


Abstract
Our objective was to study 2 radioligands for visualization of σ-receptors with PET. Methods: Two radioligands—σ1-selective 11C-1-(3,4-dimethoxyphenethyl)-4-(3-phenylpropyl)piperazine (11C-SA4503) and nonsubtype-selective 1-(4–2′-18F-fluoroethoxy-3-methoxyphenethyl)-4-(3-(4-fluorophenyl)propyl)piperazine (18F-FE-SA5845)—were evaluated for tumor imaging. Results: Binding studies to rat glioma cells (C6) and human nonsmall cell lung cancer cells (N417) indicated interaction of 18F-FE-SA5845 with 2 sites and interaction of 11C-SA4503 with a single site. Specific binding of 18F-FE-SA5845 was 93% ± 2% and that of 11C-SA4503 was 78% ± 6% of the total cellular uptake of radioactivity. Uptake of the 18F-labeled ligand, but not that of the 11C-labeled ligand, appeared to be related to the growth phase of the cells. Biodistribution experiments in C6 tumor-bearing nude rats (Ham HSD RNU rnu) indicated tumor-to-plasma ratios of 13.3 for 11C-SA4503 and 8.0 for 18F-FE-SA5845 and tumor-to-muscle ratios of 5.0 for 11C-SA4503 and 4.9 for 18F-FE-SA5845, 60 min after injection, which were reduced to values ranging from 1.4 to 2.0 after pretreatment of animals with haloperidol (2 μmol/kg). Tumor uptake of 18F-FE-SA5845 showed a negative correlation with tumor size (P < 0.0001), in contrast to that of 11C-SA4503, suggesting that tissue binding of the former ligand is related to cellular proliferation. A study with 11C-SA4503 in a human volunteer indicated high uptake in liver, kidney, and heart but relatively low background in thorax and lower abdomen. Conclusion: Both 18F-FE-SA5845 and 11C-SA4503 demonstrate specific binding to σ-receptors in vivo and may be useful for the detection of pulmonary and abdominal tumors. However, the 18F-labeled compound may be better for tumor staging than the 11C-labeled drug.
The most widely used radiopharmaceutical in clinical oncology is 18F-FDG. Although the applications of 18F-FDG PET in tumor detection, staging, and therapy evaluation are rapidly expanding, 18F-FDG uptake is not tumor specific. Various forms of inflammatory lesions also take up 18F-FDG and are a major cause of false-positive results. A decreased uptake of 18F-FDG is seen in hyperglycemic patients, which can cause false-negative scan data. Because of the urinary excretion of 18F-FDG, tumors in the lower abdomen (e.g., prostate tumors) are relatively poorly visualized. Thus, there is opportunity for the development of novel tumor imaging agents that may lack some of the disadvantages of 18F-FDG.
σ-Receptors are unique proteins with a high affinity for neuroleptics such as haloperidol (1). They are strongly overexpressed in a large variety of human tumors—for example, glioma, neuroblastoma, melanoma, and breast cancer (2–4). The endogenous ligands for these sites have not yet been identified, although neuroactive steroids such as progesterone have a high affinity for σ-receptors (5,6).
Two subtypes of the σ-receptor have been discerned: σ1 and σ2, based on their opposite enantioselectivity for benzomorphans and different molecular masses (7). The σ2-receptor density of tumors is a biomarker of cellular proliferation (8–10). Activation of intracellular σ2-receptors increases cytosolic Ca2+ and induces apoptosis via a caspase- and p53-independent mechanism (1,11,12). Therefore, σ-agonists may be effective antineoplastic agents and chemosensitizers (12–14)
A radioligand for visualization of σ-receptors with PET could be useful for (a) selective detection of primary tumors and their metastases, (b) noninvasive assessment of tumor proliferative status, and (c) measurement of the σ-receptor occupancy of novel and established antineoplastic drugs. We have evaluated 2 radioligands for these purposes.
SA4503 has a relatively high affinity for σ1-receptors (50% inhibitory concentration [IC50], 17.4 nmol/L) but negligible affinity for the σ2-subtype (IC50, 1,784 nmol/L) and for 36 other neuroreceptors, ion channels, and second messenger systems (15). In contrast, its fluoroethyl analog, FE-SA5845, has a high affinity to both σ1- and σ2-receptors (IC50 values, 3.1 and 6.8 nmol/L, respectively) (16). The lipophilicity of both drugs is about optimal for entry of the central nervous system (log P values, +2.5 and +2.7, respectively) (16,17). In fact, 11C-SA4503 has been successfully used for PET studies of σ-receptors in cat, monkey, and human brain (18–21). Monkey studies with 18F-FE-SA5845 have also been performed (22).
