


Abstract
This trial was performed to determine the maximum tolerated whole-body radiation-absorbed dose of fractionated 131I-cG250. Methods: This was a phase 1 dose escalation trial. Dose escalation refers here to the escalation of average whole-body absorbed dose. Fifteen patients with measurable metastatic renal cancer were studied. For each treatment cycle, patients initially received a “scout” administration consisting of 5 mg of cG250 antibody labeled with 185 MBq (5 mCi) of 131I. Whole-body and serum activity was measured for 1 wk, and a simple pharmacokinetic model was fitted to the measured data. The pharmacokinetic model was used to calculate the required activities, administered in a fractionated pattern with 2–3 d between fractions, projected to deliver the prescribed whole-body absorbed dose. The initial cohort of 3 patients was prescribed an average whole-body absorbed dose of 0.50 Gy. In subsequent cohorts this was increased in 0.25-Gy increments. The first fraction in each cycle was 1,110 MBq (30 mCi) of 131I conjugated to 5 mg of antibody. Subsequent fractions consisted of variable activities depending on the patient-specific whole-body clearance rates and the times between fractions. Patients without evidence of disease progression were retreated after recovery from toxicity if there was no evidence of altered pharmacokinetics or serum human antichimeric antibody titers, for a total of no more than 3 treatments. Results: For the initial treatment course, the pharmacokinetics of the scout dose accurately predicted the pharmacokinetics of fractionated 131I-cG250 therapy. In 2 patients, altered clearance accurately predicted development of human antichimeric antibody. Targeting to known disease ≥ 2 cm in diameter was noted in all patients. Dose-limiting toxicity was hematopoietic, and the maximum tolerated dose per cycle was 0.75 Gy. Conclusion: Measurements of whole-body and serum clearance of cG250 antibody can be used to accurately predict the clearance of subsequent administrations, thus enabling rational treatment planning. An additional practical benefit of real-time pharmacokinetic monitoring is that therapy can be altered dynamically to reduce toxic side effects. However, there was no evidence for fractionation-induced sparing of the hematopoietic system in this study.
Annual estimates of new cases of renal cell carcinoma in the United States have not increased appreciably over the years (1). In 2003, this estimate was 31,900 new cases, resulting in 11,900 deaths. Overt metastatic disease is present at the time of diagnosis in 25% of patients, and median survival is 10 mo in these patients (2). When the disease appears to be localized, the treatment of choice is radical nephrectomy. Twenty-five percent of all patients with renal cancer will later manifest metastatic disease and ultimately succumb to their cancer (3).
No drug regimen of the more than 30 chemotherapeutic agents and other drugs evaluated alone or in combination has demonstrated a response rate greater than 15%–20% or prolonged survival (3). Indirect evidence suggests that host immune mechanisms play a role in the natural history of renal cell carcinoma and justifies programs of immunotherapy for renal cell carcinoma patients. Both interferon-α and interleukin-2 show antitumor activity in 20%–30% of patients but have considerable toxicity (4–6).
Radioimmunotherapy has entered the cancer therapeutics armamentarium with the approval of agents against cluster designation 20 antigen-positive lymphomas (7). Targeting of primary and metastatic renal cell cancer by monoclonal antibody G250 has been among the highest (expressed as percentage injected dose per gram) demonstrated in solid tumors (8), and radioimmunotherapy with this agent has seemed promising (9). Radioimmunotherapy with 131I-labeled murine G250 established hematotoxicity as being dose limiting; the invariable development of a host immune response (manifested by altered G250 serum clearance, lack of targeting, and presence of serum human antimouse antibody) precluded repeated therapy. Chimeric (murine Fv-grafted human IgG1κ) G250 (cG250) was developed to circumvent the immunogenicity inherent in the murine antibody, and clinical studies with 131I-labeled cG250 demonstrated tumor targeting comparable to murine G250 without evidence of immunogenicity (10). Therapeutic trials with 131I-cG250 were therefore initiated.
