


Abstract
P-glycoprotein (P-gp) regulates the ability of endogenous and exogenous compounds to cross the blood–brain barrier. We investigated whether PET tracers used clinically for studying brain function are affected by P-gp. Methods: Modulation of 8 radioligands by P-gp was assayed in mice by evaluating the effect of treatment with cyclosporine A (CsA) on uptake into the brain (assay 1) and the effect of treatment with a cold ligand of the corresponding radioligand on uptake of 11C-verapamil, a representative radioligand for P-gp (assay 2). Brain-to-blood ratios were also examined as the other index to correct the delivery of radioligands. The radioligands investigated were 11C-TMSX (adenosine A2A receptor), 11C-MPDX (adenosine A1 receptor), 11C-PK11195 (peripheral benzodiazepine receptor), 11C-flumazenil (central benzodiazepine receptor), 11C-raclopride (dopamine D2-like receptor), 11C-pyrilamine (histamine H1 receptor), 11C-PIB (amyloid plaque), and 11C-donepezil (acetylcholine esterase). Results: In assay 1, CsA treatment increased both the uptake and the brain-to-blood ratio of 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil among the 8 radioligands. In assay 2, in which 4 cold ligands were examined, cold verapamil slightly increased the brain-to-blood ratio of 11C-verapamil, but TMSX, MPDX, and MPPF did not increase either parameter. Conclusion: Assay 1 was suitable for evaluating the P-gp modulation of radioligands. Among the 8 radioligands investigated, 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil were modulated by P-gp.
The permeability of endogenous and exogenous compounds across the blood–brain barrier is regulated by transporters such as P-glycoprotein (P-gp), multidrug-resistance–associated protein, and organic anion transporters. Furthermore, these transporters can also be expressed on the membrane of tumor cells. Chemotherapeutic treatment of cancer patients is often unsuccessful because of the involvement of various mechanisms leading to multidrug resistance, in which P-gp is involved in effluxion of drugs. PET and SPECT can be used to investigate the functionality of these transporters in the human brain and in tumors in vivo (1–4). So far, several radioligands for PET and SPECT have been proposed (5–7), and 11C-verapamil and 99mTc-sestamibi have been used in clinical studies (8–13). On the other hand, some radioligands for mapping neuroreceptors are also substrates for P-gp: 11C-carazolol and 18F-fluorocarazolol for β-adrenergic receptor (14) and 18F-MPPF for serotonin 5-HT1A receptor (15). These radioligands showed a relatively fast clearance of radioactivity from the brain of rodents; however, the uptake was remarkably enhanced on treatment with cyclosporine A (CsA), a so-called modulator of P-gp (Table 1). These phenomena result from inhibition of radioligand efflux from the endothelial membrane into plasma. When we compared the in vivo brain kinetics of 2 σ1-receptor radioligands, 3H-pentazocine and 11C-SA4503, in mice, the former showed a much faster clearance and a much lower specific binding than the latter despite similar affinities for σ1-receptors (16). The different kinetics were explained by the finding that 3H-pentazocine was modulated by P-gp but that 11C-SA4503 was not. In the case of 11C-GR218231, a dopamine D3 receptor antagonist, brain uptake was low because of P-gp, and no receptor-specific binding was found (17). Therefore, it is important to the development of new radioligands for PET or SPECT whether they are modulated by P-gp. It is also of interest whether the radiotracers presently used in clinical studies are modulated by P-gp.
Effects of CsA on Brain Uptake of Radioactivity After Injection of Each Radioligand into Mice
So far, the modulation of radioligands by P-gp in vivo has been evaluated in rodents treated with CsA. In this study, we investigated 2 assays for evaluating P-gp modulation of radioligands in vivo. Assay 1 was a standard method previously used (8,14–17). Mice were intravenously treated with CsA, and the brain uptake of a radioligand was examined to see whether it could be enhanced. In assay 2, we examined whether uptake of 11C-verapamil, a representative probe for P-gp, was affected by cold ligand. When cold ligands directly modulate P-gp or compete with 11C-verapamil in drug efflux by P-gp, uptake of 11C-verapamil will be enhanced. If assay 2 could be used for the evaluation of candidate PET radioligands, they could be screened in vivo before radiolabeling.