Here, we report the results of in vitro and in vivo experiments in which 18F-FE-SA5845 and 11C-SA4503 were evaluated for tumor imaging. 18F-FE-SA5845 offers the advantages of a longer half-life of the radionuclide (109.8 min for 18F vs. 20.3 min for 11C) and a high affinity to σ2-receptors, which are the major subtype in tumor cells (3). In vivo assays of σ2-receptor density may allow tumor staging (8–10). Since not only σ2-receptors but also σ1-receptors are present in most tumors (3) and the relationship between σ1-receptor density and proliferative status is unknown, 11C-SA4503 could also be a successful imaging agent.
MATERIALS AND METHODS
Culture Media and Drugs
(+)-Pentazocine and (S)-(−)-raclopride were purchased from Research Biochemicals International. Haloperidol was a product of Sigma. Unlabeled SA4503 was a gift from Santen Pharmaceutical Co. (S)-Ketamine (Ketanest) was purchased from Parke-Davis; Matrigel Basement Membrane Matrix was from BD Biosciences; and Dulbecco’s Minimum Essential Medium (DMEM), fetal calf serum (FCS), and trypsin were from Invitrogen.
Radioligands
18F-FE-SA5845 and 11C-SA4503 were prepared by reaction of 18F-fluoroethyl tosylate and 11C-methyl iodide, respectively, with the appropriate 4-O-demethyl compound, as described previously (16,17). The decay-corrected radiochemical yields of 18F-FE-SA5845 and 11C-SA4503 were 4%–7% and 9%–11%, respectively. Synthesis times were 80 and 45 min, respectively. The specific radioactivities of 18F-FE-SA5845 and 11C-SA4503 were >74 and >10 TBq/mmol at the time of injection. Radiochemical purities were >95%.
Cell Culture
C6 rat glioma cells obtained from the American Type Culture Collection were cultured in monolayers in DMEM supplemented with 5% FCS in a humidified atmosphere of 5% CO2/95% air at 37°C. Before each experiment, C6 cells were seeded in 12-well plates (Costar) with 1.5 mL of DMEM supplemented with 5% FCS per well. After 24 h at 37°C, monolayers had grown.
N417 human nonsmall cell lung cancer cells were grown in suspension in 25-cm2 plastic flasks (Corning Co.) and cultured in DMEM with 10% FCS. After harvesting by centrifugation and resuspending in fresh medium, 1-mL samples of cell suspension in 15-mL test tubes were used for binding studies. All incubations were performed at 37°C in a shaking waterbath (Heidolph).
Binding Studies: C6 Cells
Binding studies to C6 rat glioma cells were performed in monolayers grown at the bottom of 12-well plates. In competition studies, various concentrations of an unlabeled competitor (haloperidol, (+)-pentazocine, raclopride, or ketamine; range, 10−12 to 10−3 mol/L) were dispensed to the culture medium in the wells. At time zero, 2 MBq of radioligand in 50 μL of saline were added to each well. At the end of incubation (60 min), the medium was quickly removed and the monolayer was washed 3 times with phosphate-buffered saline (PBS). Cells were then treated with 0.25 mL of trypsin. When the cells had detached from the bottom of the well, 1 mL of DMEM was added to stop the proteolytic action. Cell clumps were removed by repeated (at least 10-fold) pipetting of the trypsin/DMEM mixture. Radioactivity in the cell suspension (1.2 mL) was assessed using a γ-counter (Compugamma 1282 CS; LKB-Wallac). A 50-μL sample of the suspension was mixed with 50 μL of trypan blue (0.4% solution in PBS) and used for cell counting. Cell numbers were manually determined, using a phase- contrast microscope (Zeiss), a Bürker bright-line chamber (depth, 0.1 mm; 0.0025-mm2 squares), and a hand-tally counter. Nonspecific binding of the radioligand was defined as residual binding to the cells in the presence of an unlabeled competitor (5 × 10−4 mol/L (+)-pentazocine, haloperidol, or unlabeled SA4503).
