


Abstract
While searching for a PET method to determine the density and occupancy of the dopamine D3 receptor, we found evidence that suggested that the dopamine D3 antagonist GR218231 could be a substrate of the P-glycoprotein efflux pump. P-glycoprotein protects the brain against toxic substances and xenobiotics, but it also hampers the delivery of various drugs into the brain. In this study, we aimed to explore whether radiolabeled GR218231 could be applied as a PET tracer for monitoring P-glycoprotein activity in the blood–brain barrier. Such an imaging technique could be useful for the development of new drugs and novel strategies to deliver drugs to the brain and for identification of undesirable drug–drug interactions. Methods: As a potential PET tracer, GR218231 was labeled with 11C by reaction of the newly synthesized desmethyl precursor with 11C-methyl triflate. The biodistribution of 11C-GR218231 was determined in rats. To assess specific binding to the dopamine D3 receptor, blocking experiments with unlabeled GR218231 (0.2 and 2.5 mg/kg) were performed. To demonstrate the influence of P-glycoprotein on cerebral uptake of 11C-GR218231, the efflux pump was modulated with 50 mg/kg cyclosporine A. The sensitivity of 11C-GR218231 for P-glycoprotein modulation was assessed in dose–response studies, using escalating cyclosporine A dosages. Results: 11C-GR218231 was prepared in 53% ± 8% decay-corrected radiochemical yield and had a specific activity of 15 ± 10 GBq/μmol (mean ± SD). Biodistribution studies in rats revealed a low and homogeneous uptake in the brain. Pretreatment of the animals with unlabeled GR218231 did not demonstrate any specific binding. Modulation of P-glycoprotein with cyclosporine A caused a 12-fold higher 11C-GR218231 uptake in the brain, indicating that the low cerebral tracer uptake was caused by the P-glycoprotein efflux pump in the blood–brain barrier. Cyclosporine A dose-escalation studies showed a dose-dependent sigmoidal increase in 11C-GR218231 uptake in brain and spleen (median effective dose [ED50], 23.3 ± 0.6 and 38.4 ± 2.4 mg/kg, respectively), whereas a dose-dependent decrease was observed in the pancreas (ED50, 36.0 ± 4.4 mg/kg). Conclusion: Although 11C-GR218231 is unsuited for dopamine D3 receptor imaging with PET, it appears to be an attractive PET tracer for visualization and quantification of P-glycoprotein activity in the blood–brain barrier.
The blood–brain barrier protects the brain from harmful substances and xenobiotics from the environment (1). The access of large and polar molecules to the brain is restricted by the brain microvasculature, which lacks fenestrations and has close tight junctions with a high electrical resistance. Consequently, passive diffusion across the brain endothelium is only possible for small lipophilic molecules, typically with an octanol–water partition coefficient (logP) between 0.9 and 2.5 (2). Efflux pumps, such as the ABC transporters P-glycoprotein and multidrug resistance–associated protein, also contribute to the blood–brain barrier by extruding a variety of small lipophilic substances from the brain endothelium back into the circulation. The integrity of the blood–brain barrier is of crucial importance for the brain, because failure of the blood–brain barrier leads to exposure of the brain to toxic agents that can cause degeneration of brain tissue.
Since the blood–brain barrier prevents the accumulation of potentially harmful compounds in the brain, it can also be a major obstacle for the delivery of drugs to the brain. P-glycoprotein is an adenosine triphosphate–driven efflux pump that has a major contribution to the blood–brain barrier. Many structurally unrelated drugs, including various anticancer drugs, antidepressants, steroids, and HIV protease inhibitors, are substrates of P-glycoprotein and are extruded from the brain by the efflux pump (3). Consequently, drug levels in the brain remain below therapeutic levels, resulting in poor therapy findings. Coadministration of another P-glycoprotein substrate (modulator), such as cyclosporine A, can induce an increase in the concentration of the therapeutic drug in the brain, due to competition between both substrates for the efflux pump (4). Thus, modulation of P-glycoprotein could be applied to improve the efficacy of chemotherapeutic treatment. However, unintentional modulation of P-glycoprotein that can occur when multiple drugs are combined can also lead to an unexpected increase in drug levels in the brain, which might cause neurotoxic side effects.
Nuclear imaging techniques, such as PET, enable the measurement of the tissue concentrations of radiolabeled P-glycoprotein substrates as a function of time. Such an imaging technique allows noninvasive monitoring of P-glycoprotein activity in the blood–brain barrier and therefore could be an attractive tool for the development of novel strategies to improve the delivery of drugs into the brain. For example, PET could be applied to evaluate the efficacy of novel P-glycoprotein modulators and to optimize the dosing schedule. Another interesting application could be monitoring of P-glycoprotein functionality in psychiatric and neurologic disorders, which could implicate the efflux pump as a possible cause in these diseases (5). Currently, several radiolabeled drugs, including 11C-verapamil and 11C-carvedilol, are under investigation as PET tracers for P-glycoprotein (3). Applications of these tracers in human modulation studies, however, is hampered by the lack of potent P-glycoprotein modulators that can be administered safely to humans at the dosage that is required for sufficient modulation to achieve maximum tracer uptake. The aim of this study was to evaluate 11C-GR218231 (Fig. 1) as a novel PET tracer for measuring P-glycoprotein activity.

