


Abstract
Detection of epidermal growth factor receptor (EGFR) overexpression in many carcinomas provides important diagnostic information, which can influence patient management. The use of PET may enable such detection in vivo by a noninvasive procedure with high sensitivity. The aim of this study was to develop a method for preparation of a positron-emitting tracer based on a natural ligand to EGFR, the recombinant human epidermal growth factor (hEGF), and to perform a preclinical evaluation of the tracer. Methods: DOTA-hEGF (DOTA is 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid) was prepared by coupling of a N-sulfosuccinimide ester of DOTA to hEGF. The conjugate was labeled with a generator-produced positron-emitting nuclide, 68Ga (half-life = 68 min), using microwave heating. Binding specificity, affinity, internalization, and retention of 68Ga-DOTA-hEGF was studied in 2 EGFR-expressing cell lines, U343 glioma cells and A431 cervical carcinoma cells. Biodistribution and microPET visualization studies were performed in BALB/c nu/nu mice bearing A431 carcinoma xenografts. Results: A 1-min-long microwave-assisted labeling provided radioactivity incorporation of 77% ± 4%. Both cell lines demonstrated receptor-specific uptake of the conjugate, rapid internalization of the tracer, and good retention of radioactivity. Binding to both cell lines occurred with high affinity, approximately 2 nmol/L. The biodistribution study demonstrated accumulation of radioactivity in xenografts and in EGFR-expressing organs. The microPET imaging study enabled visualization of tumors and demonstrated quick—within 5 min—localization of radioactivity in tumors. Conclusion: 68Ga-DOTA-hEGF has potential for imaging EGFR overexpression in tumors.
The epidermal growth factor receptor (EGFR, HER1, ErbB-1) is a transmembrane protein of the tyrosine kinase receptor family. Activation of EGFR causes signaling that may lead to cell division, increasing motility and suppression of apoptosis (1). In several carcinomas, amplification or translocation of EGFR genes causes an increased transcription and a subsequent high level of EGFR expression (2,3). Such overexpression is documented in, for example, carcinomas of breast and lung (4–6). A high level of EGFR expression provides malignant cells with an advantage in survival by increasing cell proliferation and metastatic spread and decreased apoptosis. For the moment, several approaches to suppress tumor growth by inactivation of EGFR signaling are in clinical use or under evaluation. These approaches are based on either blocking ligand binding to the EGFR extracellular domain using anti-EGFR antibodies or preventing intracellular signaling with selective tyrosine kinase inhibitors (7).
EGFR expression in tumors has documented prognostic and predictive value. It has been shown that such overexpression is associated with poor survival and recurrences in non–small cell lung cancer (NSCLC) and breast cancer (5,8,9). Apparently, detection of EGFR in clinical practice might influence patient management, including questions of the relevance of the use of EGFR-targeted drugs.
Detection of EGFR is possible in surgical samples or samples of fine-needle biopsies using immunohistochemical or fluorescent in situ hybridization techniques. However, we believe that nuclear medicine visualization may provide advantages because of evaluation of the whole volume of both the primary tumor and the metastases, avoiding false-negative results associated with sampling errors and heterogeneity of EGFR expression.
The 111In-labeled anti-EGFR antibody 425 was successfully used for detection of malignant gliomas (10). 99mTc-Labeled anti-EGFR humanized antibodies hR3 and C225 are under clinical evaluation (11,12). It should be noted, however, that the use of a bulky antibody might complicate radioconjugate diffusion through healthy tissues and into tumors. An alternative to anti-EGFR antibodies might be the use of a natural ligand, human epidermal growth factor (hEGF), as a targeting vector for delivery of radionuclides to tumor cells (13). The molecular weight of hEGF, 6.2 kDa, might enable fast tumor penetration and fast blood clearance, providing good contrast of the image. Earlier, 131I-labeled hEGF has been used successfully for visualization of lung cancer (14). However, poor cellular retention of radiohalogens might lead to decreased tumor accumulation and suboptimal imaging contrast, and the use of radiometals might be a better choice for labeling of hEGF (15). Thus, single-photon radiometal labels such as 111In (half-life [t1/2] = 2.8 d) and 99mTc (t1/2 = 6 h) have previously been attached to hEGF (15–20). It might be advantageous, however, to use a positron-emitting label for hEGF, as PET is a superior detection technique in sensitivity, resolution, and quantification compared with SPECT (21,22).
An attractive positron-emitting label for hEGF might be 68Ga (t1/2 = 68 min). The half-life of this nuclide is compatible with the quick blood clearance of hEGF. 68Ga possesses high positron emission (89%) and is readily available from a commercial 68Ge/68Ga generator (68Ge, t1/2 = 270.8 d) (23,24). The use of derivatives of macrocyclic chelators provided stable gallium labeling of somatostatin analogs and oligonucleotides (24–26).
Because of the short half-life of 68Ga, the labeling time is important. To accelerate the chelating, microwave heating was used. This technique was shown previously to be efficient for 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA)-mediated 68Ga labeling of oligonucleotides and peptides (24,26,27).
This study was performed with the intention of creating a positron-emitting 68Ga-labeled conjugate of hEGF and evaluating the feasibility for the visualization of EGFR- expressing tumors.
MATERIALS AND METHODS
Materials
hEGF was purchased from Chemicon. Sodium acetate (99.995%), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), and double-distilled hydrochloric acid (Riedel de Haën) were obtained from Sigma-Aldrich Sweden. Sodium dihydrogen phosphate, disodium hydrogen phosphate, and trifluoroacetic acid (TFA) were obtained from Merck. The N-hydroxysuccinimide ester of DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) was purchased from Macrocyclics. The purchased chemicals were used without further purification. Deionized water (18.2 MΩ), produced with a Purelab Maxima Elga system was used in all reactions. 68Ga was obtained from a commercial 68Ge/68Ga generator with 1.85 GBq 68Ge-loaded activity and a 2- to 3-y shelf-life (Cyclotron C; Obninsk).
Preparation of 68Ga-DOTA-hEGF
hEGF (32–70 nmol, 80–180 μL) in 0.08 mol/L borate buffer, pH 9.4, was added to dry N-hydroxysulfosuccinimide ester of DOTA (10- to 20-fold excess) under stirring, and the pH was further adjusted to 9.0 by adding borate buffer (240–340 μL). The mixture was left at room temperature for 3–4 h or overnight. The conjugate was purified on a Bio-Select RP C18 SPE column (Vydac). The reaction mixtures were passed slowly though the extraction disk, which was then washed with 2 mL of 0.1% TFA. The product was eluted in 1 mL of 70% acetonitrile with 0.1% TFA. The solvent was evaporated using a vacuum centrifuge (Labconco CentriVap Console), operated at 50°C, and the dry purified product was stored at a temperature below zero. Alternatively, the conjugate was purified and concentrated by ultracentrifugation through a filter with a molecular weight cutoff of 3,000 Da (Centricon-3; Amicon) against 3 changes of 12 mmol/L HEPES buffer. The filtration fractions were analyzed by reversed-phase high-performance liquid chromatography (RP-HPLC).