To study the influence of cell density on ligand binding, different numbers of C6 cells were seeded in various wells. After 1–4 d of growth in 12-well plates, cell densities ranged from 55,000 to 2,600,000 cells per well. Two megabecquerels of 18F-FE-SA5845 or 11C-SA4503 in 50 μL of saline were then added. At the end of incubation (60 min), medium was quickly removed. Cells were washed, trypsinized, and counted as described.
Binding Studies: N417 Cells
Binding studies to N417 cells were performed in test tubes containing a suspension of ∼106 cells/mL. In competition studies, various concentrations of an unlabeled competitor (haloperidol, (+)-pentazocine, ketamine, or SA4503; range, 10−12 to 10−3 mol/L) were dispensed to the culture medium in the tubes. At time zero, 2 MBq of radioligand in 50 μL saline were added to each tube. At the end of incubation (60 min in most studies), the cells were harvested by centrifugation (10 s in a Hettich Micro 20 centrifuge at maximum speed, 14,000g). The cells were washed 3 times by resuspending them in PBS followed by repeated centrifugation. Removal of cell clumps, radioactivity, and cell counting were then performed as described.
Biodistribution Experiments
The experiments were performed by licensed investigators in accordance with the Law on Animal Experiments of The Netherlands (“Wet op de Dierproeven” 1977 + Amendments 1996, “Dierproevenbesluit” 1985 + Amendments 1996). Female nude rats (HSD Ham RNU rnu; 150–200 g body weight) were obtained from Harlan. After 1 wk of acclimatization, C6 glioma cells (2.5 × 106, in a 1:1 v/v mixture of Matrigel and DMEM containing 5% FCS) were subcutaneously injected into both flanks. Matrigel was included to avoid migration of tumor cells to other sites than the place of injection. Biodistribution studies were done after 9–10 d, when solid tumor nodules of 0.7- to 1.7-cm diameter had grown.
The rats were anesthetized using sodium pentobarbital (60 mg/kg intraperitoneal). Animals were kept under anesthesia for the rest of the experiment. The radiopharmaceutical (10 MBq 18F-FE-SA5845 or 20 MBq 11C-SA4503) was intravenously administered through a cannula inserted into a lateral tail vein. In blocking experiments, haloperidol (2 μmol/kg in saline containing <10% ethanol) was administered 2 min before injection of the radioligand, through the same cannula, whereas control rats received saline only. The animals were sacrificed 60 min after radiotracer injection by extirpation of the heart (under general pentobarbital anesthesia). Blood was collected and normal tissues (brain, fat, bone, heart, intestines, kidney, liver, lung, skeletal muscle, pancreas, spleen, submandibular gland, and urinary bladder) were excised. Urine was collected and plasma plus a red cell fraction were obtained from blood centrifugation (5 min at 1,000g). The complete tumor was excised and carefully separated from muscle and skin. All samples were weighed, and the radioactivity was measured using the γ-counter, applying a decay correction. The results are expressed as dimensionless standardized uptake values (SUVs): cpm measured per gram of tissue/cpm injected per gram of body weight. Tumor-to-plasma and tumor-to-muscle concentration ratios of radioactivity were also calculated.
Statistical Analysis
Statistical analysis was performed using the software package Statistix (NH Analytic Software). Differences between the various groups (control and haloperidol pretreated) were tested for statistical significance using the 2-sided Student t test for independent samples. P < 0.05 was considered significant.
Human Study
A pilot study with 11C-SA4503 was performed on a volunteer. The study was approved by the Ethical Committee of the Tokyo Metropolitan Institute of Gerontology. A female subject (76 y old, 45.5 kg, 157.3 cm) participated, after written informed consent had been obtained. She had no clinical abnormality. The PET camera used was a SET-2400W type having an axial field of view of 20 cm (Shimadzu Co.) (23). Thirty-one minutes after intravenous injection of 11C-SA4503 (560 MBq/9.7 nmol) into the subject, a whole-body scan, spanning from head to legs, was performed in 2-dimensional mode (5 overlapping bed positions, 7-min emission time per position, with attenuation correction). Data were reconstructed using an ordered-subsets expectation maximization method (subsets, 16; iterations, 2). Pixel counts of the whole-body image were calibrated to activity concentration (Bq/mL).
RESULTS
Binding Studies in Tumor Cells
Initial in vitro studies showed rapid association of 18F-FE-SA5845 and 11C-SA4503 with tumor cells, equilibrium being reached after 20 min of incubation and remaining for >120 min at 37°C (values not shown). Therefore, an incubation time of 60 min was used in all subsequent experiments (24).