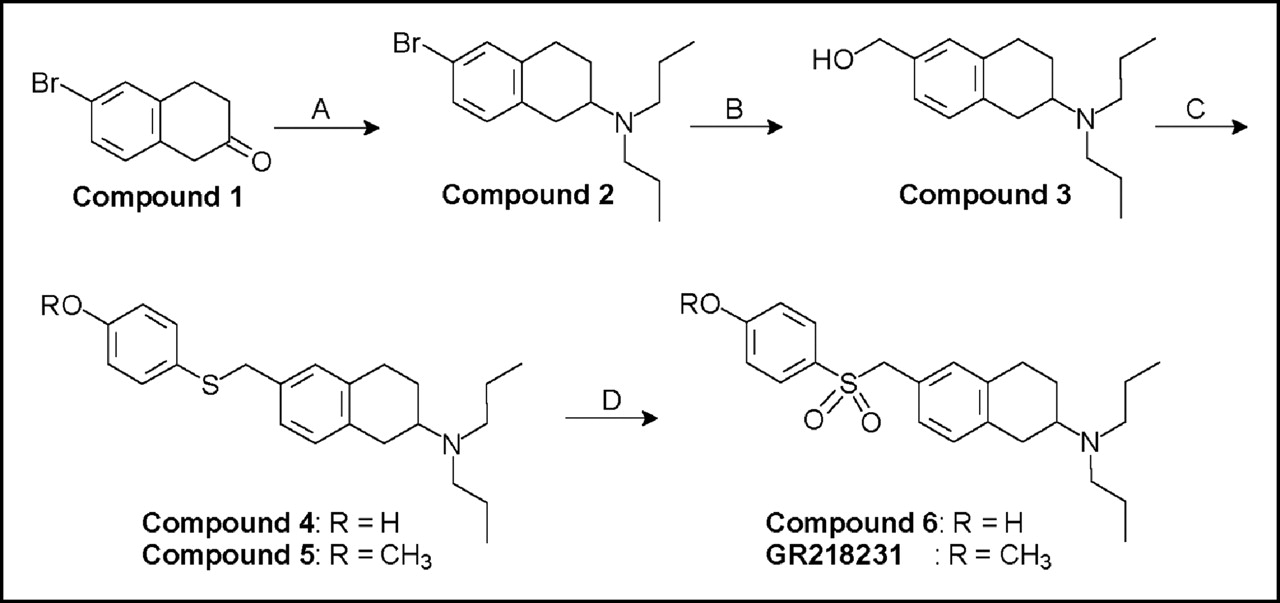
Synthesis of GR218231 and its labeling precursor, Compound 6, using the following reagents and conditions: (A) (CH3CH2CH2)2NH, NaCNBH3, MeOH; (B) (i) n-BuLi, tetrahydrofuran, −78°C; (ii) N,N-dimethylformamide; (iii) NaBH4, MeOH; (C) (i) SOCl2, CHCl3; (ii) ROC6H4SH, K2CO3, Kryptofix 2.2.2., CHCl3; (D) (i) HCl, EtOH; (ii) m-chloroperbenzoic acid, CH2Cl2.
GR218231 is an antagonist of the dopamine D3 receptor, which was selected as a potential radiotracer for measuring the density and occupation of the dopamine D3 receptor in the brain with PET. In vitro, GR218231 exhibits a high affinity and selectivity for the dopamine D3 receptor (6,7). The (R)-isomer of GR218231 (pKi = 8.9) is slightly more potent than the racemic mixture (pKi = 8.8) and the compound is sufficiently lipophilic for penetration of the blood–brain barrier (logD = 2.2) (7). In vivo, GR218231 behaves as an antagonist at the dopamine D3 receptor but is inactive in animal models for D2 receptor–mediated effects, such as models for antipsychotic and extrapyramidal activity (6,8). The course of the evaluation studies of radiolabeled GR218231 as a PET tracer for the dopamine D3 receptor, however, led us to believe that this compound is a substrate of the P-glycoprotein pump. Previously, other dopamine receptor agonists and antagonists, such as domperidone (9), bromocriptine (10), and flupentixol (11), have also been identified as substrates of the P-glycoprotein pump. The efflux pump can reduce brain uptake of radiotracers and, thus, hampers successful imaging by decreasing the signal-to-noise ratio (12,13). This prompted us to investigate a new and interesting application of radiolabeled GR218231: as PET tracer for imaging of P-glycoprotein activity.
This article describes the radiolabeling of racemic dopamine D3 receptor antagonist GR218231 with the positron emitter 11C and the investigation of its normal brain uptake and biodistribution in rats. In addition, the effect of modulation of the P-glycoprotein efflux pump on uptake of 11C-GR218231 in the brain is described.
MATERIALS AND METHODS
Materials
Solvents and reagents were obtained from commercial suppliers and used without further purification. 1H NMR (200 MHz) and 13C NMR (50 MHz) spectra were recorded on a Varian Gemini 200 spectrometer. 1H and 13C chemical shifts were determined relative to the signal of the solvent, converted to the tetramethylsilane (TMS) scale and expressed in δ-units (ppm) downfield from TMS. Melting points were measured with a Mettler FP61 apparatus. High-performance liquid chromatographic (HPLC) analyses and preparative separations were performed on a Waters system, consisting of a 515 isocratic pump, a 486 multiwavelength ultraviolet detector operated at 254 nm, and a Bicron Geiger–Müller radioactivity detector.
Chemistry
6-Bromo-2-(N,N-Di-n-Propylamino)Tetralin (Compound 2).
To a solution of 5.0 g (22 mmol) 6-bromo-2-tetralone (compound 1) in 200 mL methanol on 4-Å molecular sieves, 15 mL (111 mmol, 5 eq) di-n-propylamine were added. The reaction mixture was stirred at room temperature for 1 h, after which 7.0 g (111 mmol, 5 eq) NaBH3CN were slowly added. The pH of the reaction mixture was adjusted to 6 by adding glacial acetic acid. After stirring at room temperature for 72 h, the reaction mixture was filtered. The filtrate was concentrated in vacuo. The residue was dissolved in a mixture of 200 mL 1N HCl, 100 mL water, and 150 mL ethyl acetate. The layers were separated and the water layer was washed with 150 mL ethyl acetate. The combined organic layers were extracted 5 times with 75 mL 1N HCl and subsequently discarded. All aqueous layers were combined and solid NaOH was added until the pH was >10. The basic water layer was extracted 3 times with 150 mL ethyl acetate. The combined organic layers were dried on MgSO4 and concentrated in vacuo, affording 2.6 g (38%) of compound 2 as yellow oil. 1H NMR (200 MHz, CDCl3): δ 7.22 (s, 1H, Ar), 7.19 (d, 1H, J = 9.0 Hz, Ar), 6.94 (d, 1H, J = 9.0 Hz, Ar), 3.00–2.65 (m, 5H, CH2+CHN), 2.47 (t, 4H, J = 7.6 Hz, CH2N), 2.06–1.97 (m, 1H, CH2), 1.69–1.38 (m, 5H, CH2), 0.88 (t, 6H, J = 7.3 Hz, CH3).
2-(N,N-Di-n-Propylamino)-6-(Hydroxymethyl)Tetralin (Compound 3).