For the assays, we selected 8 radioligands for PET that have been used clinically at the Tokyo Metropolitan Institute of Gerontology (TMIG) or Tohoku University: 11C-TMSX, 11C-MPDX, 11C-PK11195, 11C-flumazenil, 11C-raclopride, 11C-pyrilamine, 11C-PIB, and 11C-donepezil (Fig. 1). These 8 radioligands were compared with 11C-verapamil and other tracers previously reported. 11C-TMSX and 11C-MPDX are xanthine-type compounds for mapping adenosine A2A and A1 receptors, respectively, and were originally developed at the TMIG (18–20). 11C-Pyrilamine has been used for the imaging of histamine H1 receptors in at least 2 PET centers, including Tohoku University (21,22). 11C-Donepezil PET is being tested clinically for imaging acetylcholine esterase at Tohoku University (23). The labeled position of the 11C-donepezil used clinically differs from that of the 11C-donepezil prepared by De Vos et al. (24). 11C-PIB has been developed for imaging amyloid plaque (25) and used at many PET centers, and the other 3 tracers, 11C-PK11195, 11C-flumazenil, and 11C-raclopride, have been widely used. All of the selected radioligands show relatively fast brain kinetics.
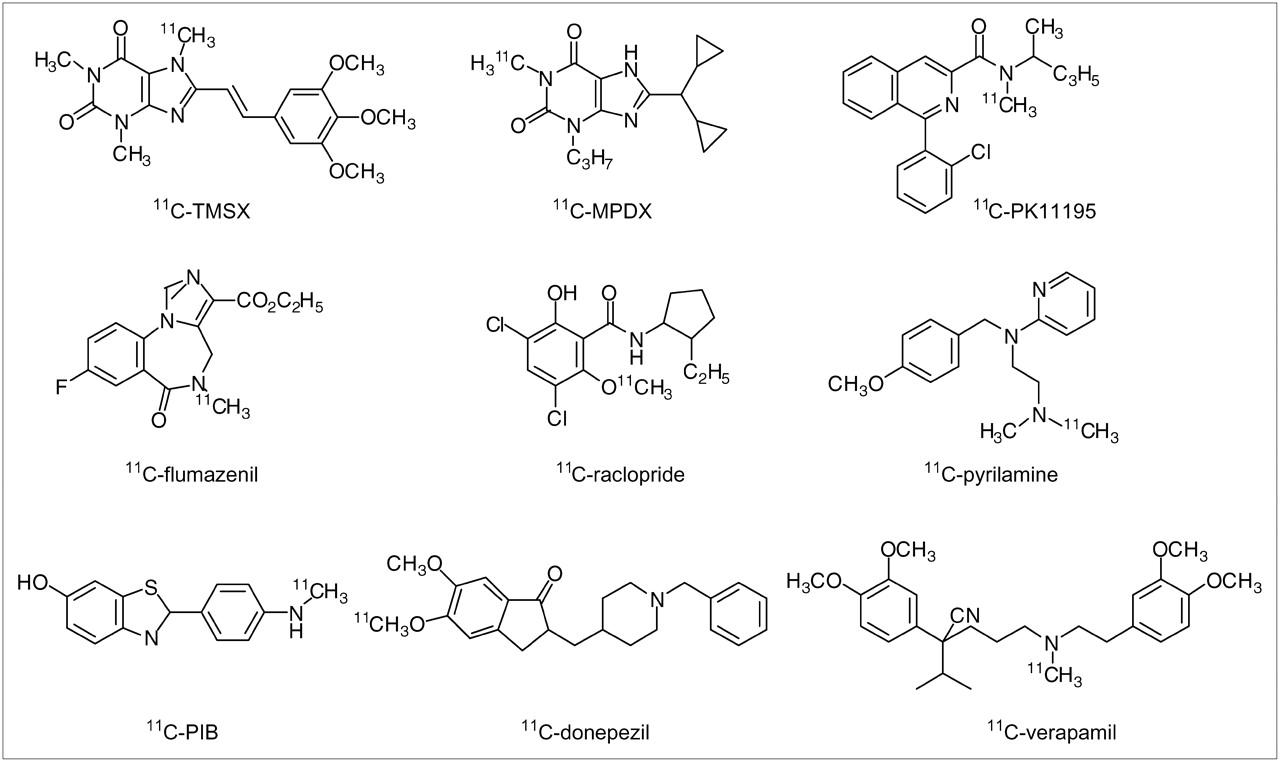
Chemical structures of radioligands investigated.