Although the availability of cG250 enables repetitive treatments, it is not immediately apparent how this may best be achieved in a clinical setting. One option is to deliver a single therapeutic administration that may be repeated after recovery from hemopoietic toxicity. Another option is to use a course of multiple therapeutic administrations in which individual administrations are separated by a relatively short interval. Courses of this type of fractionated therapy may then be repeated after recovery from hemopoietic toxicity. In either case the timing between courses of treatment should be similar, as it is governed by the rate of recovery of the hemopoietic system.
Theoretic arguments may be advanced that favor fractionated treatments. Multiple administrations may allow some degree of proliferative recovery by bone marrow, enabling higher absorbed doses to be delivered. In addition, the effects of preceding therapeutic administrations on tumor architecture may enable subsequent administrations to target tumor regions inaccessible to single administrations. This would improve the uniformity of absorbed dose distributions in tumors and thereby increase therapeutic effectiveness (11). Fractionation may also allow hypoxic tumor regions to reoxygenate during treatment and thus render the tumor more sensitive to radiation. Data from animal experiments suggest that a series of relatively small administrations, each separated by several days, is both intrinsically more tumoricidal and less toxic to bone marrow than is a large single administration (12–15).
However, there is, as yet, no clinical evidence of a therapeutic benefit from fractionation of radioimmunotherapy. We therefore decided to explore both therapeutic strategies. Accordingly, 2 phase 1 radioimmunotherapy studies were initiated: a single-dose trial with escalating amounts of 131I-cG250, performed at the University of Nijmegen, Holland (16), and a fractionated-dose trial performed at Memorial Hospital for Cancer and Allied Diseases, New York.
Numerous studies have shown lack of a clear correlation between amount of administered radioactivity and toxicity. In patients with limited prior chemotherapy treated with 131I-labeled antibodies that do not specifically target the hemopoietic system, we have found the best predictor of dose-limiting hematologic toxicity to be absorbed radiation dose (either whole body or red marrow) (17). We therefore decided to base dose escalation of fractionated radioimmunotherapy on the whole-body radiation-absorbed dose. The clinical results of the trial are presented here. Subsequent articles will describe in detail the methodology for treatment planning and verification and the comparative results between the single-dose and fractionated studies.
MATERIALS AND METHODS
The phase 1 dose escalation study was performed under an Investigational New Drug application (BB-IND-8241) submitted to the Food and Drug Administration by the Ludwig Institute for Cancer Research. The protocol was approved by the Institutional Review Board at Memorial Sloan-Kettering Cancer Center, the sole study site.
Patients
Between November 1998 and October 1999, 15 patients were enrolled in the study. All patients had clear cell renal carcinoma confirmed histopathologically at Memorial Sloan-Kettering Cancer Center. All patients had progressive disease, as assessed by serial CT scans within 8 wk of each other (with the last scan having been obtained within 4 wk before study entry).
Patients were required to sign an informed consent form and be at least 16 y of age with renal cell carcinoma proven by histology, a clinical presentation consistent with metastatic renal carcinoma, and disease bidimensionally measurable by conventional anatomic imaging methods including radiography, ultrasound, CT, or others. Lesions seen on skeletal scintigraphy alone were not considered measurable. Patients had not received chemotherapy or immunotherapy for at least 6 wk before entry into the study. Women of child-bearing age tested negatively for pregnancy the day of and before receiving therapy and were asked to use effective contraception during the study. Patients were required to be ambulatory, to have a Karnofsky Performance Status ≥ 70%, and to have a serum creatinine level ≤ 2 mg/dL, a serum bilirubin level ≤ 1 mg/dL, a white blood cell count ≥ 3,500/mm3, a platelet count ≥ 100,000/mm3, and a prothrombin time < 1.3 × control. Patients who had received significant prior radiotherapy to the entire pelvis or lumbosacral spine or who had clinically significant cardiac disease (New York Heart Association Class III/IV), serious infection requiring treatment with antibiotics, or other serious illness were excluded. Women who were pregnant or lactating were ineligible. Any patient who had a survival expectancy of <6 wk, had evidence of an active central nervous system tumor or hypercalcemia > 12.5 mg/100 mL, or was symptomatic was excluded.