Under an argon atmosphere, 12.1 mL 1.6N (19.4 mmol, 2 eq) n-butyllithium in hexane were added to a solution of 3.0 g (9.7 mmol) of compound 2 in 60 mL of freshly distilled (from sodium/benzophenone) tetrahydrofuran (THF) at −80°C. After stirring at −80°C for 15 min, 3.7 mL (48 mmol, 5 eq) anhydrous N,N-dimethylformamide (DMF) were added. The reaction mixture was allowed to warm to room temperature in 1 h, after which stirring was continued for 1 h. The reaction was quenched by the addition of 150 mL water and the reaction mixture was extracted 3 times with 100 mL ethyl acetate. The combined organic layers were washed twice with 200 mL of water, dried on Na2SO4, and concentrated in vacuo. The crude aldehyde (2.68 g) was dissolved in 100 mL methanol. To this solution, 0.8 g (20 mmol, 2 eq) NaBH4 was slowly added. The reaction mixture was stirred at room temperature for 1 h and subsequently concentrated in vacuo to a volume of approximately 50 mL. The residue was diluted with 250 mL water and extracted 3 times with 100 mL ethyl acetate. The combined organic layers were washed with 200 mL water, dried on MgSO4, and concentrated in vacuo. The crude product was purified by flash column chromatography over a silica gel 60 column, using ethyl acetate/n-hexane/triethylamine (20:78:2) as the eluent. This yielded 1.35 g (53%) of compound 3, as a white solid. Melting point: 53°C. 1H NMR (200 MHz, CDCl3): δ 7.08 (s, 3H, Ar), 4.61 (s, 2H, CH2O), 3.04–2.65 (m, 5H, CH2+CHN), 2.48 (t, 4H, J = 7.6 Hz, CH2N), 2.07–1.98 (m, 1H, CH2), 1.79 (bs, 1H, OH), 1.70–1.38 (m, 5H, CH2), 0.89 (t, 6H, J = 7.8 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 136.8, 135.2, 134.6, 128.2, 125.8, 123.0, 63.7, 55.4, 51.1, 30.4, 28.4, 24.4, 20.5, 10.4.
2-(N,N-Di-n-Propylamino)-6-(4-Hydroxyphenylthiomethyl)Tetralin (Compound 4).
Thionyl chloride (340 μL, 4.6 mmol, 4 eq) was slowly added to a solution of 300 mg (1.16 mmol) of compound 3 in 10 mL CHCl3. The reaction mixture was stirred at ambient temperature for 2 h. The mixture was poured into 50 mL water and extracted twice with 25 mL CHCl3. The combined organic layers were washed with 50 mL 0.5 mol/L K2CO3, dried on MgSO4, and concentrated in vacuo. To remove traces of SOCl2, the residue was dissolved in 50 mL 1,2-dichloroethane and concentrated again. The crude chloride (0.53 g) was dissolved in 20 mL CHCl3. To this solution, 0.30 g (2.4 mmol, 2 eq) 4-mercaptophenol, 0.45 g (3.3 mmol, 2.8 eq) K2CO3, and 25 mg (66 μmol, 0.06 eq) Kryptofix 2.2.2. (Merck) were subsequently added. After the reaction mixture was stirred at room temperature for 18 h, 25 mL water were added. The layers were separated and the water layer was extracted twice with 20 mL CHCl3. The combined organic layers were washed twice with 25 mL water, dried on Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography over a silica gel 60 column, using ethyl acetate/n-hexane/triethylamine (40:58:2) as the eluent. The desired product was obtained as colorless glassy oil in 71% yield (300 mg). 1H NMR (200 MHz, CDCl3): δ 7.22 (d, 2H, J = 8.6 Hz, Ar), 6.99–6.89 (m, 3H, Ar), 6.72 (d, 2H, J = 8.6 Hz, Ar), 3.92 (s, 2H, CH2S), 3.00–2.82 (m, 5H, CH2+CHN), 2.50 (t, 4H, J = 7.7 Hz, CH2N), 2.0 (m, 1H, CH2), 1.65–1.40 (m, 5H, CH2), 0.88 (t, 6H, J = 7.8 Hz, CH3).
2-(N,N-Di-n-Propylamino)-6-(4-Methoxyphenylthiomethyl)Tetralin (Compound 5).
Compound 5 was prepared in the same manner as was described for compound 4, using 4-mercaptoanisol instead of 4-mercaptophenol. The crude product was purified by flash column chromatography over a silica gel 60 column, using ethyl acetate/n-hexane/triethylamine (10:88:2) as the eluent. For further purification, the product was dissolved in 25 mL 1N HCl and 25 mL diethyl ether. The layers were separated and the organic layer was extracted twice with 25 mL 1N HCl. Solid NaOH was added to the combined aqueous layers until the pH was >10. The basic solution was extracted twice with 25 mL diethyl ether. The organic layers were dried on MgSO4 and concentrated in vacuo, affording 318 mg (72%) of compound 5 as a white solid. Melting point: 46°C. 1H NMR (200 MHz, CDCl3): δ 7.28 (d, 2H, J = 8.9 Hz, Ar), 7.01–6.92 (m, 3H, Ar), 6.81 (d, 2H, J = 8.9 Hz, Ar), 3.94 (s, 2H, CH2S), 3.79 (s, 3H, CH3O), 2.97–2.69 (m, 5H, CH2+CHN), 2.47 (t, 4H, J = 7.6 Hz, CH2N), 2.0 (m, 1H, CH2), 1.69–1.38 (m, 5H, CH2), 0.89 (t, 6H, J = 7.5 Hz, CH3).
2-(N,N-Di-n-Propylamino)-6-(4-Hydroxyphenylsulfonylmethyl)-Tetralin (Compound 6). To a solution of 300 mg (0.82 mmol) of compound 4 in 50 mL absolute ethanol, 37% HCl was added drop by drop until the pH was <1. The solvent was evaporated in vacuo and the residue was dissolved in 10 mL CH2Cl2. To this solution, 0.47 g (1.8 mmol, 2 eq) 70%–75% m-chloroperoxybenzoic acid was added and the reaction mixture was stirred overnight at room temperature. The mixture was washed with 50 mL 0.5N NaHCO3. The water layer was extracted twice with 20 mL CH2Cl2. The combined organic layers were washed with 40 mL water, dried on Na2SO4, and concentrated in vacuo. The crude product was purified by flash column chromatography over a silica gel 60 column, using methanol/CH2Cl2/triethylamine (4:95:1) as the eluent. For further purification, the product was dissolved in 50 mL 1N HCl and 25 mL ethyl acetate. The layers were separated and the organic layer was extracted twice with 25 mL 1N HCl. The combined aqueous layers were neutralized first with solid NaOH to pH 4 and then with NaHCO3 to pH 8. The solution was extracted twice with 25 mL diethyl ether. The organic layers were dried on Na2SO4 and concentrated in vacuo, affording 140 mg (43%) of compound 6 as a white solid. Melting point: 67°C. 1H NMR (200 MHz, CDCl3): δ 7.41 (d, 2H, J = 8.8 Hz, Ar), 6.95–6.75 (m, 5H, Ar), 4.18 (s, 2H, CH2S), 3.07–2.56 (m, 9H, CH2+CHN), 2.0 (m, 1H, CH2), 1.66–1.51 (m, 5H, CH2), 0.91 (t, 6H, J = 7.2 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 162.4, 134.6, 134.4, 129.5, 129.4, 128.0, 126.7, 125.1, 124.5, 114.9, 61.4, 56.0, 51.0, 29.7, 27.8, 23.7, 19.3, 10.3. Combustion analysis: calculated for C23H31NO3S: C 68.8, H 7.8, N 3.5; found: C 66.6, H 7.9, N 3.8.