Competition studies using the σ1-subtype-selective agonist (+)-pentazocine indicated binding of 18F-FE-SA5845 to 2 sites, both in the rodent and human cells. Biphasic competition was also observed for the nonsubtype-selective σ-ligand haloperidol (Table 1). The N-methyl-d-aspartate (NMDA) antagonist (S)-ketamine (which is often used for rodent anesthesia) and the dopamine D2-receptor antagonist (S)-(−)-raclopride inhibited the binding of 18F-FE-SA5845 only at high concentrations (IC50 values in the 10−5 to 10−4 mol/L range; Table 1).
Drug Interactions with Binding of σ-Receptor Ligands to Intact Tumor Cells
In contrast to 18F-FE-SA5845, (+)-pentazocine competition indicated binding of 11C-SA4503 to a single site, both in C6 and N417 cells. Monophasic inhibition of 11C-SA4503 binding was also observed in experiments using haloperidol and unlabeled SA4503 (Table 1). The NMDA antagonist (S)-ketamine inhibited the binding of 11C-SA4503 only at high concentrations (IC50 values in the 10−5 to 10−4 mol/L range; Table 1).
When 18F-FE-SA5845 was added to cells that either were actively growing or had become confluent, the equilibrium binding of the radioligand was found to be dependent on the growth phase. Rapidly growing cells showed a high uptake of 18F, but uptake in confluent cells was >5-fold reduced (Fig. 1). Different results were obtained with 11C-SA4503. Uptake of this tracer in confluent cells was only slightly (∼2-fold) reduced compared with that of actively growing cells (Fig. 1).
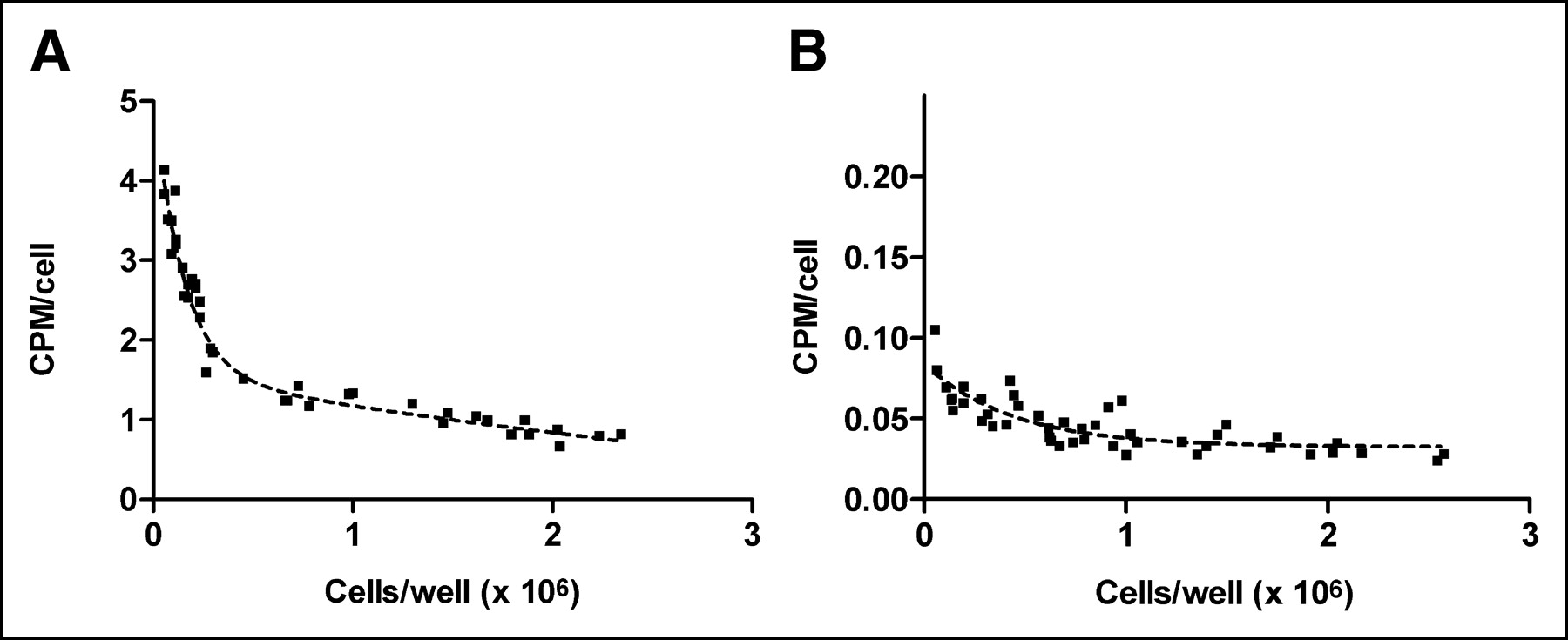
Relationship between growth stage (number of C6 cells per well) and cellular uptake of 18F-FE-SA5845 (A) and 11C-SA4503 (B). Ordinate indicates total bound radioactivity within cells. Dashed line is an exponential curve fit.