The labeling of the conjugate was performed using either nonconcentrated 68Ga-eluate or eluate preconcentrated, as described previously (24). In some cases, the eluates from 2 generators were preconcentrated to increase the amount of 68Ga used in the labeling reaction. The amount of DOTA-hEGF used in the labeling reaction was 6–10 and 2–5 nmol, respectively, when using nonconcentrated and preconcentrated 68Ga-eluate, respectively. Sodium acetate buffer, pH 5.0–5.5, was used for labeling with nonconcentrated 68Ga, and HEPES buffer, pH 4.6–4.8, was used for preconcentrated eluate. The labeling was performed at 90°C ± 5°C under microwave heating for 1 min. The product was purified on a Bio-Select RP C18 SPE column as described. The solvent was then changed to phosphate-buffered saline (PBS) on NAP-5 columns (Sephadex G-25; Amersham Pharmacia Biotech AB). The radiochemical purity of 68Ga-DOTA-hEGF was assessed by UV-radio-HPLC, and the concentration of the conjugate and the tracer was determined from UV-HPLC standard plots.
To verify that the binding of 68Ga to hEGF was DOTA mediated, a blank experiment was performed. The procedures were the same as described, but nonconjugated hEGF was used.
69,71Ga of natural isotope composition was complexed to DOTA-hEGF using the same protocol. 69,71Ga-DOTA-hEGF, characterized with liquid chromatography electrospray ionization mass spectrometry (LC-ESI-MS), was used for the identification of the radio-HPLC chromatogram signals.
HPLC Analysis
Analytic LC was performed using an HPLC system from Beckman consisting of a 126 pump, a 166 UV detector, and a radiation detector coupled in series. Data acquisition and handling were performed using the Beckman System Gold Nouveau Chromatography Software Package. The column used was a Vydac RP 300-Å HPLC column (Vydac) with the dimensions 150 × 4.6 mm, 5-μm particle size. The applied gradient elution had the following parameters: A = 10 mmol/L TFA; B = 70% acetonitrile (MeCN), 30% H2O, 10 mmol/L TFA with UV detection at 220 nm; the flow rate was 1.2 mL/min; 0–2 min isocratic 20% B, 20%–90% B linear gradient 8 min, 90%–20% B linear gradient 2 min. The quantity of 68Ga-DOTA-hEGF and radioimpurities retained on the column were controlled.
Microwave Heating
Microwave heating was performed in a SmithCreator monomodal microwave cavity producing continuous irradiation at 2,450 MHz (Biotage AB).
LC-ESI-MS Analysis
LC-ESI-MS was performed using the Waters Micromass Quattro Premier Mass Spectrometer (Micromass) and an HPLC system from Alliance (Waters 269) with a Photodiode Array UV detector. The column used was an Atlantis, dC 18, RP-HPLC column with the dimensions 100 × 2.1 mm, 3-μm particle size. Isocratic elution was applied with the following parameters: A = 10 mmol/L formic acid; B = 100% MeCN, with UV detection at 210–400 nm; the flow rate was 0.3 mL/min. LC-ESI-MS was performed with positive-mode scanning and selected ion recording detecting [M+6H]6+, [M+7H]7+, and [M+8H]8+ ions of hEGF, (DOTA)n-hEGF, and (Ga-DOTA)n-hEGF, where n = 1;2;3. Reconstitution of the data gave molecular weight of 6,244.67 ± 1.15; 6,629.95 ± 0.05; 7,016 ± 0.08; 7,402 ± 0.1; 6,699.95 ± 0.05; 7,157.55 ± 2.47; 7,612.31 ± 0.05, respectively, for hEGF, (DOTA)n-hEGF and (Ga-DOTA)n-hEGF.
Cell Cultures
The human squamous carcinoma cell line A431 (CLR 1555; American Type Culture Collection) and the malignant glioma cell line U343MGaCl2:6 (28) (denoted U343) were used in all cell experiments. This A431 cell line is reported to express approximately 2 × 106 EGFRs per cell, and the U343 cell line expresses approximately 5.5 × 105 EGFRs per cell. The cells were cultured in Ham’s F10 medium (Biochrom Kg), supplemented with 10% fetal calf serum (Sigma), l-glutamine (2 mmol/L), and PEST (penicillin 100 IU/mL and streptomycin 100 μg/mL), both from Biochrom Kg. During cell culture and cell experiments (unless otherwise stated), cells were grown at 37°C in incubators with humidified air, equilibrated with 5% CO2. The cells were trypsinized with trypsin–ethylenediaminetetraacetic acid (EDTA) (0.25% trypsin, 0.02% EDTA in PBS without Ca and Mg) from Biochrom Kg.
Binding of 68Ga-DOTA-hEGF to Cells
A431 and U343 cells were cultured in 3-cm Petri dishes (approximately 3.5 × 105 and 1.9 × 105 cells per dish, respectively). Triplicate cell dishes were used for each measuring point. After washing the cells once, 1 mL of 68Ga-DOTA-hEGF in cell culture medium (35 ng/dish, 50 kBq/dish for A431 cells and 5 ng/dish, 20 kBq/dish for U343 cells) was added. To some dishes, a molar excess of hEGF (5 or 3 μg/dish) was added together with the labeled conjugate to estimate the binding specificity of the 68Ga-DOTA-hEGF conjugate. After 0.5- to 6-h incubation at 37°C, the cells were washed 6 times with cold serum-free medium, and they were then harvested using 0.5 mL of trypsin-EDTA (15 min, 37°C). The trypsination was terminated with addition of 1 mL of cell culture medium, and part of the cell suspension (0.5 mL) was used for cell counting, whereas the rest was measured in a γ-counter.
To estimate the cellular internalization of the 68Ga-DOTA-hEGF conjugate, several additional cell dishes were used during the binding study to separate the membrane-bound fraction of the conjugate from internalized radioactivity. Instead of trypsinizing the cells, treatment with 0.5 mL of ice-cold 0.1 mol/L glycine-HCl buffer, pH 2.5, for 6 min at 0°C was used to extract the membrane-bound fraction of the conjugate. An additional 0.5 mL of the glycine-HCl buffer was used to wash the cells once. The remaining radioactivity, considered to be internalized radioactivity, was collected by treatment with 0.5 mL of 1 mol/L NaOH solution at 37°C for about 60 min. Another 0.5 mL of NaOH solution was used for washing. The collected fractions were measured in an automated γ-counter.