Radiolabeling of cG250 with 131I
cG250 was radiolabeled at Memorial Sloan-Kettering Cancer Center using sterile techniques and pyrogen-free glassware or plasticware. Buffered cG250 at pH 7.4 was mixed with ≤1,295 MBq (35 mCi) of 131I in an IODO-GEN (Pierce)–coated sterile glass vial. The mixture was incubated at room temperature for 15 min and then purified by being passed through an ion-exchange resin. Terminal sterilization was completed using a 0.22-μm Millipore filter. Instant thin-layer chromatography of an aliquot of the final preparation was performed, and the preparation was administered to the patient only if the amount of protein-bound radioactivity was ≥95%. Prior to injection, the total radioactivity of the radiolabeled antibody was measured in a radioactive dose calibrator.
Purified radiolabeled antibody (131I-cG250) immunoreactivity was tested by the cell adsorption method. 131I-cG250 antibody was incubated at 4°C with an excess of antigen on viable renal carcinoma cells (SK-RC-52), and the percentage of bound radiolabeled antibody was determined using serial cell pellets.
Treatment Planning and Verification
Fractionated treatment consisted of a series of administrations of 131I-cG250 with 2–3 d between each administration. The administered activity for the first treatment fraction was 1,110 MBq (30 mCi). In subsequent fractions, administered activities were determined by the requirement to “top up” the whole-body activity to 1,110 MBq. The goal of the treatment planning process was to calculate the activity for each administration in order to deliver the prescribed value of whole-body absorbed dose based on patient-specific pharmacokinetic measurements.
Initially, retained activity in the whole body and serum was measured after a pretherapy “scout” administration of 185 MBq (5 mCi) of 131I conjugated to 5 mg of cG250 antibody. These measurements were then used to fit 2 different pharmacokinetic models. The first of these was the previously described (18) simple 2-compartment model illustrated in Figure 1. Four model parameters were derived: the coefficients representing transfer from serum to rest of body, transfer from rest of body to serum, excretion from rest of body, and the serum volume of distribution. This was the primary model used to calculate the treatment pre-scription. The second model fitted was a monoexponential whole-body clearance together with a biexponential serum clearance. In this model, 6 parameters were derived: clearance rates and time-zero intercepts for the whole body (2 parameters) plus clearance rates and time-zero intercepts for the serum (4 parameters). The serum fit enabled concurrent estimation of the volume of distribution. Parallel treatment-planning calculations were performed independently with this model as a cross-check on the reliability of the primary prescription.

Two-compartment model used to describe cG250 pharmacokinetics and to calculate fraction numbers and activity for treatment.
Using a computer spreadsheet, we ran a treatment simulation for each patient and calculated the predicted whole-body and red marrow absorbed doses. Whole-body absorbed doses were calculated on the basis of the area under the whole-body time–activity curve multiplied by a whole-body–to–whole-body S-factor derived from MIRDOSE3 (19) and adjusted for patient mass. Red marrow absorbed doses were calculated on the basis of the method described by Sgouros (20) using S factors derived from MIRDOSE3 for red marrow to red marrow and rest of body to red marrow adjusted for patient mass. The fractionation pattern that came closest to delivering the target dose was selected and treatment prescribed accordingly. Usually, the expected whole-body absorbed dose was either slightly greater or slightly less than the target value. This is reflected in the set of prescribed values seen in Table 1.
Whole-Body Radiation-Absorbed Dose and Administered Activity
Whole-body and serum activity was measured daily from the initiation of treatment until 1 wk after the last fraction. Areas under the whole-body and serum time–activity curve were derived by trapezoidal integration, and the actual whole-body and red marrow absorbed doses were estimated using the same methods described above. These are presented in Table 1 under the heading “Delivered.”