2-(N,N-Di-n-Propylamino)-6-(4-Methoxyphenylsulfonylmethyl)-Tetralin (GR218231). GR218231 was prepared in the same manner as was described for compound 6, using compound 5 as the starting material. After the reaction was completed, the reaction mixture was washed with 50 mL 1N NaOH. The water layer was extracted twice with 25 mL CH2Cl2. The combined organic layers were washed with 25 mL water, dried on MgSO4, and concentrated in vacuo. The crude product was dissolved in 50 mL absolute ethanol and acidified with 37% HCl until the pH was <1. The solvent was evaporated in vacuo. The residue was taken up in 75 mL diethyl ether and stirred for 72 h. The white suspension was filtered and washed with ether. The residue was further purified by flash column chromatography over a silica gel 60 column, using CH2Cl2/n-hexane/triethylamine (20:78:2) as the eluent. The product was obtained as a white solid in 50% yield. Melting point: 97°C (free base). λmax (40% acetonitrile in 0.1N NaH2PO4): 243 nm. 1H NMR (200 MHz, CDCl3): δ 7.57 (d, 2H, J = 9.0 Hz, Ar), 6.99–6.75 (m, 5H, Ar), 4.19 (s, 2H, CH2S), 3.87 (s, 3H, CH3O), 3.00–2.71 (m, 5H, CH2+CHN), 2.47 (t, 4H, J = 7.5 Hz, CH2N), 2.0 (m, 1H, CH2), 1.62–1.38 (m, 5H, CH2), 0.88 (t, 6H, J = 7.3 Hz, CH3). 13C NMR (50 MHz, CDCl3): δ 162.2, 135.8, 135.3, 129.5, 129.3, 128.3, 128.1, 126.4, 123.9, 112.5, 61.3, 55.3, 54.1, 51.1, 30.4, 28.1, 24.1, 20.5, 10.4. Combustion analysis: calculated for C24H33NO3S: C 69.4, H 8.0, N 3.4; found: C 69.1, H 7.9, N 3.4.
Radiochemistry
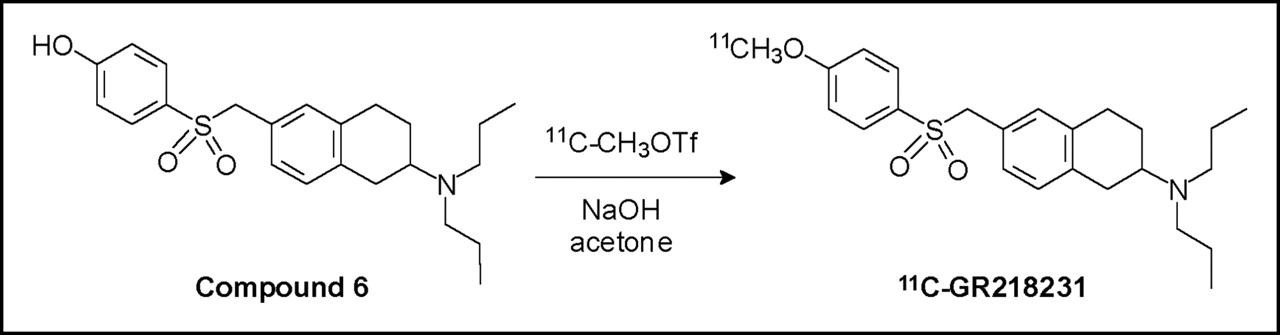
2-(N,N-Di-n-Propylamino)-6-(4-11C-Methoxyphenylsulfonyl- methyl)Tetralin (11C-GR218231). 11C-Methane was produced by the 14N(p,α)11C nuclear reaction, using a Scanditronix MC17 cyclotron to irradiate the N2 + 5% H2 target gas with 17-MeV protons. 11C-Methyl iodide was prepared from 11C-methane (14) and converted into 11C-methyl triflate (15) via procedures that have been described previously in the literature. 11C-Methyl triflate was transported by a stream of argon (20 mL/min) into a minivial containing 0.5 mg precursor 6 and 2.5 μL 0.5N NaOH dissolved in 300 μL methanol at room temperature. After the trapping of 11C-methyl triflate was complete, the solvent was evaporated at 80°C with the aid of an argon flow. The residue was dissolved in 1.5 mL 40% acetonitrile in 0.1 mol/L NaH2PO4 and injected into a semipreparative reversed-phase HPLC column. Purification of the product was accomplished, using an alphabond C18 column (10 μm, 7.8 × 300 mm) with 40% acetonitrile in NaH2PO4 as the eluent at 3 mL/min flow rate. The radioactive product with a retention time of 12 min was collected (retention time of compound 6, 7 min). The product was diluted with 15 mL water and transferred to an Oasis SepPak cartridge (Waters). The cartridge was washed with 5 mL water and eluted with 0.5 mL ethanol followed by 4.5 mL 0.9% NaCl solution, affording the tracer solution ready for injection in animal experiments. In this manner, 11C-GR218231 with a specific activity of 15 ± 10 GBq/μmol was prepared in 53% ± 8% (n = 18) decay-corrected radiochemical yield (based on 11C-methyl triflate). The total synthesis time, including the preparation of 11C-methyl triflate, HPLC purification, and formulation, was approximately 40 min. The identity of the product was confirmed by coinjection with a reference sample on reversed-phase HPLC. The radiochemical purity of the product was >99%, as determined by analytic reversed-phase HPLC using a Platinum C18 column (5 μm, 4.6 × 250 mm) and 60% MeOH in 0.1 mol/L NaH2PO4 as the eluent at a flow rate of 1 mL/min (retention time, 11.5–12 min).