The binding of 68Ga-DOTA-hEGF to A431 cells and U343 cells on ice was also studied to determine the time required for binding in the saturation study. Cell dishes placed on ice were incubated with ice-cold 68Ga-DOTA-hEGF solution for 0.5–4 h. The cells were then washed, trypsinized, and counted as described.
Cellular Retention of Radioactivity
The cellular retention of radioactivity was studied after 1 h of incubation with 68Ga-DOTA-hEGF. After the incubation was interrupted, the cells were washed thoroughly to eliminate unbound conjugate, and the incubation was then continued in fresh cell culture medium. After 0.5–4 h, the cells were trypsinized, counted, and measured for radioactivity, as described.
Saturation Assay and Estimation of Dissociation Constant, Kd
The equilibrium Kd was determined from a saturation study with 68Ga-DOTA-hEGF on A431 cells and U343 cells. Cells cultured in 24-well plates (approximately 3.1 × 104 A431 cells/well and 7.8 × 104 U343 cells/well) were placed on ice, and ice-cold 68Ga-DOTA-hEGF solutions of different concentrations (0.26–16.9 nmol/L for A431 and 0.14–36 nmol/L for U343, 0.5 mL/well) were added. For each concentration, the nonspecific background binding was studied by adding a 100 times excess of unlabeled hEGF to some wells. After 2 h of incubation (the time was determined from the results of the uptake study on ice), the cells were washed 6 times with cold serum-free medium. The cells were then trypsinized with 0.5 mL of trypsin-EDTA (15 min at 37°C), and the cells were counted and measured for radioactivity in a γ-counter.
Animal Tumor Model
The in vivo studies were performed in adult female BALB/c nu/nu mice (21–25 g) (Möllegård) with tumor xenografts. All animals were handled according to the guidelines by the Swedish Animal Welfare Agency, and the experiments were approved by the local Ethics Committee for Animal Research. The mice were injected subcutaneously with A431 tumor cells (approximately 7 million cells per tumor in 100 μL of cell culture medium) in both front legs. The tumors were allowed to grow for 12–13 d before the experiments were performed and had then reached a weight of 0.1–0.8 g.
Biodistribution in Mice with A431 Tumor Xenografts
Mice with A431 tumor xenografts were injected intravenously with 50 μL of 68Ga-DOTA-hEGF solution (0.16 nmol or 0.016 nmol in PBS per animal; 4 animals in each group), and 30 min after injection the animals were sacrificed and dissected. The mice were anesthetized by an intraperitoneal injection of a mixture of Rompun (Bayer Co.; 1 mg/mL) and Ketalar (Parke Davis; 10 mg/mL), 0.2 mL/10 g of animal weight, and killed by heart puncture. In addition to the tumors, the blood, heart, pancreas, spleen, stomach, liver, kidneys, lungs, small and large intestine, muscle, bone, and salivary gland were collected, weighed, and measured in an automated γ-counter. The tails were also measured for radioactive content to determine the accuracy of the injections. Organ values were calculated as the percentage of injected dose per gram of organ (%ID/g).
microPET
Imaging was performed on a microPET R4 scanner (Concorde Microsystems, Inc.). Mice with A431 tumor zenografts were anesthetized with isoflurane (Baxter Medical AB) inhalation. 68Ga-DOTA-hEGF was injected via a tail vein (2.0 ± 0.5 MBq in 100 μL followed by 50 μL of saline). The mice were imaged (1 bed position; filtered-backprojection reconstruction; image resolution of 2 mm) for 30 or 120 min. microPET images were corrected for decay and attenuation. Regions of interest (ROIs) were drawn on liver, kidney, bladder, salivary gland, and tumor using ASIpro software (Concorde Microsystems, Inc.). Pharmacokinetic curves, representing the radioactivity concentrations (Bq/cm3 of tissue) versus time after injection, were acquired by drawing ROIs in the selected organs. The uptake index was calculated as radioactivity in organ (kBq/mL)/injected activity (kBq) × 100%.
RESULTS
Chemistry and Radiochemistry of 68Ga-DOTA-hEGF Preparation
68Ga-DOTA-hEGF was synthesized by a 2-step procedure, where hEGF was initially conjugated to a bifunctional chelator, DOTA, and thereafter labeled with 68Ga via a complexation reaction of 68Ga with the chelator (24,27). In the conjugation step, one of the carboxylic groups of the DOTA chelate was coupled to an amine functionality of the peptide, forming an amide bond (Fig. 1). hEGF contains 1 terminal and 2 lysine amino groups. Consequently, the conjugation reaction of hEGF resulted in the formation of a mixture of molecules with 1, 2, and 3 DOTA fragments, as determined by LC-ESI-MS analysis.
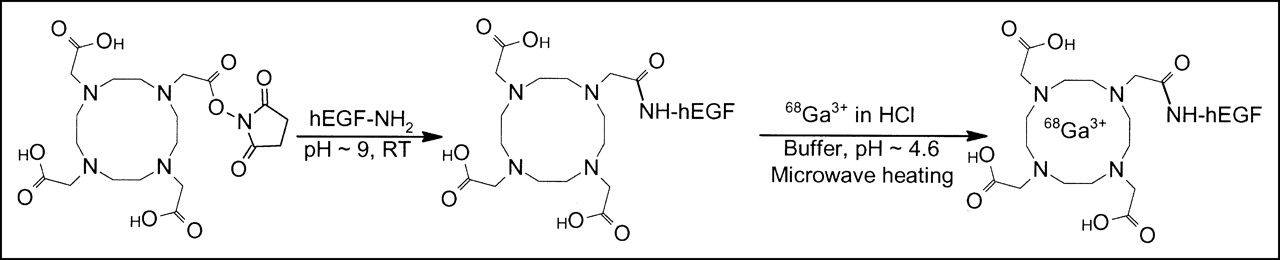
Reaction scheme for conjugation of DOTA to hEGF using a commercial N-hydroxysulfosuccinimide ester of DOTA and subsequent microwave-accelerated complexation of 68Ga with DOTA-hEGF. RT = room temperature.
The microwave-accelerated labeling of the conjugates (Fig. 1) was performed by use of a nonconcentrated or a preconcentrated generator 68Ga-eluate (24) with respective radioactivity incorporation of 60% ± 10% (n = 3) and 77% ± 4% (n = 3). The attachment of 68Ga to hEGF was DOTA mediated, as the same treatment of nonconjugated hEGF did not provide any labeled peptide. The radiochemical purity of the tracers in the study exceeded 95%. The tracer proved to be stable in the PBS during the stability assay of 4 h with no additional radio-HPLC signals.