Treatment and Dose-Escalation Schema
The initial group of patients was treated so as to deliver to the whole body as close to an absorbed dose of 0.50 Gy as possible. Subsequent treatments were in 0.25-Gy increments. At least 3 patients per dose level were followed for up to 8 wk or until recovery from all toxicity, using imaging, biochemical tests, and hematologic tests for toxicity. CT was performed at baseline and no more than 6 wk after the last fraction for response assessment. In the absence of disease progression and after recovery from toxicity, patients were eligible for retreatment no sooner than 8 wk after the last fraction of the previous series, for a total of no more than 3 courses of treatments.
To prevent uptake of 131I by the thyroid, 10 drops of a saturated solution of potassium iodide and potassium perchlorate, 200 mg, were given orally daily beginning 24 h before the 131I-cG250 scout dose and continuing for 2 wk after the last treatment fraction.
All 131I-cG250 infusions were in 50 mL of 5% human serum albumin in normal saline, administered intravenously over 10 min. Patients initially received an 131I-cG250 antibody concentration of 185 MBq/5 mg (scout dose). Therapeutic 131I-cG250 was administered the following week, starting with 1,110 MBq/5 mg. Subsequent fractions were administered at 2- to 3-d intervals. The total amount administered was calculated on the basis of clearance of the scout dose as described above. Patients were treated as outpatients. After each therapeutic infusion, a handheld radiation survey meter was used to confirm acceptable radiation for outpatient procedures.
The maximum tolerated dose was defined as the highest dose at which no more than a third of the patients had grade 3 or greater toxicity.
Whole-Body and Serum Pharmacokinetics
All patients underwent daily anterior and posterior whole-body radiation measurements, obtained with a 12.7-cm (5 in.) scintillation crystal detector placed 3.05 m (10 ft) from the patient, starting immediately after the scout infusion and continuing for a week after the last therapy infusion. (Measurements were not performed on Sundays.) Probe measurements were performed for each orientation in duplicate for 1 min. Blood was obtained at multiple times after each infusion, and aliquots of serum were measured in a γ-well counter (LKB Wallac, Inc.) along with appropriate standards (known amounts of 131I and a 133Ba standard calibrated according to the method of the National Institute of Standards and Technology). Geometric means of whole-body counts per minute were used to obtain time–activity curves of whole-body clearance. The measured serum radioactivity was used to calculate serum clearance.