Biodistribution Studies
Male Wistar rats (236–330 g) were anesthetized by intraperitonial injection of a mixture of ketamine (25 mg/kg) and medetomidine (0.2 mg/kg). The rats were injected into the tail vein with 28 ± 11 MBq of 11C-GR218231. After 45 min of tracer distribution, the animals were sacrificed by extirpation of the heart. The relevant tissues were excised and weighed. Tracer uptake in the tissues was measured using a γ-counter and expressed as standardized uptake value (SUV), which was defined as:
 Values were statistically analyzed using the 1-way ANOVA procedure followed by a Bonferroni multiple comparisons post hoc test. Differences between groups were considered statistically significant at P < 0.01.
Values were statistically analyzed using the 1-way ANOVA procedure followed by a Bonferroni multiple comparisons post hoc test. Differences between groups were considered statistically significant at P < 0.01.
In the blocking experiments, the specific binding sites were saturated by injection of 0.2 mg/kg or 2.5 mg/kg unlabeled GR218231 into the tail vein 5 min before administration of the tracer.
In modulation experiments, the P-glycoprotein pump was inhibited by injection of 50 mg/kg cyclosporine A into the tail vein 30 min before tracer injection.
The cyclosporine A dose-escalation studies were performed several months after the other experiments on a different batch of Wistar rats. In these studies, P-glycoprotein was modulated with 0, 10, 20, 30, 40 and 60 mg/kg cyclosporine A.
All animal studies were performed in compliance with the national law on animal experiments. The protocols were approved by the Animal Ethics Committee of the Groningen University.
Phosphor Storage Imaging
After 45 min of 11C-GR218231 distribution, isolated rat brains were frozen in liquid nitrogen and stored on dry ice. The frozen brain samples were cut into 80-μm-thick slices, using a microtome at −6°C. Brain slices were placed on slides and covered with a multipurpose Cyclone Storage Phosphor Screen (Packard Instruments). After exposure for 24 h, the phosphor screens were scanned using a Cyclone imaging system (Packard Instruments).
RESULTS
Chemistry
The labeling precursor (compound 6) and a reference sample of GR218231 were prepared as outlined in Figure 1. First, 6-bromo-2-tetralone (compound 1) was converted into compound 2 via a reductive alkylation with di-n-propylamine. Although reductive alkylations of ketones with secondary amines can be difficult due to steric hindrance (16), compound 2 could be produced in this manner in a yield of 38%. Next, compound 2 was lithiated via a metallo-dehalogenation reaction with n-butyllithium. The organolithium intermediate was carbonylated in situ with DMF and the resulting aldehyde was reduced with NaBH4. This 3-step procedure gave compound 3 in 53% yield. Alcohol function in compound 3 was converted into a chloride atom by reaction with thionyl chloride. The crude chloride was reacted with either 4-mercaptophenol or 4-mercaptoanisol in the presence of K2CO3 and Kryptofix 2.2.2. as a phase-transfer catalyst. This 2-step reaction produced compounds 4 and 5 in 71% and 72% yield, respectively. 4-Mercaptophenol contains a hydroxyl and a thiol group that, in theory, can act as the nucleophile in this substitution reaction. Formation of the S-substituted product in the reaction with 4-mercaptophenol was confirmed by the 1H NMR chemical shifts of the methylene group adjacent to the sulfur atom, which was almost identical to the signal of the corresponding protons in compound 5 (δ 3.92 vs. 3.94). Under the reaction conditions applied, no evidence for the formation of the O-substituted product could be observed in the 1H NMR spectrum of crude compound 4. Finally, compounds 4 and 5 were oxidized with m-chloroperbenzoic acid. When this reaction was performed on the free amine of compounds 4 or 5, oxidation predominantly occurred at the nitrogen atom, yielding unidentified oxidation products, which probably were N-oxides, hydroxylamines, and nitroso compounds. This obstacle, however, could easily be overcome by protonation of the amine group with HCl before the oxidation reaction. The oxidation of the ammonium salts of compounds 4 and 5 resulted in the formation of the labeling precursor (compound 6) and reference compound GR218231 in 43% and 50% yield, respectively.
Radiochemistry
The labeling of 11C-GR218231 was accomplished by alkylation of compound 6 with 11C-methyl triflate, as depicted in Figure 2. After the phenol group of compound 6 was deprotonated with NaOH, 11C-methyl triflate was bubbled through the precursor solution, the solvent was evaporated, and the product was purified by reversed-phase HPLC. For in vivo studies, the product was formulated in 10% ethanol in saline using a C18 solid-phase-extraction cartridge. The entire labeling, purification, and formulation procedure required approximately 40 min from the end of bombardment. 11C-GR218231 was prepared with a specific activity of 15 ± 10 GBq/μmol (at the end of synthesis) in 53% ± 8% decay-corrected radiochemical yield (n = 18, based on 11C-methyl triflate). Analytic HPLC showed that the labeled product had a radiochemical purity of >99%.

Radiolabeling of 11C-GR218231 via methylation of desmethyl precursor with 11C-methyl triflate.
Biodistribution
The resolution of our clinical PET camera does not allow the discrimination of the relevant organs and individual brain regions. Therefore, 11C-GR218231 was further evaluated in biodistribution studies instead of PET studies. A 45-min tracer distribution interval was chosen for the biodistribution studies. This time point should offer sufficient distribution time to allow clearance of unbound tracer, while maintaining reliable counting statistics of the short-lived radionuclide in the small organs and brain areas of the rat. Table 1 presents the distribution of 11C-GR218231 in various peripheral tissues and brain regions at 45 min after intravenous injection of the tracer, as determined by ex vivo γ-counting. Highest tracer uptake was observed in stomach, pancreas, kidney, and submandibular gland. The uptake of 11C-GR218231 in the brain was low and homogeneously distributed, as the SUV in all brain areas ranged from 0.19 to 0.23. To determine whether the tracer uptake was due to specific binding to the receptor, the receptor was saturated with unlabeled drug by treating the animals with 0.2 or 2.5 mg/kg GR218231 5 min before administration of the tracer. Pretreatment of the animals with 0.2 mg/kg unlabeled GR218231 did not result in any significant reduction in tracer uptake in any peripheral tissue or brain area (P = 1.0). Increasing the dose of unlabeled GR218231 to 2.5 mg/kg did not significantly affect the peripheral or cerebral tracer uptake either (P = 1.0 vs. controls and P = 1.0 vs. the 0.2 mg/kg group; data not shown) and, thus, specific binding could not be demonstrated.