MATERIALS AND METHODS
General
CsA (Sandimmun) was purchased from Novartis Pharma; verapamil hydrochloride, from Sigma-Aldrich; MPPF dihydrochloride, from Research Biochemicals International; and dimethyl sulfoxide (DMSO), from Wako Pure Chemical Industries. 11C-TMSX (26), 11C-MPDX (27), 11C-PK11195 (28), 11C-flumazenil (29), 11C-raclopride (30), 11C-pyrilamine (31), 11C-PIB (32), 11C-donepezil (23), and 11C-verapamil (33) were prepared at the TMIG as described previously (23,26–33).
Male ddY mice were purchased from Tokyo Laboratory Animals. All experiments with animals were approved by the Animal Care and Use Committee of the TMIG.
Assay 1
The effects of CsA on uptake of radioactivity into the brain were investigated for all 8 radioligands—11C-TMSX, 11C-MPDX, 11C-PK11195, 11C-flumazenil, 11C-raclopride, 11C-pyrilamine, 11C-PIB, and 11C-donepezil—and for 11C-verapamil, a standard radioligand for P-gp, as a reference. Assay 1 is a standard method (8,14–17), but the time course of the effect of CsA has not been reported. Therefore, first, the time course of the effect of CsA was investigated using 11C-verapamil to determine the optimal period before injection of the tracer. The mice received an intravenous injection of CsA, which was dissolved at a concentration of 10 mg/mL in physiologic saline. The dose of CsA was 50 mg/kg, and the volume of the mixture was 5 mL/kg. Thirty, 60, 120, and 240 min later, 11C-verapamil was injected via the tail into 4 groups of mice. Control mice were treated with physiologic saline at 5 mL/kg and then given 11C-verapamil 30 min later. They were killed by cervical dislocation 30 min after the injection. The 30-min period was chosen because uptake of 11C-verapamil into the brain remained constant for the first 60 min after injection (6). Blood was collected by heart puncture using a syringe, and the brain was harvested. The samples were measured for 11C-radioactivity with an auto-γ-counter and weighed. Tissue uptake of radioactivity was expressed as a percentage of the injected dose per gram of tissue (%ID/g).
In the same way, each of the other 8 radioligands was intravenously injected into the mice 30 min after intravenous injection of CsA. Control mice were given physiologic saline. Fifteen minutes after injection of each radioligand, the mice were killed by cervical dislocation and blood and brain were obtained as described above. The 15-min period was chosen because all 8 radioligands showed a similar pattern of clearance, that is, the highest uptake was observed within 15 min after injection. In the case of 11C-TMSX, the target tissue (rich in adenosine A2A receptors) is the striatum (18,26), as it is in the case of 11C-raclopride. Peripherally, 11C-TMSX specifically bound to the adenosine A2A receptors in the heart and muscle (34). Therefore, the heart and muscle were also obtained. The brains of the 11C-TMSX– or 11C-raclopride–injected mice were further divided into the cerebral cortex, striatum, cerebellum, and remainder, and those of the 11C-MPDX-injected mice, into the cerebral cortex, hippocampus, cerebellum, and remainder. Uptake of 11C-MPDX was relatively high in the hippocampus (27,35). Tissue uptake of radioactivity was expressed as %ID/g.
Assay 2
The effects of cold ligands on uptake of 11C-verapamil were investigated. We selected verapamil, TMSX, and MPDX as cold ligands and MPPF as a reference ligand that was modulated by P-gp (15). Cold verapamil was dissolved at 0.2 or 1.0 mg/mL in physiologic saline, and TMSX and the other 2 ligands were dissolved at 0.1 and 0.2 mg/mL, respectively, in a mixture of physiologic saline and DMSO (1/1, v/v). The dose of verapamil was 1 or 5 mg/kg, and the doses of TMSX and the other 2 ligands were 0.5 and 1 mg/kg. The volume injected was 5 mL/kg for all ligands.