Cell-Binding and Retention Experiments
The binding specificity of 68Ga-DOTA-EGF to EGFR-expressing cervical carcinoma A431 and glioma U343 cell lines in vitro is shown in Figure 2. To demonstrate that the binding was receptor specific, a large amount of unlabeled hEGF was added to cells in the control experiments to saturate the EGFR. Results of the binding specificity experiments demonstrated that the binding of 68Ga-DOTA-hEGF to both cell lines could be prevented by receptor saturation at all tested data points.

Specificity of 68Ga-DOTA-hEGF binding to A431 carcinoma (A) and U343 glioma (B) cell lines. At all time points, EGFR binding was blocked with excess of unlabeled hEGF. Binding was specific, as it could be suppressed. Data are presented as mean ± SD (n = 3).
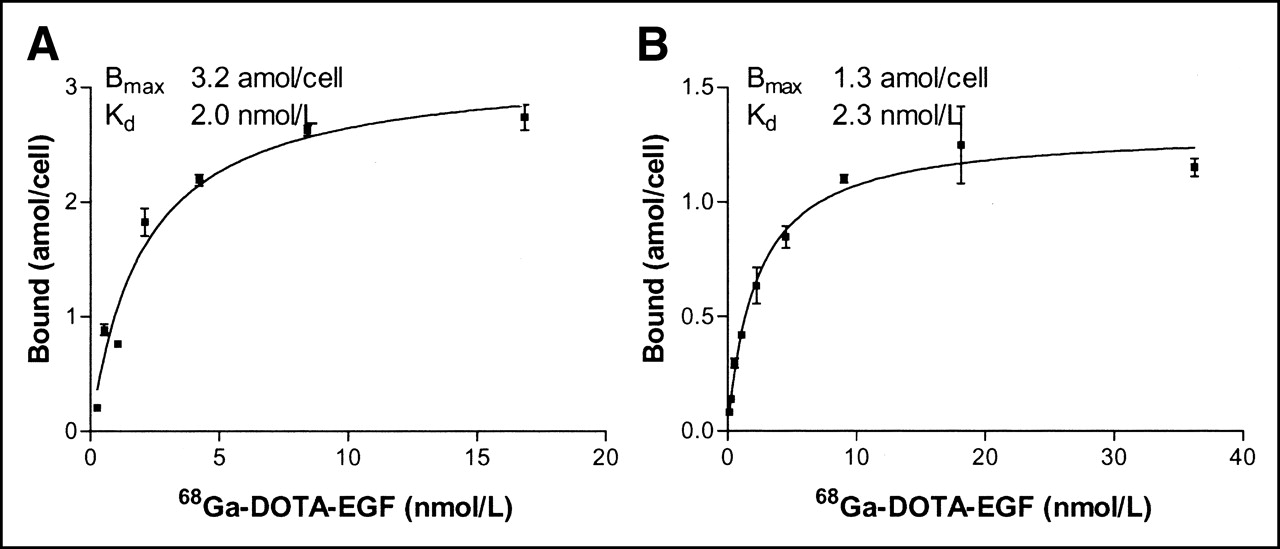
The results of the saturation experiments with 68Ga-DOTA-hEGF on cervical carcinoma A431 and glioma U343 cells are shown in Figures 3A and 3B, respectively. The specific binding (amol/cell) was plotted against the total molar concentration of added radiolabeled conjugate, and the result was analyzed by nonlinear regression using the GraphPad Prism Software. Both curves seem to have reached a maximum value, indicating saturation. The obtained Kd values were in an excellent agreement, 2.0 nmol/L for A431 cells and 2.3 nmol/L for U343 cells. The number of binding sites per cell could be calculated from the Bmax value. The obtained number, 7.8 × 105 sites per U343 cell, corresponds reasonably well with 5.4 × 105 as previously determined for a 111In-Bz-DTPA-hEGF conjugate (DTPA is diethylenetriaminepentaacetic acid) (20). The number of binding sites for A431, 1.9 × 106 EGFR per cell, was also in good agreement with literature data.
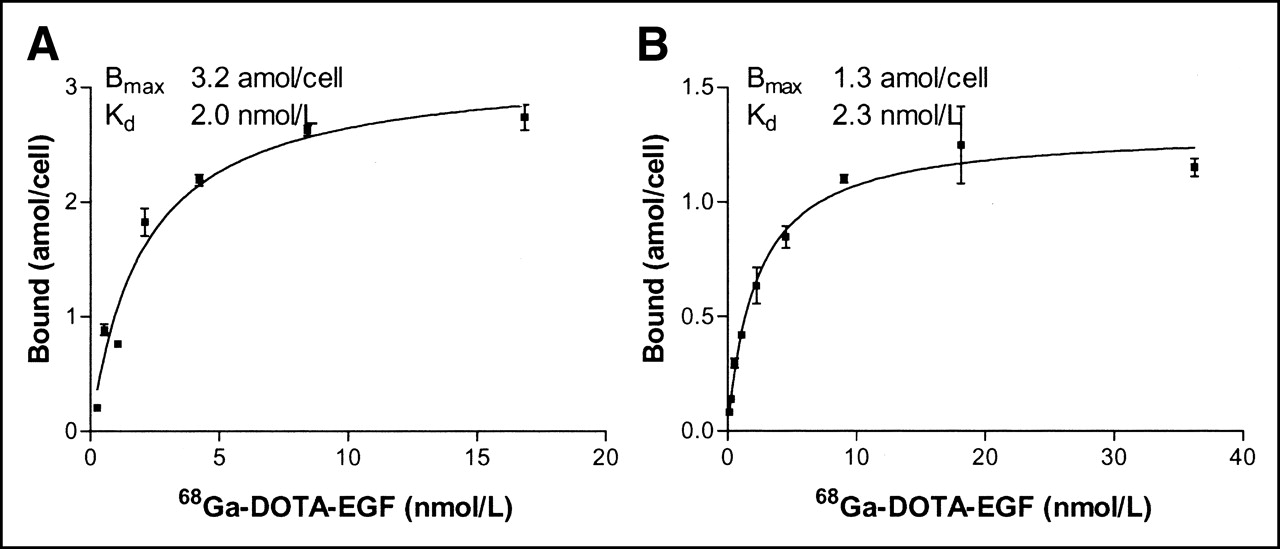
Saturation of 68Ga-DOTA-hEGF binding to cultured carcinoma A431 (A) and glioma U343 (B) cells. Cells were incubated with different concentrations of 68Ga-DOTA-hEGF (0.26–16.9 nmol/L for A431 cells and 0.14–36 nmol/L for U343 cells) for 2 h on ice. Data are presented as mean ± SD (n = 3).
The degree of internalization was estimated by acid wash. Radioactivity that was removed from cells by an acidic buffer was considered membrane bound and the rest was considered internalized. The results summarized in Figure 4 show that internalization of 68Ga-DOTA-hEGF was a rapid process in both cell lines. However, the internalization rate was faster in glioma U343 cells than in A431 cells. More than 50% of the radioactivity was internalized 30 min after the start of incubation in glioma U343 cells.