Distribution of 11C-GR218231 in Various Peripheral Organs and Brain Regions 45 Minutes After Injection of Tracer in Wistar Rats (n = 4)
Modulation of P-Glycoprotein
To test whether the low cerebral uptake of 11C-GR218231 was due to the fact that the tracer is a substrate of the P-glycoprotein pump, the effect of inhibition of the efflux pump on the uptake of 11C-GR218231 was investigated. For this purpose, Wistar rats were treated with the P-glycoprotein modulator, cyclosporine A (50 mg/kg), 30 min before injection of the tracer. As shown in Table 1, modulation of P-glycoprotein with cyclosporine A caused a tremendous increase in cerebral uptake of 11C-GR218231 in all brain areas (P < 0.001). In the whole brain, overall tracer uptake increased by 12-fold, whereas regional increases as high as 14-fold were observed. After modulation of the P-glycoprotein pump, highest tracer uptake was found in striatum and neocortex. The differences in tracer uptake between brain areas, however, were not statistically significant (P > 0.08). In peripheral organs, tracer uptake was significantly enhanced in spleen (+80%, P = 0.001), kidney (+57%, P = 0.001), and heart (+38%, P = 0.005) after treatment with cyclosporine A.
Phosphor Storage Imaging
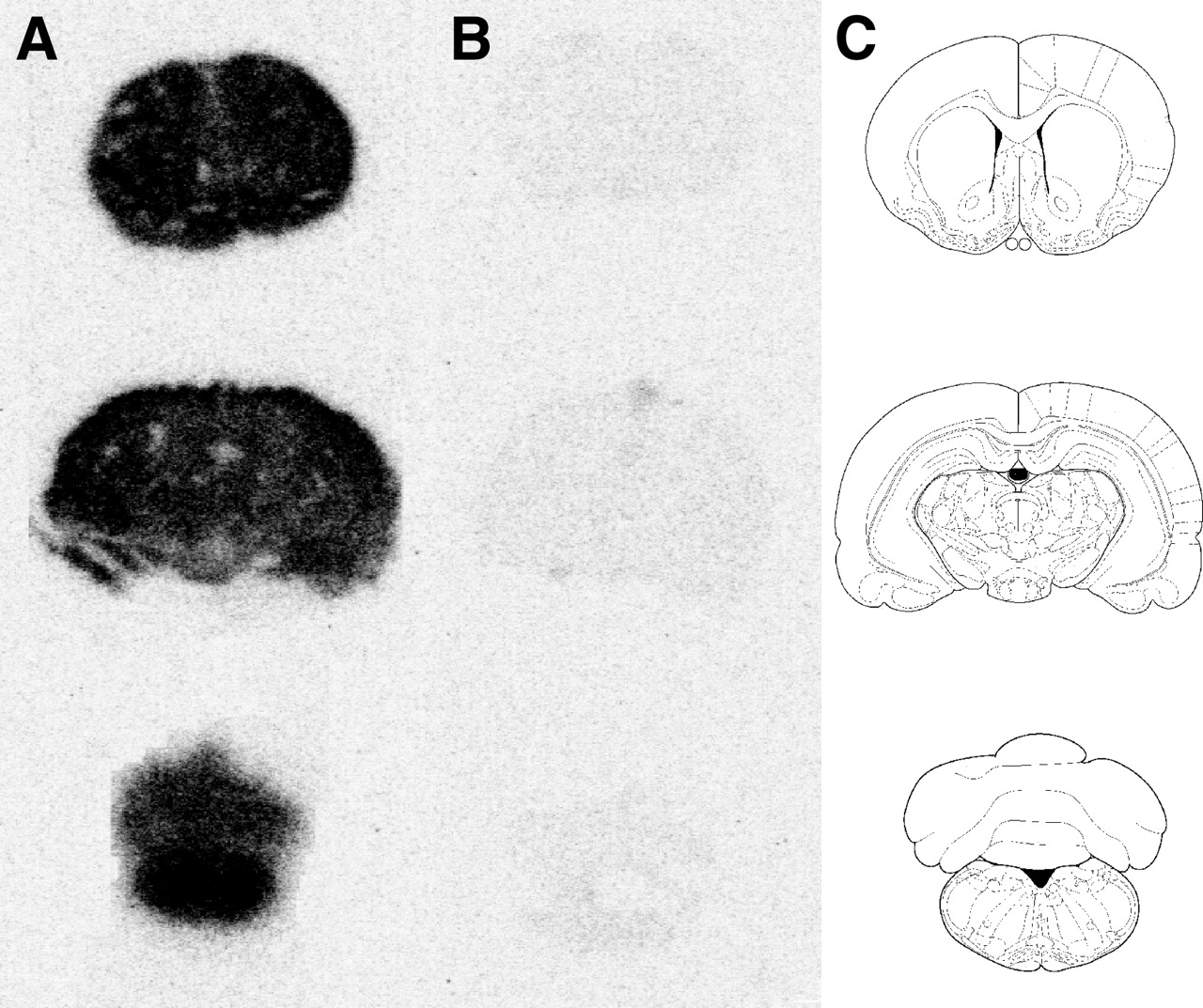
Representative phosphor storage images of the 11C-GR218231 uptake in the brain of control rats and in the brain of rats that were treated with cyclosporine A are depicted in Figure 3. This figure clearly demonstrates the large increase in cerebral tracer uptake after modulation of P-glycoprotein. Both in control rats and in P-glycoprotein–modulated rats, the tracer is homogeneously distributed in the brain.

Phosphor images of 11C-GR218231 binding in coronal 80-μm-thick brain slices of 50 mg/kg cyclosporine A–treated (A) and control (B) rats. Each row depicts slices at different Bregma levels (1.0, −4.8, and −12.8) and the corresponding template (C).