The mice received an intravenous injection of each of the 4 cold ligands. Control mice for the verapamil group received physiologic saline, and control mice for the other groups received a mixture of physiologic saline and DMSO (1/1, v/v) at a concentration of 5 mL/kg. Thirty or 10 min after injection of the verapamil and each of the other ligands, 11C-verapamil was intravenously injected. The 30-min period was selected for verapamil because uptake of 11C-verapamil remained constant for the first 60 min (6), and the 10-min period was selected for the others because 11C-TMSX, 11C-MPDX, and 18F-MPPF rapidly washed out from the brain after the injection (15,26,27). The mice were killed by cervical dislocation 30 min after injection, and blood and brain were obtained. In the case of TMSX, the heart and muscle were also obtained. The brains of TMSX-pretreated mice were divided into the cerebral cortex, striatum, cerebellum, and remainder, and those of the MPDX- or MPPF-pretreated mice, into the cerebral cortex, hippocampus, cerebellum, and remainder. Tissue uptake of radioactivity was expressed as %ID/g.
RESULTS
Assay 1
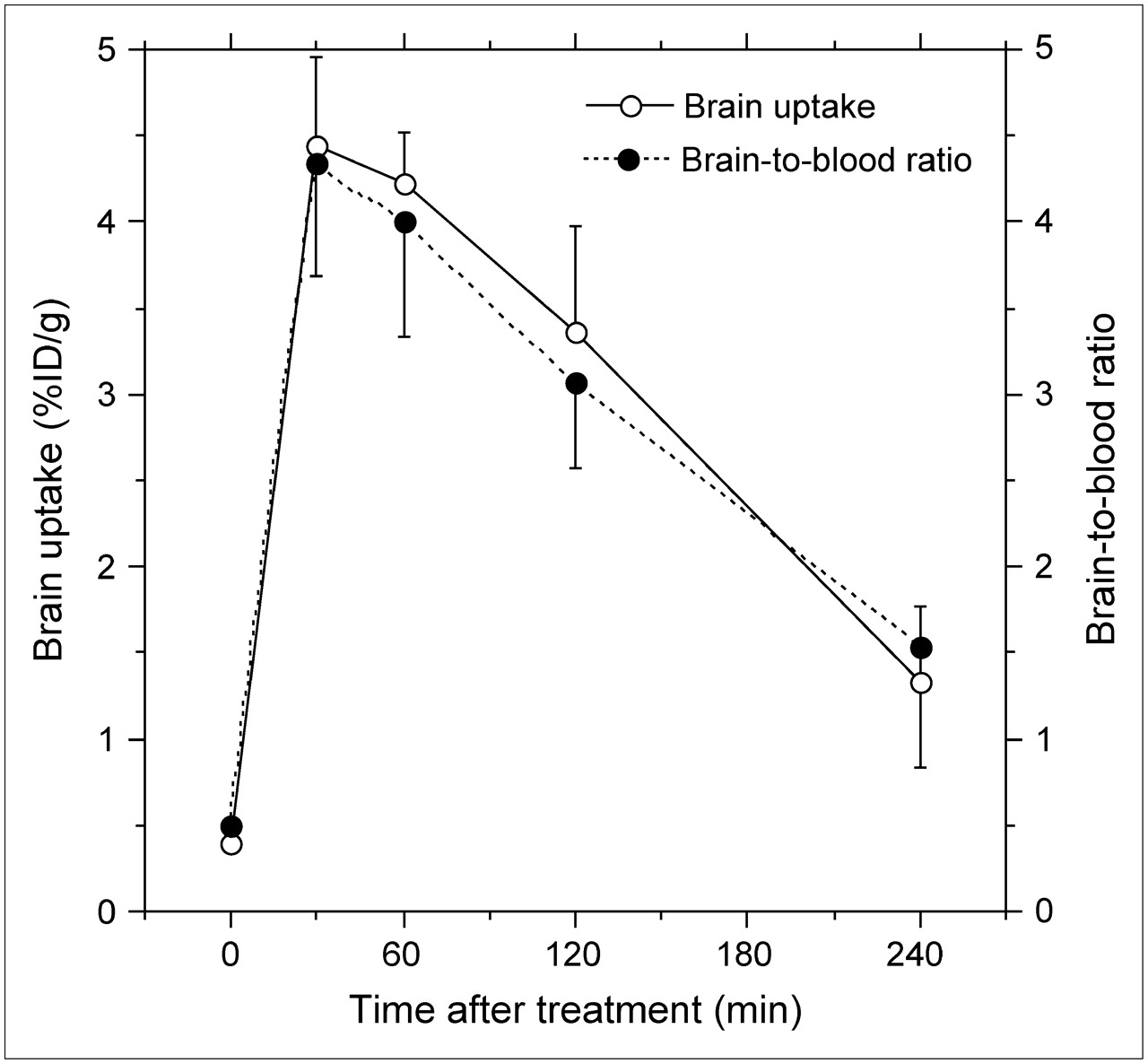
To determine the optimal time after treatment with CsA, the time course of the effects of CsA on uptake of 11C-verapamil was investigated 4 h after the treatment. Figure 2 shows that the largest effect was observed at 30 min (1,105% of the control) and that the effects gradually decreased (293% of the control at 4 h). The effects on the ratio of brain to blood were slightly less than those on uptake into the brain.