Biodistribution of 18F-FE-SA5845
After injection of 18F-FE-SA5845 in tumor-bearing rats, the highest uptake of radioactivity was observed in liver, followed by kidney, pancreas, and lung. Submandibular gland, intestines, spleen, urinary bladder, brain, and C6 tumor showed moderate tracer uptake, whereas low levels of radioactivity were found in heart, adipose tissue, bone, muscle, plasma, and red blood cells (Table 2).
Biodistribution of 18F-FE-SA5845 in Tumor-Bearing Rats 60 Minutes After Injection
Pretreatment of animals with haloperidol (2 μmol/kg) caused a strong reduction of tissue uptake in brain, intestines, kidney, liver, and C6 tumors, whereas radioactivity in plasma and blood cells was increased. Radioactivity in spleen and submandibular gland seemed also to be decreased by haloperidol, but statistical significance (P < 0.05) was not reached in these organs because of a relatively high interindividual variation. Control animals showed tumor-to-plasma and tumor-to-muscle ratios of 8.0 and 4.9, respectively, at 60 min after injection. These were reduced to low values (1.4–1.5) in pretreated rats (Table 2).
Biodistribution of 11C-SA4503
The σ1-subtype-selective radioligand 11C-SA4503 showed a biodistribution similar to that of nonsubtype-selective 18F-FE-SA5845. The highest uptake of radioactivity was observed in liver, followed by pancreas, small intestine, and kidney. Moderate tracer uptake was found in spleen, submandibular gland, large intestine, lung, urinary bladder, and the C6 tumor. Low uptake of 11C was observed in heart, bone, adipose tissue, muscle, plasma, and red blood cells (Table 3). Uptake of 11C in C6 tumors after injection of 11C-SA4503 tended to be lower than the uptake of 18F after administration of 18F-FE-SA5845 (SUV values in Table 2 compared with those in Table 3).
Biodistribution of 11C-SA4503 in Tumor-Bearing Rats 60 Minutes After Injection
Pretreatment of animals with haloperidol (2 μmol/kg) suppressed the uptake of 11C-SA4503 in target tissues. A reduction of tissue radioactivity was observed in brain, intestines, kidney, liver, and C6 tumors, whereas radioactivity in plasma, red blood cells, and myocardium was increased. However, radioactivity levels in spleen and submandibular gland were not reduced after haloperidol pretreatment.
Although tumor SUVs of 11C-SA4503 appeared to be lower than those of 18F-FE-SA5845, similar target-to-nontarget ratios were measured for the 2 tracers. Control animals showed tumor-to-plasma ratios of 13.3 and tumor-to-muscle ratios of 5.0, 60 min after injection of 11C-SA4503. These were reduced to low values (1.9–2.0) in pretreated rats (Table 3).