Dose-Escalation Studies
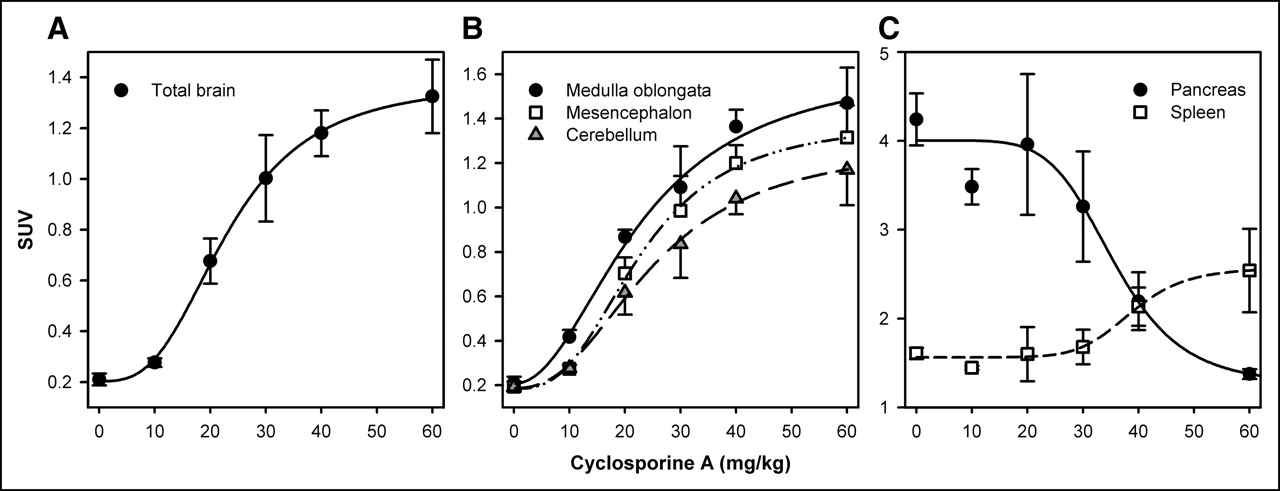
As is depicted in Figure 4A, uptake of 11C-GR218231 in the brain showed a sigmoidal relation with cyclosporine A dosage. The cyclosporine A dosage at which 50% of the maximum P-glycoprotein modulation was achieved (ED50) was 23.3 ± 0.6 mg/kg for the total brain. Only small differences in the effect of cyclosporine A dosage on the magnitude of 11C-GR218231 uptake were observed between individual brain regions (Fig. 4B). The ED50 showed little variation between brain regions and ranged from 21.7 ± 0.1 mg/kg for striatum to 26.4 ± 3.4 mg/kg for the olfactory bulbs (Table 1). In peripheral organs, sigmoidal relations between tracer uptake and modulator dosage were observed only in pancreas and in spleen (Fig. 4C), indicating that tracer uptake is influenced by P-glycoprotein in these organs. In spleen, tracer uptake increased with increasing cyclosporine A dosage (ED50, 38.4 ± 2.4 mg/kg). Remarkably, 11C-GR218231 uptake decreased with increasing mod-ulator dosage in pancreas (ED50, 36.0 ± 4.4 mg/kg). Tracer uptake in heart (P = 0.4) and kidney (P = 0.1) could not be correlated with modulator dosage by a sigmoidal fit.

Uptake of 11C-GR218231 in total brain (A), selected brain regions (B), and pancreas and spleen (C) for various dosages of cyclosporine A. Tracer uptake is expressed as the mean SUV ± SE. Plots of all tissues were fitted by a sigmoidal curve that is described by the mathematic expression: 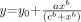 (total brain: r2 = 0.87, P < 0.0001; medulla oblongata: r2 = 0.88, P < 0.0001; mesencephalon: r2 = 0.89, P < 0.0001; cerebellum: r2 = 0.85, P < 0.0001; spleen: r2 = 0.53, P = 0.006), except for the fitted curve for pancreas, which is described by the equation:
(total brain: r2 = 0.87, P < 0.0001; medulla oblongata: r2 = 0.88, P < 0.0001; mesencephalon: r2 = 0.89, P < 0.0001; cerebellum: r2 = 0.85, P < 0.0001; spleen: r2 = 0.53, P = 0.006), except for the fitted curve for pancreas, which is described by the equation:  (r2 = 0.55, P = 0.005).
(r2 = 0.55, P = 0.005).
DISCUSSION
In an attempt to develop an imaging method for the dopamine D3 receptor using PET, we labeled the selective dopamine D3 antagonist GR218231 with 11C. 11C-GR218231 was reproducibly synthesized in good radiochemical yields that were sufficient for preclinical and clinical studies. Biodistribution studies of 11C-GR218231 in rats showed that the tracer uptake was homogeneously distributed throughout the central nervous system. The tracer uptake did not correspond to the distribution of the dopamine D3 receptor, which is heterogeneously distributed in the brain, with highest concentrations in the islands of Calleja, nucleus accumbens, ventral pallidum, substantia nigra, and lobes 9 and 10 of the cerebellum (17), which should have led to elevated uptake in the striatum, mesencephalon, and caudal cerebellum. Tracer uptake could not be blocked by pretreatment with 0.2 mg/kg unlabeled GR218231. To ensure that the dopamine D3 receptors were completely saturated with unlabeled GR218231, the blocking experiments were repeated using a 2.5 mg/kg dose of the antagonist, but even at this high dose of blocker specific binding could not be demonstrated. Similar results were obtained by Sovago et al., who investigated the ligand 11C-RGH-1756 for imaging the dopamine D3 receptors in monkey brain (18). Despite promising binding characteristics of 11C-RGH-1756 in vitro, the ligand showed a homogeneous distribution with low levels of specific binding in vivo. As a possible explanation, the authors suggested that the dopamine D3 receptors could have been occupied by endogenous dopamine, thus preventing binding of the radioligand. An alternative explanation could be the low abundance of the dopamine D3 receptor, which leads to low levels of specific binding that could have been obscured by the nonspecific binding.
The most remarkable observation in this study was the low cerebral uptake of 11C-GR218231 in control rats, which was much lower than expected on basis of the lipophilicity of the tracer (logD = 2.2 (7)) (2). These results appear to contradict pharmacologic studies that show that GR218231 can enter the brain of rats and bind to the dopamine D3 receptor, resulting in GR218231-mediated attenuation of the dopaminergic behaviors induced by the preferential dopamine D3 receptor agonists 7-OH-DPAT and PD128,907 (6,8). Apparently, GR218231 shows sufficient brain uptake at pharmacologic dosing but not at the tracer dosing that was used in this study.
A possible explanation for the low uptake of 11C-GR218231 in the rat brain at tracer dosing could be that the tracer is extruded from the brain by an efflux pump in the blood–brain barrier. The pharmacologic effect of GR218231 that was observed at a high dosing could then be due to saturation of the efflux pump, resulting in incomplete removal of drug from the brain. We hypothesized that P-glycoprotein could be the efflux pump that was responsible for the low brain uptake of 11C-GR218231. P-glycoprotein is abundant in the blood–brain barrier, where it can extrude a variety of structurally unrelated drugs from the brain.