Time course of effects of CsA on brain uptake of 11C-verapamil and brain-to-blood ratios. Data are mean ± SD (n = 5).
The effects of treatment with CsA on the 9 radioligands are summarized in Table 1. The effects on another 5 radioligands previously reported are also included as references. Uptake was significantly enhanced for 6 of the 8 radioligands, in addition to 11C-verapamil. However, the brain-to-blood ratio for 11C-raclopride was not affected, and that for 11C-pyrilamine was significantly decreased. Thus, treatment with CsA significantly enhanced both uptake into the brain and the brain-to-blood ratio for 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil.
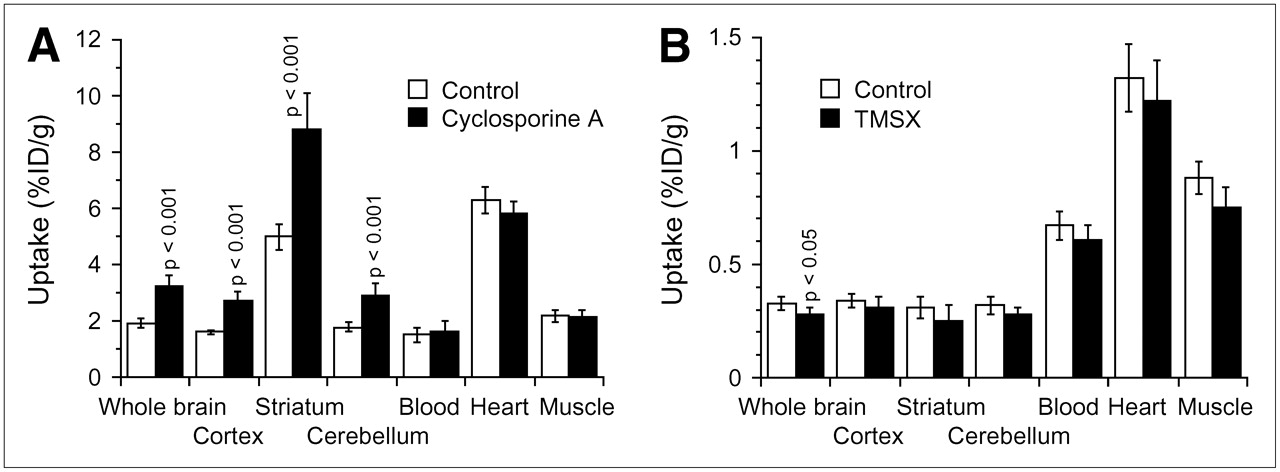
Figure 3A shows the effects of CsA on 11C-TMSX radioactivity levels in the 3 regions of the brain and in heart, muscle, and blood. In the brain, uptake of 11C-TMSX was enhanced to a similar extent both in the adenosine A2A receptor–rich striatum and in the adenosine A2A receptor–poor cerebral cortex and cerebellum but not in the heart and muscle. In the striatum, the receptor-specific uptake (%ID/g) determined by subtracting cerebellar uptake from striatal uptake significantly increased: 3.21 ± 0.33 versus 5.89 ± 0.94 (P < 0.001), whereas the target-to-nontarget ratio (striatum-to-cerebellum ratio) was not affected: 2.81 ± 0.17 versus 3.03 ± 0.23. Similar effects of CsA on the levels of radioactivity of 11C-MPDX or 11C-raclopride were also found in all 3 regions of the brain (data not shown).