Size-Dependent Tracer Uptake
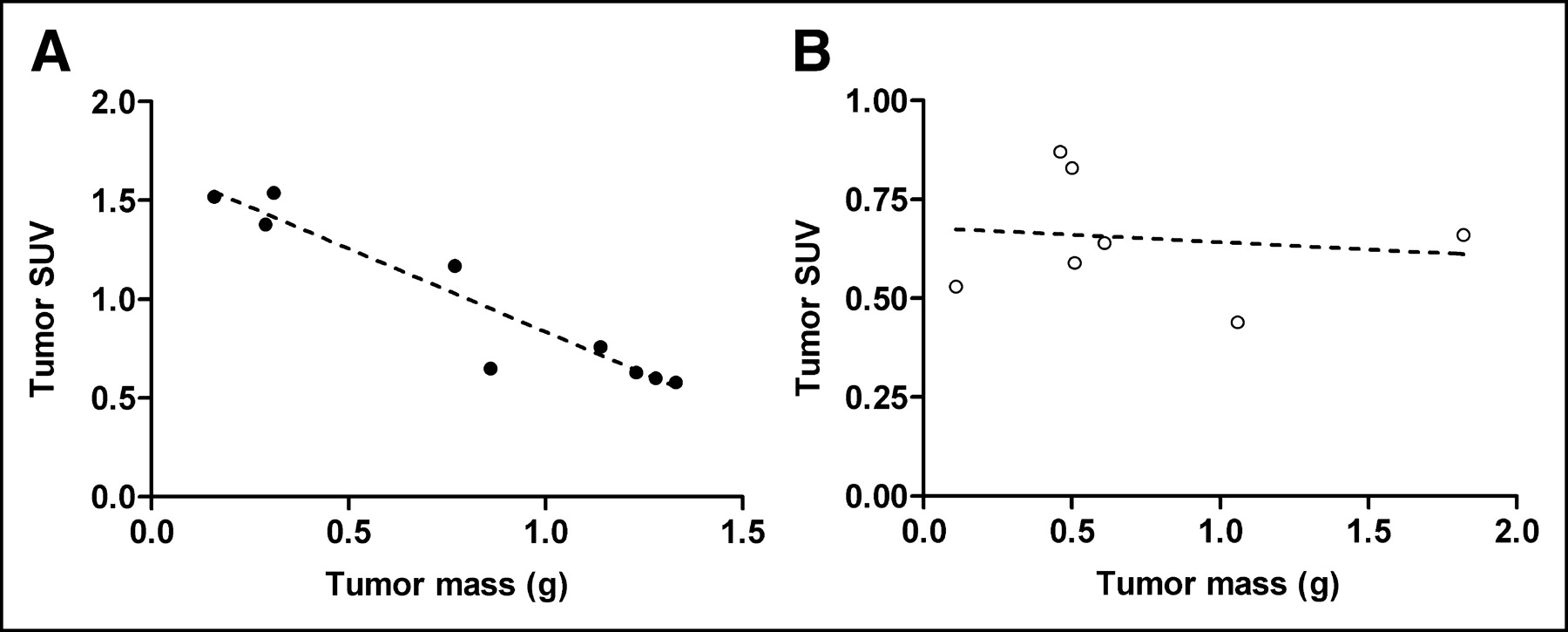
When tracer binding in individual tumors was plotted against tumor mass, the uptake of 18F-FE-SA5845 was found to be negatively correlated with tumor size (slope, −0.85 ± 0.10; r = 0.95, P < 0.0001; Fig. 2). Different results were observed for the in vivo uptake of 11C-SA4503. The uptake of this tracer did not show a significant relationship with tumor size (slope, −0.04 ± 0.12; r = 0.14, P = not significant; Fig. 2).

Relationship between tumor mass (in vivo C6 tumors grown in nude rats) and tumor uptake of 18F-FE-SA5845 (A) and 11C-SA4503 (B). Solid and open symbols are data points of individual tumors; dashed line is result of a linear regression analysis.
Human Study
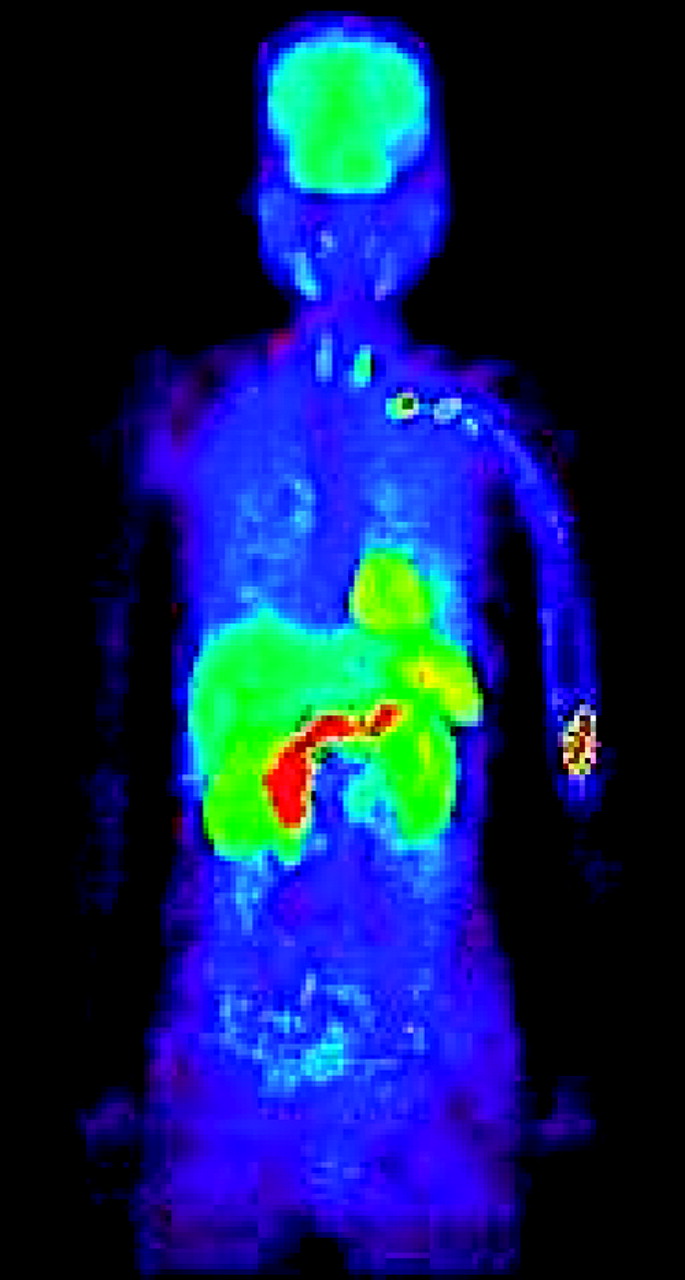
The human pilot study with 11C-SA4503 in a single volunteer (Fig. 3) indicated high uptake of the tracer in brain, heart, liver and kidneys, but a relatively low background of radioactivity in the thorax, extremities, and lower abdomen.

Whole-body scan of human volunteer, made 31–66 min after injection of 11C-SA4503.
DISCUSSION
This study confirms that the nonsubtype-selective ligand 18F-FE-SA5845 binds to 2 sites and the σ1-subtype-selective ligand 11C-SA4503 binds to a single site in intact tumor cells (Table 1). Probably, the 2 binding sites of 18F-FE-SA5845 are σ1- and σ2-receptors. In vitro competition experiments with the established σ1-receptor agonists (+)-pentazocine and SA4503, or the σ-receptor antagonist haloperidol, indicated that the specific binding of 18F-FE-SA5845 was 93% ± 2% and of 11C-SA4503 was 78% ± 6% of the total cellular uptake of radioactivity, both in rodent and human cells (Table 1). Thus, the in vitro experiments suggest better signal-to-noise ratios for 18F-FE-SA5845 than for 11C-SA4503. 