Image-Based Biodistribution and Dosimetry
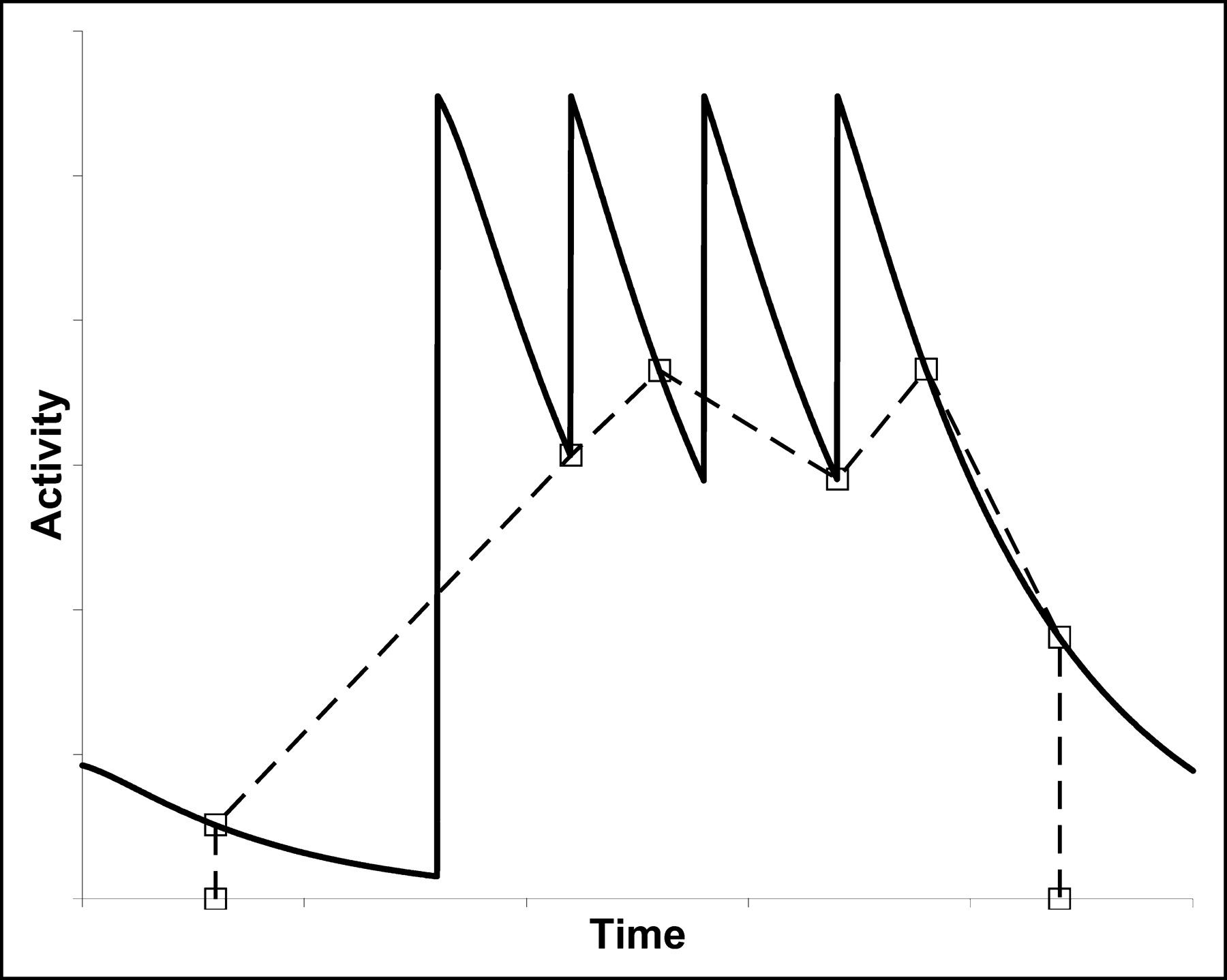
Absorbed dose to the liver and to selected index lesions was estimated on the basis of scintigraphic imaging. In general, anterior and posterior whole-body images were obtained 2–3 d after each infusion using a γ-camera (ADAC Labs. Inc.) equipped with a high-energy collimator. However, because of the fractionated nature of the treatment, determining the time course of activity in body regions using these incomplete image sets was not a straightforward process, as there was no general clearance curve that could be fitted to the data. The problem of estimating the area under the curve (AUC) is illustrated schematically in Figure 2. To address this problem, for 2 patients (patients 3 and 6) we acquired comprehensive image sets comprising whole-body images obtained every day (excluding Sundays) from immediately after the scout administration to approximately 1 wk after the last therapeutic administration. Patient 3 had 3 courses of treatment and acquisition of images during each course, for a total of 63 images over approximately 7 mo. Patient 6 had only 1 course of treatment, with acquisition of a total of 21 images. The comprehensive image sets were used to correct the AUC derived from the more usual incomplete image sets.

Trapezoidal integration of an incomplete set of pharmacokinetic data (□) for fractionated treatment leads to an inaccurate estimate of AUC. For the patients in the study, correction factors were derived by comparison of comprehensive and incomplete datasets.
The primary data used for estimating whole-body activity as a function of time were the probe measurements. Because we had extensive probe data, we did not use an activity calibration standard for the whole-body images. At each time point, camera sensitivity was determined from the geometric mean of counts in whole-body regions of interest, corrected for attenuation and scatter, and the whole-body activities estimated from the probe measurements. Additional regions of interest were drawn to encompass an index lesion and a portion of the liver, together with normal tissue and couch backgrounds. Attenuation correction was based on CT-derived patient, liver, and lesion thicknesses. The activity per gram in the index lesion and liver was estimated from the geometric mean of counts in the appropriate region of interest, corrected for tissue background and attenuation.