Internalization of 68Ga-DOTA-hEGF after binding to carcinoma A431 and glioma U343 cells. Internalization was determined by acid wash. y-Axis presents percentage of the total cell-associated radioactivity. Data are presented as mean ± SD (n = 3).
The retention pattern of radioactivity after interrupted incubation with 68Ga-DOTA-hEGF for A431 and U343 cells was similar for both cell lines (Fig. 5). An initial drop of radioactivity, which was most probably due to dissociation of membrane-bound conjugates, was followed by a relatively constant amount of cell-bound 68Ga. Both cell lines demonstrated good retention, with >70% of the radioactivity still cell associated 4 h (3 half-lives of the label) after interrupted incubation.
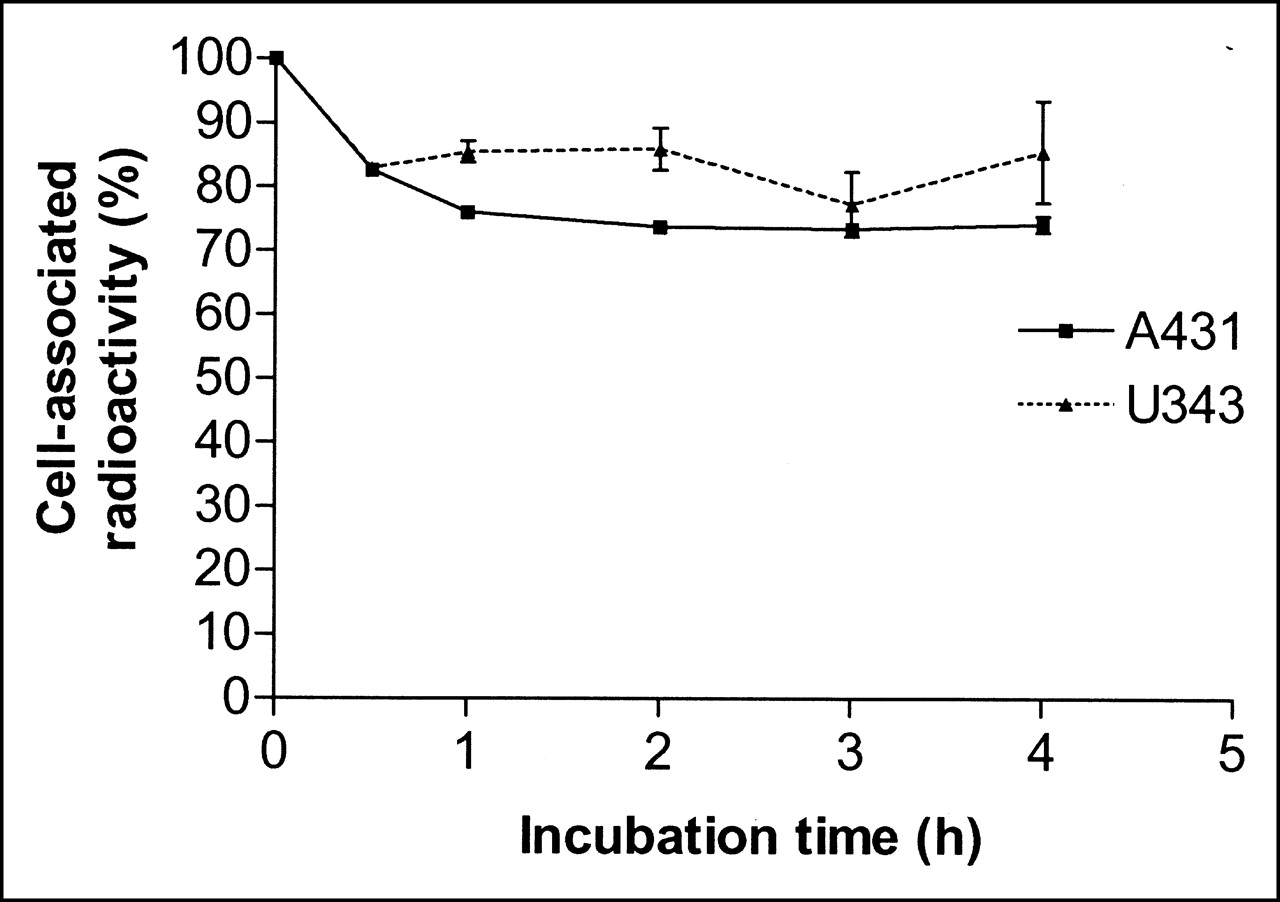
Cell-associated 68Ga radioactivity as function of time after interrupted incubation of A431 (solid line) and U343 (dotted line) cells with 68Ga-DOTA-hEGF. Cell-associated radioactivity at time zero after interrupted incubation was considered as 100%. Data are presented as mean ± SD (n = 3). Both A431 and U343 cell cultures were incubated with 68Ga-DOTA-hEGF for 4 h.