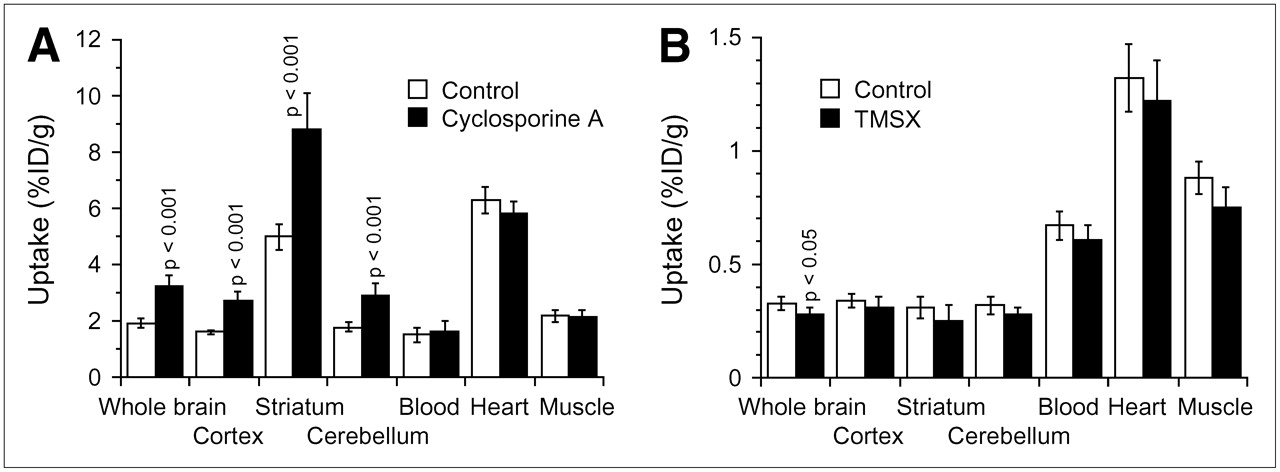
Uptake of radiotracers in brain, heart, and muscle in the 2 assays for evaluating P-gp modulation. (A) Assay 1: effect of treatment with CsA on uptake of 11C-TMSX. (B) Assay 2: effect of treatment with cold TMSX on uptake of 11C-verapamil. Data are mean ± SD (n = 5–6). Student t test was performed for comparison with control data.
Assay 2
Table 2 summarizes the effects of 4 cold ligands (verapamil, TMSX, MPDX, and MPPF) on uptake into the brain and the brain-to-blood ratio for 11C-verapamil. Cold verapamil at a dose of 5 mg/kg slightly increased the brain-to-blood ratio but not uptake. Uptake was slightly decreased by cold TMSX, but the brain-to-blood ratio was not affected. MPDX and MPPF had no effect on either parameter.
Effects of Cold Ligands on Brain Uptake of Radioactivity 30 Minutes After Injection of 11C-Verapamil into Mice
Figure 3B shows the effects of cold TMSX on the levels of radioactivity of 11C-verapamil in the 3 regions of the brain and in heart, muscle, and blood. There was a tendency for levels to decrease slightly in all tissues investigated, but the decrease was not significant. The other 2 cold ligands had no effect on the levels of radioactivity of 11C-verapamil in the 3 regions of the brain (data not shown).