To test our hypothesis, the influence of P-glycoprotein on cerebral tracer uptake was investigated. P-glycoprotein modulation induced a 12-fold increase in the brain uptake of 11C-GR218231, proving that GR218231 is a substrate of the P-glycoprotein pump and that the low cerebral tracer uptake in control animals was indeed caused by the efflux activity of this pump. Although modulation of the P-glycoprotein pump dramatically enhanced brain uptake of 11C-GR218231, it did not result in any specific binding (data not shown).
To further evaluate the feasibility of 11C-GR218231 as a PET tracer for imaging of P-glycoprotein functionality, the sensitivity of tracer uptake for cyclosporine A dosage was investigated. Cerebral 11C-GR218231 uptake showed a sigmoidal correlation with the dosage of cyclosporine A, indicating that P-glycoprotein responsible for the efflux of the tracer from the brain and that P-glycoprotein–mediated transport is saturable. The ED50 of cyclosporine A for modulation of 11C-GR218231 uptake in the rat brain was 23.3 ± 0.6 mg/kg and varied little between individual brain regions. This could imply that P-glycoprotein is rather homogeneously distributed throughout the blood–brain barrier. The results of 11C-GR218231 are in agreement with previous animal experiments with the P-glycoprotein tracers 11C-verapamil and 11C-carvedilol, in which similar ED50 values were observed (19,20).
Modulation of P-glycoprotein with cyclosporine A caused a significant increase in tracer uptake not only in the brain but also in the spleen, kidney, and heart. In contrast, P-glycoprotein modulation resulted in a significant decrease in 11C-GR218231 uptake in the pancreas. A sigmoidal relationship between 11C-GR218231 uptake and cyclosporine A dosage was observed for the spleen and pancreas. Low levels of P-glycoprotein messenger RNA have been detected in these organs (21). Splenic P-glycoprotein supposedly is involved in the protection of the spleen by extrusion of toxic unconjugated bilirubin, which is formed during hemolysis (22). Thus, cyclosporine A–induced inhibition of P-glycoprotein could explain the increase in tracer uptake in the spleen in a similar manner as was described for the brain. In the pancreas, P-glycoprotein is located on the apical surface of small pancreatic ductules, where it is responsible for the excretion physiologic metabolites and chemotherapeutic drugs into the pancreatic duct (23). When secretion of the tracer from the small pancreatic tubules into the intestines is relatively slow, 11C-GR218231 will accumulate in the pancreatic duct, resulting in the high pancreatic uptake that was observed in control animals. Inhibition of P-glycoprotein would prevent the accumulation of the tracer in the pancreatic duct, causing an overall reduction in tracer uptake in the pancreas. This would explain the sigmoidal decrease in pancreatic tracer uptake in response to P-glycoprotein modulation.
In contrast to brain, spleen, and pancreas, tracer uptake in kidney and heart did not have a sigmoidal relationship with the cyclosporine A dosage. Tracer uptake in kidney and heart showed a slight decrease after modulation with 10 and 20 mg/kg cyclosporin A but showed an increase at higher dosages. The effect of cyclosporine A pretreatment on tracer uptake in kidney and heart could be due to cyclosporine A–mediated variations in the plasma clearance (24). In fact, average plasma levels of the tracer showed a similar pattern in the response to cyclosporine A dosage as tracer uptake in kidney and heart. Therefore, 11C-GR218231 uptake does not seem to be significantly affected by P-glycoprotein transport in these organs but predominantly by cyclosporine A–mediated effects on plasma clearance.
Taken together, these results show that 11C-GR218231 is not a suitable PET tracer for dopamine D3 receptor imaging, but it could be useful for imaging of P-glycoprotein in the blood–brain barrier. A PET method for imaging of P-glycoprotein functionality could provide a useful tool to improve pharmacotherapeutic treatment with drugs that exert their activity in the central nervous system. Information about the functionality of the P-glycoprotein pump could be of major clinical and pharmaceutical importance, since therapeutic drugs can become ineffective when drug levels in the brain are reduced by the efflux activity of the P-glycoprotein. Modulation of the P-glycoprotein pump may restore the efficacy of these drugs. P-glycoprotein can also be involved in undesired drug–drug interactions when multiple drugs that are substrates of the efflux pump are administered simultaneously. Competition for the efflux pump results in increased concentrations of the drugs in the brain, which can lead to unexpected neurotoxic side effects. Identification of drugs that are substrates of P-glycoprotein could prevent these harmful drug–drug interactions.
Imaging of P-glycoprotein could also be applied to determine the status of the efflux pump—for example, in neurodegenarative diseases. Recently, Kortekaas et al. used the P-glycoprotein substrate 11C-verapamil and PET to show that P-glycoprotein activity in the affected midbrain of patients with Parkinson’s disease is reduced compared with that of healthy volunteers, suggesting a causative relation between P-glycoprotein dysfunction and Parkinson’s disease (5).
CONCLUSION
The dopamine D3 receptor antagonist GR218231 was reliably labeled with 11C in good radiochemical yield. 11C-GR218231 does not appear to be suited as a PET tracer for dopamine D3 receptor imaging, as uptake of the tracer in the brain was low, did not correspond to the regional distribution of the dopamine D3 receptor, and appeared to be nonspecific. 11C-GR218231 proved to be an excellent substrate of the P-glycoprotein efflux pump in the blood–brain barrier, resulting in rapid extrusion of the tracer from the brain. Modulation of the P-glycoprotein pump with cyclosporine A induced an enormous increase in the cerebral uptake of 11C-GR218231. These findings indicate that 11C-GR218231 might be useful as a PET tracer for the visualization and quantification of P-glycoprotein function in the blood–brain barrier. An in vivo imaging technique to monitor the P-glycoprotein function in the blood–brain barrier could aid the development of new drugs and novel strategies to deliver drugs to the brain and might be able to identify undesirable drug–drug interactions (3).
Footnotes
Received Nov. 16, 2004; revision accepted Apr. 15, 2005.
For correspondence contact: Erik F.J. de Vries, PhD, PET Center, University Medical Center Groningen, P.O. Box 30,001, 9700 RB Groningen, The Netherlands.
E-mail: e.f.j.de.vries{at}pet.umcg.nl





{kind=link}
{kind=link}
{kind=link}
{kind=link}