Biodistribution Studies
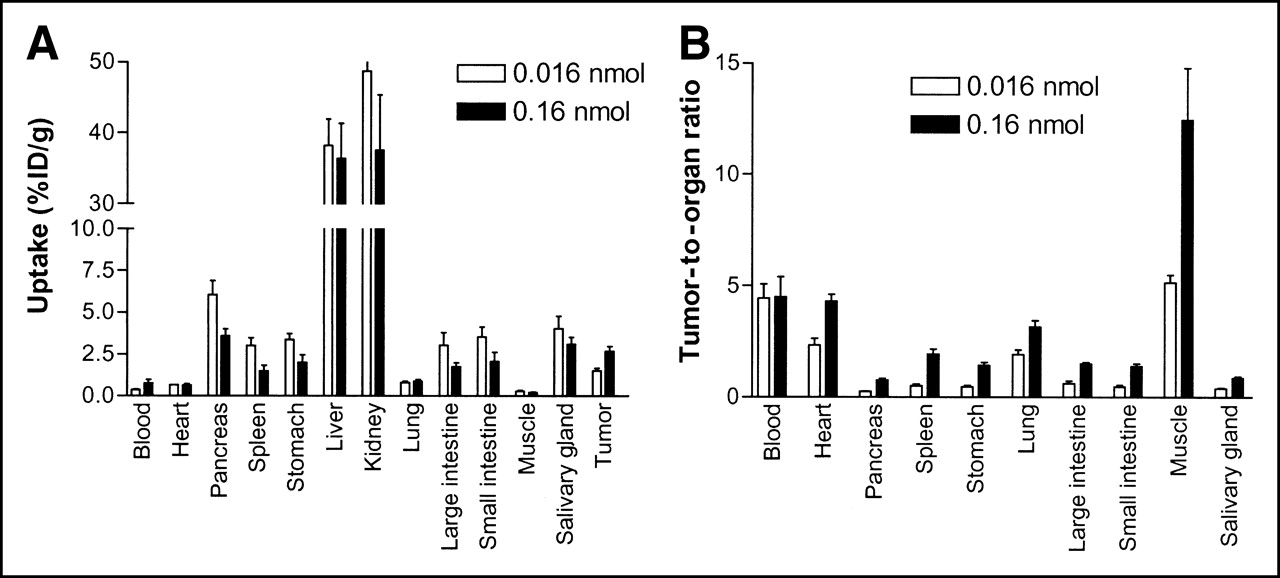
A summary of the biodistribution data for 68Ga-DOTA-hEGF in A431 tumor-bearing mice is shown in Figure 6. The measurement of the organ radioactivity 30 min after intravenous administration of 68Ga-DOTA-hEGF showed the highest values in the kidneys and liver, followed by pancreas, salivary gland, small and large intestine, stomach, and spleen. The uptake of 68Ga-DOTA-hEGF in the A431 tumor xenograft was 1.51 ± 0.16 %ID/g and 2.69 ± 0.29 %ID/g, for 0.016 and 0.16 nmol of injected conjugate, respectively (P = 0.036). The radiotracer had a rapid blood clearance, with <1 %ID/g remaining in the circulation at the 30-min time point. There was a statistically significant decrease in the radioactivity uptake in pancreas, spleen, and stomach when 0.16 nmol of conjugate was injected. There was a statistically significant increase of tumor-to-organ ratios for heart, pancreas, stomach, spleen, lungs, intestines, muscles, and salivary glands when 0.16 nmol of conjugate was injected. However, there was no difference in tumor-to-blood ratio, 4.42 ± 1.81 %ID/g and 4.50 ± 2.53 %ID/g, for 0.016 and 0.16 nmol of injected conjugate, respectively (P = 0.036).

(A) Biodistribution of 68Ga-DOTA-hEGF expressed as %ID/g tissue in tumor-bearing nude mice at 30-min time point. (B) Tumor-to-organ ratios of 68Ga-DOTA-EGF in tumor-bearing nude mice at 30-min time point. Mice were intravenously injected with either 0.016 or 0.16 nmol of radiotracer and killed at 30-min time point. Data are presented as mean ± SD (n = 4).
microPET
The localization of 68Ga-DOTA-hEGF in tumor-bearing mice as determined by microPET imaging (Fig. 7) was followed by radioactivity measurements of blood, liver, kidney, and both tumor samples collected after decapitation of the animal.

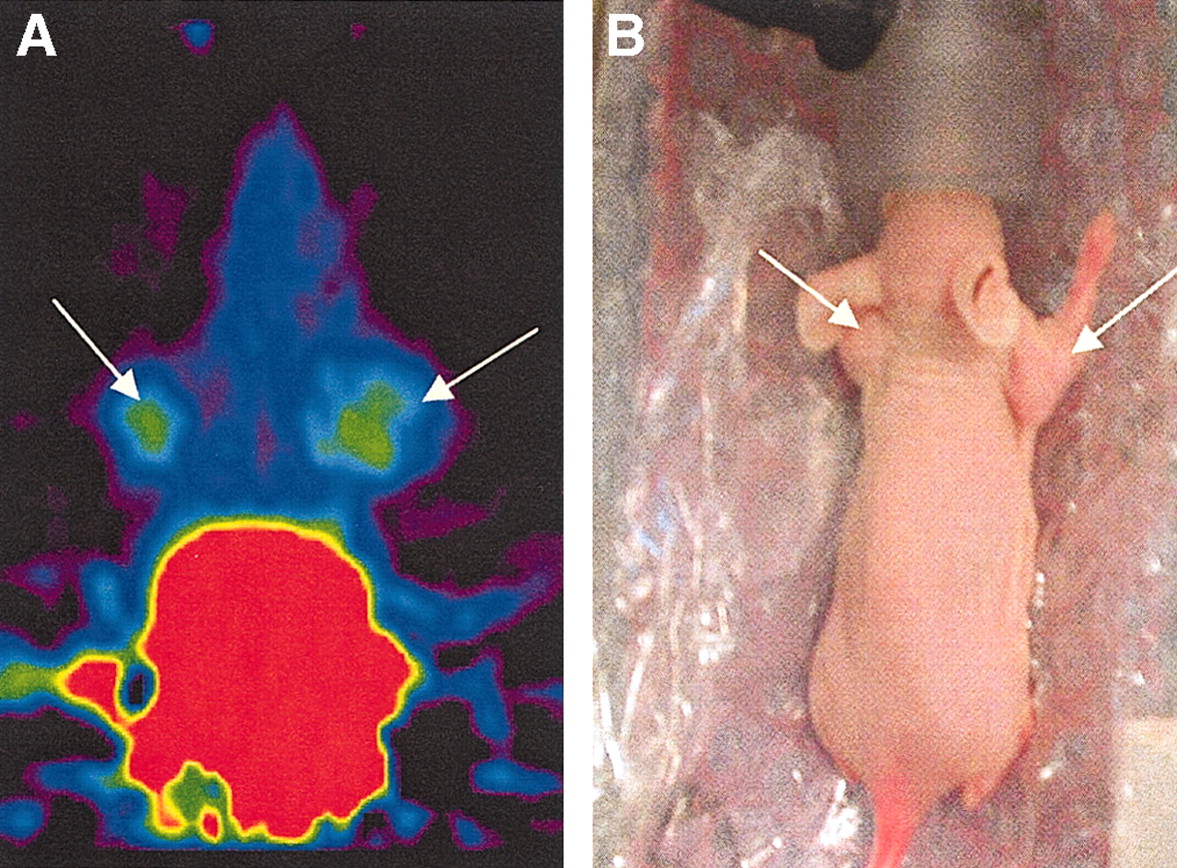
(A) Image shows summation of frames 20–24 (20–30 min after injection). Tumors (arrows) can clearly be seen at either side of head. (B) Photograph of positioning of mouse.
The image of a tumor-bearing mouse 30 min after administration of 2.0 MBq 68Ga-DOTA-hEGF with a specific radioactivity (SRA) of 12–20 MBq/nmol is shown in Figure 7A. The results of the microPET image were correlated with the radioactivity measurements of blood, liver, and kidney samples. Both right and left leg tumors were visible with clear contrast against the adjacent background. Prominent uptake was observed in the liver and kidneys, and clearance of the radioactivity through the urinary bladder was evident (Fig. 8). The distribution to tumors and salivary gland was slower. Uptake data derived from microPET and biodistribution studies were found to be in agreement with data obtained from the tissue sampling after imaging.