18F-FE-SA5845 and 11C-SA4503 do not bind to dopamine D2-receptors or NMDA receptors on intact tumor cells, since the D2/D3-receptor antagonist raclopride and the NMDA antagonist (S)-ketamine inhibited the binding only at high concentrations (10−5 to 10−3 mol/L range).
Experiments in which the effect of the growth phase on the cellular uptake of the σ-ligands was examined indicated a marked decline in the uptake of 18F-FE-SA5845, but a less marked decline in 11C-SA4503 uptake, in confluent cells (Fig. 1). 11C-SA4503 is selective for the σ1-subtype, whereas 18F-FE-SA5845 binds equally well to σ1- and σ2-receptors. Figure 1 is therefore in accordance with reports in the literature which suggest that the density of σ2-receptors is related to the proliferative status of tumor cells (8–10). Using another cell line (66 cells rather than C6 cells), Mach et al. have shown that the σ2-receptor density is >10-fold reduced in quiescent cells (8).
In biodistribution experiments (Tables 2 and 3), both 18F-FE-SA5845 and 11C-SA4503 showed specific (i.e., haloperidol sensitive) binding in C6 tumors and also in other target organs, such as brain, intestines, kidney, and liver. The high and haloperidol-sensitive uptake of 18F-FE-SA5845 and 11C-SA4503 in liver, kidney, and intestines is consistent with the reported high levels of σ1-receptor messenger RNA (25,26) and high densities of σ1- and σ2-receptors in these excretory organs (27–30). Although both σ1- and σ2-receptors have been reported to be expressed on splenocytes (31,32), only 18F-FE-SA5845 showed some specific binding in rat spleen (Table 2). Apparently, the σ1-subtype-specific radioligand ‘11C-SA4503 shows high nonspecific and little specific binding in rat spleen in vivo.