Areas under the time–activity curves up to the last measured point were estimated by trapezoidal integration. Apart from patients 3 and 6, for whom images were acquired daily, a further correction was made to compensate for the limited number of images. Using the comprehensive data from patients 3 and 6, we generated artificial sets of incomplete time–activity data, based on the actual times after administration when images were acquired. For every treatment course, 4 such artificial time–activity datasets were derived (3 from the 3-treatment courses of patient 3 and 1 from the single-treatment course of patient 6). The calculated AUC for the incomplete dataset was compared with that for the comprehensive dataset, and a correction factor was calculated for each course by taking the mean ratio of comprehensive AUC to incomplete AUC. The area under the terminal portion of the clearance curve was then added to give the total AUC.
Absorbed doses were calculated by multiplying the AUC (in MBq h/g) by the equilibrium dose constant for 131I. The photon components of the absorbed doses to the liver and lesions were ignored.
Human Antichimeric Antibody (HACA)
Patient sera were assayed for HACA by enzyme-linked immunosorbent assay using cG250 as capture and biotinylated cG250 as sensor antibody (10). Sera were analyzed in serial dilutions using pretreatment serum as negative control and antiidiotype G250 (Num 82) serially diluted in negative control serum as positive control.
Extent of Disease
Extent of disease was assessed within 1 wk before 131I-cG250 administration and no more than 6 wk after the last fraction or no more than 12 wk after entry into the protocol. Response was evaluated using standard anatomic imaging criteria and, whenever possible, was compared with pretreatment disease extent as evaluated using the same imaging modality.
Response parameters were evaluated as complete response (disappearance of all measurable disease, lasting at least 1 mo), partial response (≥50% reduction in the sum of the product of 2 perpendicular diameters of all measurable lesions and no new lesions, lasting at least 1 mo), stable disease (no change in the size of lesions, a decrease in size of lesions but less than a partial response, or no new lesions), or disease progression (the appearance of new lesions or a ≥25% increase in the size of any measurable lesion). Duration of response was measured from the start of treatment until disease progression.
RESULTS
Patient characteristics are detailed in Table 2. All patients tolerated the infusions without any acute side effects. One patient (patient 7) received the full course of treatment but died of progressive disease before he could be evaluated for toxicity; this patient was replaced. Five patients received additional cycles of treatment beyond the first cycle. Patients 4, 8, and 10 received 1 additional cycle, and patients 3 and 5 received 2 additional cycles.
Patient Characteristics and Outcome
The prescribed and delivered amounts of 131I-cG250 therapy, expressed as whole-body absorbed dose and as megabecquerels, are detailed in Table 1. The number of fractions administered per course ranged from 3 to 7 per patient, as outlined in Table 1.
Immune Response (HACA)
HACA reactivity was not detectable in any patient before the first injection of cG250 antibody. Of the 15 patients treated, 2 (patients 1 and 8) developed HACA (detectable by enzyme-linked immunosorbent assay and manifest as a change in pharmacokinetics) after therapy.
In patient 1, a low titer of HACA (1:40) developed immediately before injection of the second scout dose. HACA levels increased after administration of the second scout dose (titer, 1:40,000) and peaked (titer, ≥1:160,000) between 2 and 5 wk after termination of treatment (last injection: cycle 2, i.e., second dose fraction). Subsequently, HACA decreased to lower levels. A HACA titer of 1:2,560 was detectable in the last available serum of this patient, sampled 13 wk after termination of the treatment, or 25 wk after the first administration of the first scout dose.
In patient 8, a low-titer HACA response (titer, ≤1:160) developed. HACA was first detectable in serum sampled between treatment cycles 2 and 3. The titer decreased to baseline levels 2 d after injection of the third scout dose (last G250 injection) and increased to a titer of 1:160 on day 6 after the third scout dose (last available serum sample).
Pharmacokinetics and Targeting
Whole-body and serum kinetics varied significantly between patients. Excluding those patients in whom HACA developed, the estimated biologic clearance half-times ranged from 3.2 to 7.5 d for the whole body and from 1.3 to >5 d for serum β–half-life. This will be discussed more fully in a subsequent article describing the detailed methodology for treatment planning and verification.