DISCUSSION
Because of increased evidence that transporters have important roles in the movement of endogenous and exogenous compounds across the blood–brain barrier or the plasma membrane in tumors and other tissues, PET and SPECT have been used to investigate the functionality of transporters (1–4). However, it has also been reported that several radioligands for mapping neuroreceptors by PET are affected by P-gp (14,15), although many PET radioligands used clinically have not been evaluated for modulation by P-gp. In the present study, we investigated the possibility of modulation for 8 radioligands. All of the selected radioligands are in clinical use and show relatively fast brain kinetics, that is, an apparent equilibrium state is reached by 60 min. We excluded radioligands showing accumulation patterns, such as 11C-SCH 23390 (dopamine D1 receptor), 11C-N-methylspiperone (dopamine D2–like receptor), 11C-β-CFT (dopamine transporter), 11C-doxepin (histamine1 receptor), and 11C-deprenyl (monoamine oxidase-B inhibitor). Two assays were used to detect modulation by P-gp.
Assay 1 was a standard method (8,14–17). Because uptake of the tracer could be partially affected by treatment with CsA, modulation was evaluated using 2 indices, brain uptake and brain-to-blood ratio. The level of radioactivity in blood at a single time point does not reflect the area under the curve of the tracer in plasma; however, the brain-to-blood ratio can be used as an index as demonstrated for 11C-verapamil (8). In the present study, we also confirmed that the 2 indices of 11C-verapamil were affected to a similar extent (Fig. 2). We found that treatment with CsA significantly increased both indices for 4 radioligands: 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil. The enhancement was much less than that observed for 11C-verapamil or the other probe for P-gp, 11C-GR218231 (17), but was comparable to that induced by 11C-carazolol and 18F-fluorocarazolol (14) or 18F-MPPF (15) (Table 1). The increased uptake of 11C-TMSX or 11C-MPDX was observed to a similar degree in 3 brain regions independently of the densities of the corresponding receptors (Fig. 3A), as observed for 11C-carazolol, 18F-fluorocarazolol, and 18F-MPPF (15,16). It is known that 11C-TMSX binds peripherally to adenosine A2A receptors in the heart and muscle (34); however, uptake in these tissues was not influenced by treatment with CsA (Fig. 3A). Therefore, we concluded that 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil were modulated by P-gp.
Although assay 1 was convenient as the first screening, the limitations of that assay in the present study should be noted. For assay 1, we carefully selected a single time point for each ligand based on the previous reports; however, CsA perturbations may alter ligand kinetics in the brain and peripheral metabolism. The brain-to-blood ratio at a single time point is not an adequate index of ligand delivery. Therefore, multiple-time-point analyses or dynamic PET measurement is preferable for elaboration of each ligand (8).
In assay 2, we studied verapamil, TMSX, MPDX, and MPPF. Only verapamil at a dose of 5 mg/kg increased (slightly) the brain-to-blood ratio of 11C-verapamil, by 1.31-fold; however, a 1.32-fold uptake increase in the brain was not significant (Table 2). The findings in this assay may be due to the low doses of cold ligands used. For instance, we used 1 or 5 mg (2 or 10 μmol) of cold verapamil per kilogram of body weight to modulate P-gp, but the in vitro assay using cell cultures described below used 20 μmol of verapamil per liter to inhibit transport of substrates for P-gp (36). In assay 1, the amount of CsA used to modulate P-gp was 50 mg/kg. The modulation occurred dose-dependently, and doses of less than 10 mg/kg were not effective (4,17,37). However, because of the toxicity of cold ligands, their doses could not be increased. The median lethal dose of verapamil in mice is 8 mg/kg intravenously (38), and a preliminary experiment showed that DMSO solutions containing larger doses of the other ligands were not acceptable (data not shown). Another reason for the findings may be the modulation period between injection of each cold ligand and injection of each tracer or the interval allowed for brain uptake after injection of each tracer. These intervals were determined on the basis of the brain kinetics of radiolabeled ligands as described in the “Materials and Methods” but may not have been optimal for the assays. Optimization of this assay for each ligand and further examination of other ligands with this assay may be required, but we did not perform further studies because a small signal for possible modulation of P-gp was observed only for verapamil. Three other ligands enhanced neither the brain uptake of 11C-verapamil nor the brain-to-blood ratio at doses appropriate for blockade of many neuroreceptors in vivo, suggesting that cold ligands did not modulate P-gp at the doses used or that efflux of 11C-verapamil by P-gp was not saturated with cold ligands. We also confirmed that cold verapamil at doses of 1 and 5 mg/kg did not affect brain uptake of 11C-TMSX and 11C-MPDX (data not shown). If detailed optimization is essential for each test ligand for which the brain kinetics in vivo are unknown, assay 2 has no practical benefit. Therefore, we concluded that modulation of radioligands by P-gp could not be evaluated in vivo by assay 2.