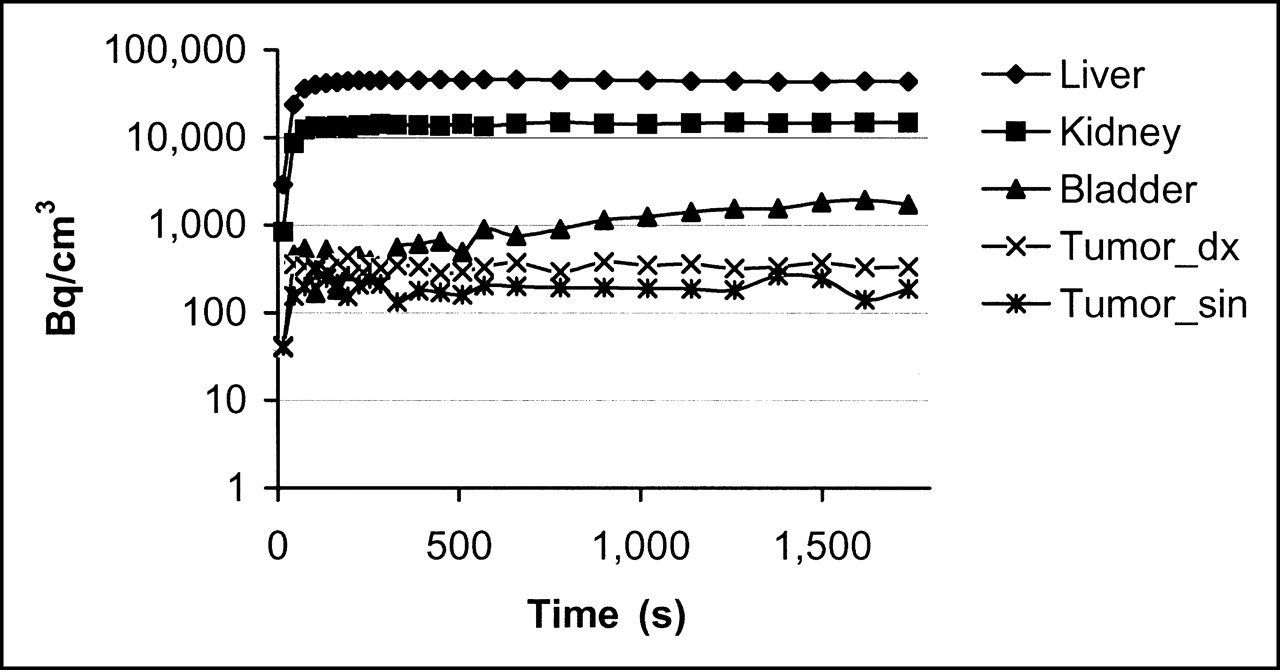
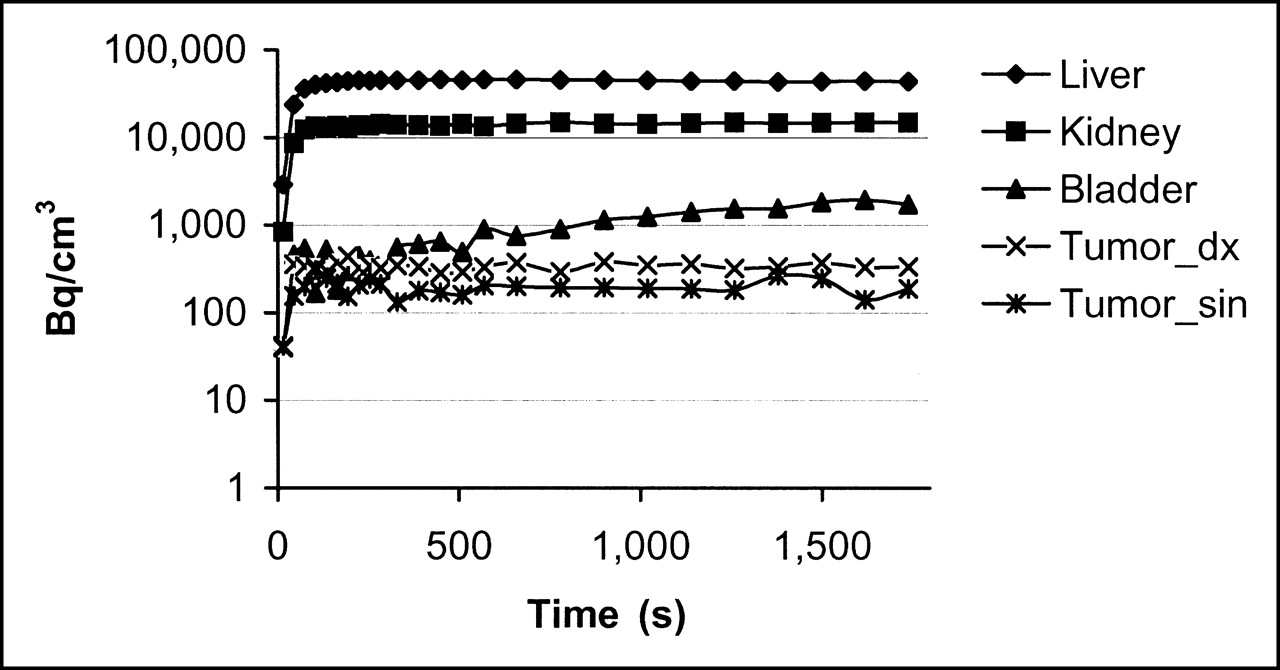
Pharmacokinetic curves show rapid distribution of 68Ga-DOTA-hEGF (0.16 nmol injected) to liver, kidney, bladder, and tumors. Tumor_dx = right side tumor; Tumor_sin = left side tumor.