The biodistribution experiments indicated tumor-to-plasma ratios of 13.3 for 11C-SA4503 and 8.0 for 18F-FE-SA5845 and tumor-to-muscle ratios of 5.0 for 11C-SA4503 and 4.9 for 18F-FE-SA5845, 60 min after injection, which were reduced to values ranging from 1.4 to 2.0 after pretreatment of animals with haloperidol (Tables 2 and 3). These tumor-to-plasma ratios are higher than or equal to those of the radiopharmaceuticals 18F-FDG and 3′-deoxy-3′-18F-fluorothymidine (18F-FLT), which were studied previously in the same in vivo tumor model (6.1 and 3.8, respectively) (33). The observed tumor-to-muscle ratios of the σ-ligands are also higher than those of 18F-FLT (3.8 ± 1.4) but lower than those of 18F-FDG (13.2 ± 3.0) (33).
Tumor uptake of the 18F-labeled ligand tended to be higher than that of the 11C-labeled agonist (SUV, 0.92 ± 0.39 vs. 0.65 ± 0.16, respectively; Tables 2 and 3). This trend is probably due to the fact that 18F-FE-SA5845 binds to both σ1- and σ2-receptors (16), whereas 11C-SA4503 interacts with σ1-receptors only (15).
Another difference between the 2 radiotracers was observed when the SUV in individual tumors was plotted against tumor mass (Fig. 2). The SUV of 18F-FE-SA5845 was negatively correlated with tumor size (P < 0.0001), in contrast to tissue uptake of 11C-SA4503. All tumors in these experiments showed only small areas of necrosis, amounting to <10% of the total tumor volume. Thus, the decline in the SUV of 18F-FE-SA5845 with increasing tumor size was not related to necrosis. Perfusion changes with increasing tumor size should have affected the uptake of both radioligands equally, since they have similar lipophilicities and the range of tumor sizes was similar in both cases (Fig. 2). Although no data on growth rate are available, the most likely explanation for the different behavior of the 2 ligands is that only the binding of 18F-FE-SA5845 is related to cellular proliferation.
The human pilot study with 11C-SA4503 indicated a relatively low background level of radioactivity in the extremities, thorax, and lower abdomen. Thus, 11C-SA4503 may be useful for the detection of sarcomas, lung tumors and abdominal tumors, using PET.
CONCLUSION
18F-FE-SA5845 and 11C-SA4503 demonstrate specific binding to σ-receptors, both in vitro and in vivo. The observed target-to-nontarget ratios suggest that these radioligands may be suitable for tumor detection and for assessment of the σ-receptor occupancy of novel therapeutic drugs. The uptake of 18F-FE-SA5845 was reduced in large tumors and quiescent cells, in contrast to the binding of 11C-SA4503. Hence, 18F-FE-SA5845 may be preferred over 11C-SA4503 for tumor staging.
Acknowledgments
This work was partially supported by a Grant-in-Aid for Scientific Research (B) no. 13557077 from the Ministry of Education, Culture, Sports, Science and Technology, Japan.
Footnotes
Received Apr. 22, 2004; revision accepted July 14, 2004.
For correspondence or reprints contact: Philip H. Elsinga, PhD, PET Center, Groningen University Hospital, P.O. Box 30001, 9700RB Groningen, The Netherlands.
E-mail: p.h.elsinga{at}pet.azg.nl
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Small-Animal PET with a {sigma}-Ligand, 11C-SA4503, Detects Spontaneous Pituitary Tumors in Aged Rats
- Assessment of Cellular Proliferation in Tumors by PET Using 18F-ISO-1
- Evaluation of 4'-[Methyl-11C]Thiothymidine in a Rodent Tumor and Inflammation Model
- Rapid Reduction of {sigma}1-Receptor Binding and 18F-FDG Uptake in Rat Gliomas After In Vivo Treatment with Doxorubicin
- Cancer Cell Cycle Modulated by a Functional Coupling between Sigma-1 Receptors and Cl- Channels
- Early Response of {sigma}-Receptor Ligands and Metabolic PET Tracers to 3 Forms of Chemotherapy: An In Vitro Study in Glioma Cells
- Synthesis and In Vivo Evaluation of 2 High-Affinity 76Br-Labeled {sigma}2-Receptor Ligands
- Comparison of Sigma-Ligands and Metabolic PET Tracers for Differentiating Tumor from Inflammation