Transcellular transport studies for determining whether compounds are substrates for the P-gp efflux pump are usually performed using P-gp–overexpressing cell lines (36,37). Before labeling candidate compounds for PET, screening of these candidates with this assay is preferable for the development of PET radioligands; however, ex vivo biodistribution studies using labeled compounds (assay 1 in the present study) may be more sensitive for detecting P-gp modulation. Bart et al. found that CsA treatment resulted in a 5-fold increase in 11C-carvedilol in rat brain and a 3-fold increase in a human small-cell lung carcinoma cell subline overexpressing P-gp (37). For the assay with ex vivo biodistribution studies, the use of P-gp knockout mice is another choice (6,14,15,37); however, assay 1 using conventional rodents and CsA is much more convenient.
When PET radioligands are modulated by P-gp, how are the PET data influenced? As reported previously (14,15) and also shown in Figure 3A, CsA treatment enhanced uptake of the radioligand in all regions of the brain, including targeted and nontargeted tissues. Thus, the receptor-specific uptake determined by subtracting uptake in nontargeted tissues from uptake in targeted tissues significantly increased, whereas PET signals revealing the specific binding ratio of each ligand to its corresponding receptor, that is, target-to-nontarget ratio, were not affected. From this point of view, distribution of the receptors represented by the radioligands may be unaffected regardless of whether they are modulated by P-gp. However, because the specific signals become much lower than those of radioligands not regulated by P-gp, subtle alterations in the brain may be missed. Furthermore, Kortekaas et al., using 11C-verapamil PET, recently found that P-gp function is reduced in the midbrain of patients with Parkinson's disease and suggested that this dysfunction is part of the pathogenesis of Parkinson's disease (9). Therefore, if a PET radioligand is modulated by P-gp, studies using that radioligand should be performed carefully and with consideration of the extent of P-gp modulation, especially in patients treated with P-gp modulators such as CsA or with P-gp substrates (3). One should keep in mind that other transporters, such as multidrug-resistance–associated protein and organic anion transporters, may also modulate the ability of radioligands to cross the blood–brain barrier and the plasma membranes of tumors and other tissues.
CONCLUSION
Modulation by P-gp of 8 PET radioligands was assayed in mice. CsA treatment increased both the brain uptake and the brain-to-blood ratio of 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil. Treatment with the cold ligand of the corresponding radioligand was not suitable for determining P-gp modulation of 11C-verapamil uptake by the brain. We concluded that of the radioligands investigated, 11C-TMSX, 11C-MPDX, 11C-flumazenil, and 11C-donepezil were modulated by P-gp.
Acknowledgments
This work was partially supported by Grant-in-Aid 13557077 for Scientific Research (B) from the Japan Society for the Promotion of Science. We thank Dr. Takashi Oda for preparing the radiotracers and Dr. Qinggeletu for performing the animal experiments.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication July 13, 2006.
- Accepted for publication October 2, 2006.





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- SARS-CoV-2 causes brain inflammation via impaired neuro-immune interactions
- Prediction of the Clinical SUV Ratio in Amyloid PET Imaging Using a Biomathematic Modeling Approach Toward the Efficient Development of a Radioligand
- Decreased intestinal acetylcholinesterase in early Parkinson disease: An 11C-donepezil PET study
- Local Drug-Drug Interaction of Donepezil with Cilostazol at Breast Cancer Resistance Protein (ABCG2) Increases Drug Accumulation in Heart
- Altered GABAA Receptor Density and Unaltered Blood-Brain Barrier Transport in a Kainate Model of Epilepsy: An In Vivo Study Using 11C-Flumazenil and PET
- Avoiding Barriers to PET Radioligand Development: Cellular Assays of Brain Efflux Transporters
- Transport of Selected PET Radiotracers by Human P-Glycoprotein (ABCB1) and Breast Cancer Resistance Protein (ABCG2): An In Vitro Screening