DISCUSSION
The experience with somatostatin analogs shows that the use of macrocyclic chelators, such as DOTA derivatives, may provide an adequate in vivo stability of the 68Ga radiolabel (25). On the other hand, macrocyclic chelators are kinetically inert, and incorporation of radiometals into DOTA requires elevated temperatures (approximately 100°C) or a long chelating procedure. Both of these approaches are undesirable because of the potential damage to the targeting peptide and the relatively short half-life of 68Ga. Microwave heating is an attractive tool that provides acceleration of the labeling. This technique has been used for labeling different organic molecules with 131I, 11C, 15O, 18F, and 13N (29). Earlier, we have demonstrated the potential of microwave heating for speeding up 68Ga complexation with DOTA bifunctional chelator coupled to oligonucleotides and peptides (24,26,27). For this reason, the use of the microwave technique was applied in this study. The method made it possible to perform labeling within 1 min with both nonconcentrated and preconcentrated generator eluate. 68Ga incorporation was satisfactory (60% ± 10%) when using the peak fraction of the nonconcentrated generator eluate. However, about 40% of the 68Ga activity was wasted. The preconcentration of the generator eluate provided use of 85%–90% of the initially available radioactivity and increased radioactivity incorporation (approximately 80%). Consequently, the SRA of the tracer was increased as well. Previously, 68Ga labeling under microwave heating was performed with small peptides with a molecular weight varying between 1.4 and 3.7 kDa (24,27). The binding properties of hEGF are determined by a more complex structure and, therefore, could be more vulnerable to the action of microwave heating. Thus, a careful characterization of the labeled conjugate was required. Initial experiments demonstrated that after microwave-assisted labeling, 68Ga-DOTA-hEGF retained its capacity to bind to the EGFR-expressing cell lines A431 and U343. This binding was receptor specific, as it could be precluded by presaturation of EGFR with an unlabeled ligand (Fig. 2). Affinity measurements showed a Kd of about 2 nmol/L for both cell lines (Fig. 3). This value was in good agreement with values obtained for hEGF-chelator conjugates labeled with indium or lutetium without microwave heating (20,30). This demonstrated that the use of microwaves did not deteriorate the receptor-binding properties of such a complex peptide as hEGF. The affinity in the low nanomolar range was also compatible with the application of 68Ga-DOTA-hEGF as a tracer for in vivo imaging.
The internalization of the targeting conjugate is considered as an important property because it prevents its eventual dissociation from cancer cells in vivo. The use of the hEGF–EGFR system for targeting should meet this requirement, as internalization of the receptor–ligand complex is well documented for this system. Indeed, the internalization study demonstrated that 68Ga-DOTA-hEGF was internalized by both model cell lines (Fig. 4). Internalization by U343 cells seems to be more rapid than internalization by A431 cells, which might be explained by the difference in cellular processing machinery of these 2 cell lines.
Good intracellular retention of radiometals after internalization was considered when we designed the EGFR-targeting conjugate. The retention studies (Fig. 5) confirmed good retention of 68Ga-DOTA-hEGF by both cell lines. The high cellular retention of 68Ga-DOTA-hEGF was also in good agreement with data relating to 111In- and 177Lu-labeled hEGF-chelator conjugates (15,20,30).
Although the in vitro properties of 68Ga-DOTA-hEGF were promising, an ultimate proof of the concept may be provided only by in vivo experiments such as microPET imaging and biodistribution. The feasibility of EGFR-expressing tumor imaging using 68Ga-DOTA-hEGF was demonstrated by microPET (Fig. 7). The tracer localization was rapid, and the plateau of accumulation in tumors, liver, and kidneys was reached within 10 min after injection. Besides tumors, high radioactivity uptake was observed in kidneys and liver. A high kidney uptake is typical for all radioconjugates, which are small enough to pass through glomerular membranes and hydrophilic enough to undergo renal clearance (31). A predominant renal clearance was also confirmed in this study by the high level of radioactivity accumulation in the urinary bladder. Radioactivity accumulation in the urinary bladder peaked at about 30 min after injection, indicating that, at this moment, the blood elimination phase was essentially accomplished. Another organ with high radioactivity uptake was the liver. It has been demonstrated earlier in several studies that liver uptake is EGFR mediated (30,32). High liver uptake is one of the limitations of 68Ga-DOTA-hEGF because it might prevent imaging of EGFR expression in hepatic metastases. However, we believe that 68Ga-DOTA-hEGF might be helpful for imaging of EGFR expression in primary tumors of NSCLC. The lesion is in this case placed at some distance from liver, and EGFR expression in NSCLC has been imaged earlier using 131I-hEGF. Expression of EGFR is a precondition for EGFR-targeting therapy using, for example, cetuximab (33). Moreover, heterogenicity of EGFR expression in NSCLC makes biopsy a less-reliable procedure (33). The use of 68Ga-DOTA-hEGF in combination with PET/CT might help to determine the eligibility of a given patient for cetuximab therapy by evaluation of EGFR overexpression in the whole tumor. The biodistribution studies revealed the influence of SRA on radioactivity uptake in tumors and normal organs. The injection of 68Ga-DOTA-hEGF with lower SRA resulted in the tumor uptake of 2.7 ± 0.3 %ID/g, which is comparable with the accumulation in murine xenografts of such a well-established radiopharmaceutical as 111In-OctreoScan (about 3 %ID/g) (34). However, it remains to be investigated if it is possible to block the liver uptake (32) by manipulating the SRA of 68Ga-DOTA-hEGF. High SRA, achieved by using a combination of the preconcentration of 68Ga and microwave heating, enables such investigation of the gaussian distribution of radioactivity uptake as a function of SRA (35,36). Furthermore, the optimization of SRA might be performed for future patient studies.
CONCLUSION
hEGF conjugated with a macrocyclic DOTA-chelating group and radiolabeled with the positron-emitting radionuclide 68Ga showed high EGFR-binding affinity and specificity and a rapid internalization in both carcinoma A431 and glioma U343 cells. The localization of 68Ga-DOTA-hEGF in A431 tumor xenografts and EGFR-positive tissues was shown in both biodistribution and microPET imaging studies. 68Ga-DOTA-hEGF as a peptide-based tracer met the major requirements, such as tissue permeability, high-affinity receptor binding, and a rapid clearance from the body. Further investigations are required to determine the optimal SRA for imaging. 68Ga-DOTA-hEGF might be useful for the selection of patients for EGFR-targeting therapy as well as evaluation of response to such therapy.
Acknowledgments
The Swedish Research Council is acknowledged for its support (grant K3464). Financial support was partially given by the Swedish Cancer Society. The authors thank the staff of the Unit of Biomedical Radiation Sciences, Uppsala University, for helpful assistance in the biodistribution studies.
Footnotes
Received Mar. 14, 2005; revision accepted Jul. 15, 2005.
For correspondence or reprints contact: Bengt Långström, PhD, Uppsala Imanet, P.O. Box 967, SE-751 09 Uppsala, Sweden.
E-mail: Bengt.Langstrom{at}uppsala.imanet.se
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Dual-Receptor-Targeted Radioimmunotherapy of Human Breast Cancer Xenografts in Athymic Mice Coexpressing HER2 and EGFR Using 177Lu- or 111In-Labeled Bispecific Radioimmunoconjugates
- Specific biomarkers of receptors, pathways of inhibition and targeted therapies: pre-clinical developments
- Affibody Molecules for Epidermal Growth Factor Receptor Targeting In Vivo: Aspects of Dimerization and Labeling Chemistry
- Tumor Receptor Imaging
- Comparison of the Biodistribution and Tumor Targeting of Two 99mTc-Labeled Anti-EGFR Nanobodies in Mice, Using Pinhole SPECT/Micro-CT