009.
- Accepted for publication August 20, 2009.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Pretargeting: A Path Forward for Radioimmunotherapy
- A New Tri-Fab Bispecific Antibody for Pretargeting Trop-2-Expressing Epithelial Cancers
- Antibody-Radionuclide Conjugates for Cancer Therapy: Historical Considerations and New Trends
- Combination Radioimmunotherapy and Chemoimmunotherapy Involving Different or the Same Targets Improves Therapy of Human Pancreatic Carcinoma Xenograft Models
- Pretargeted 177Lu Radioimmunotherapy of Carcinoembryonic Antigen-Expressing Human Colonic Tumors in Mice
- Epidermal Growth Factor Receptor-Targeted Radioimmunotherapy of Human Head and Neck Cancer Xenografts Using 90Y-Labeled Fully Human Antibody Panitumumab