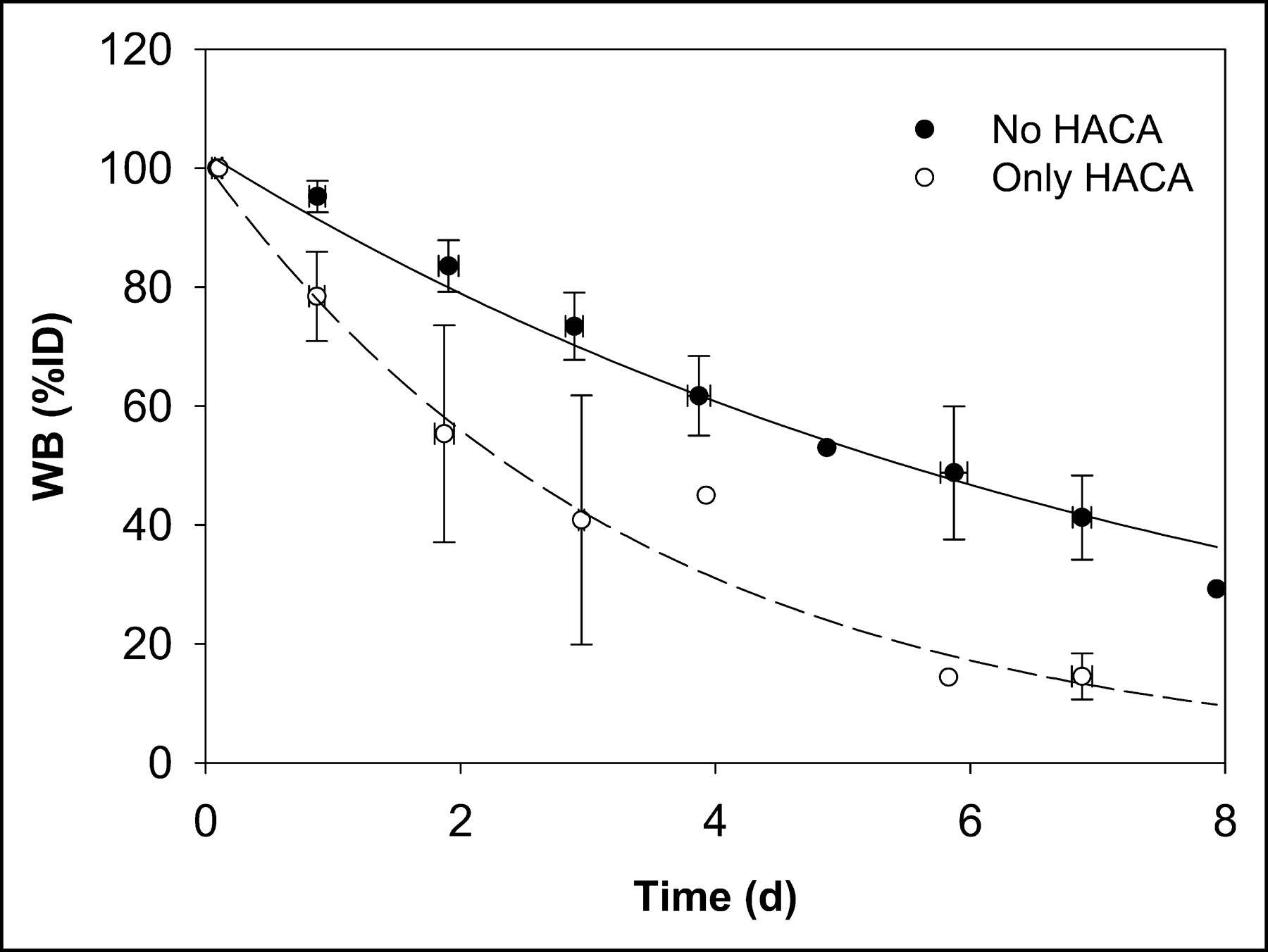
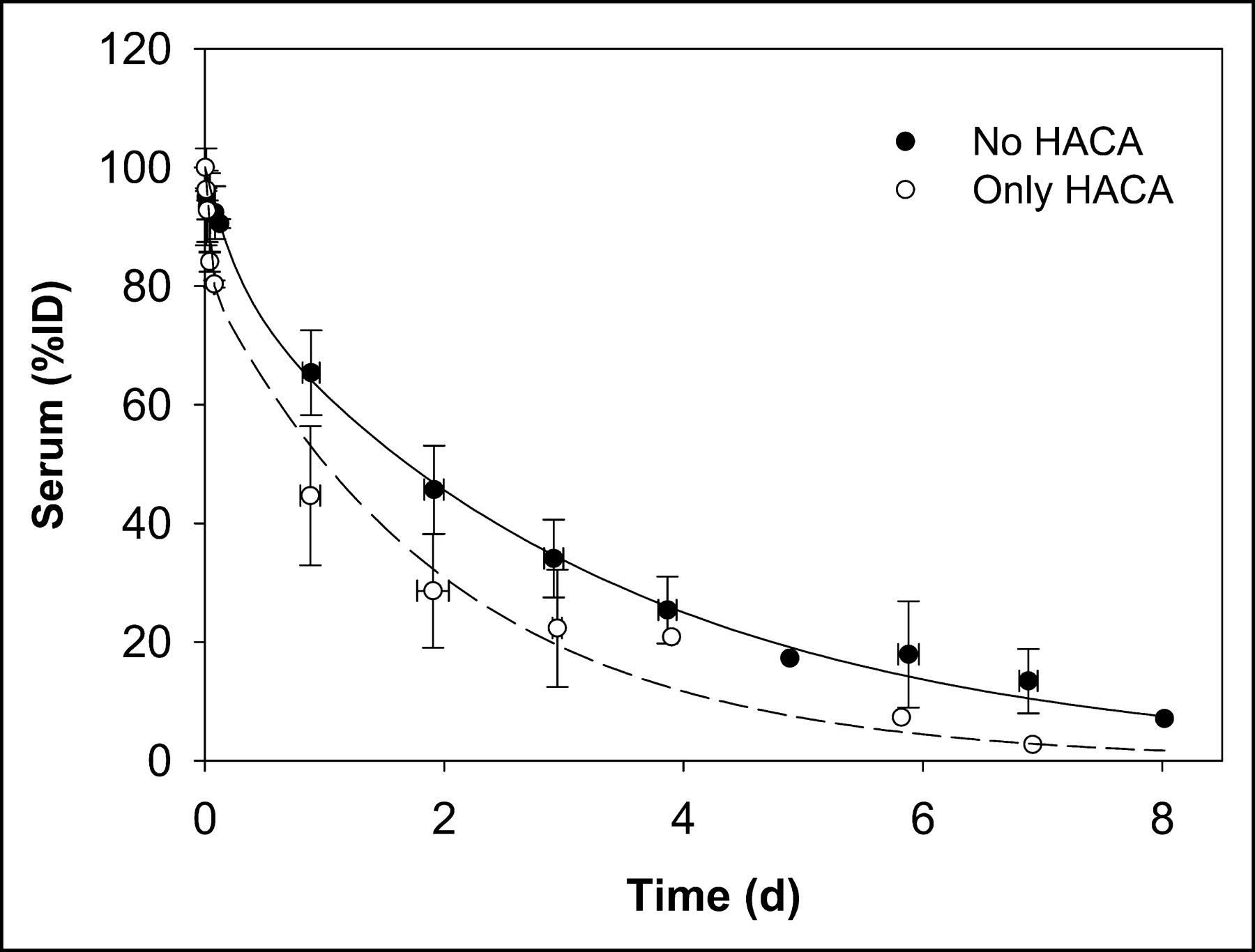
An average whole-body biologic clearance curve was generated by combining all data and fitting a monoexponential function (not shown). Figure 3 shows the whole-body clearance curve for all data excluding the 2 administrations in 2 patients in whom HACA developed (patient 1, scout 2, and patient 8, scout 3), and separately for those administrations with altered kinetics (represented as “only HACA” in Fig. 3).
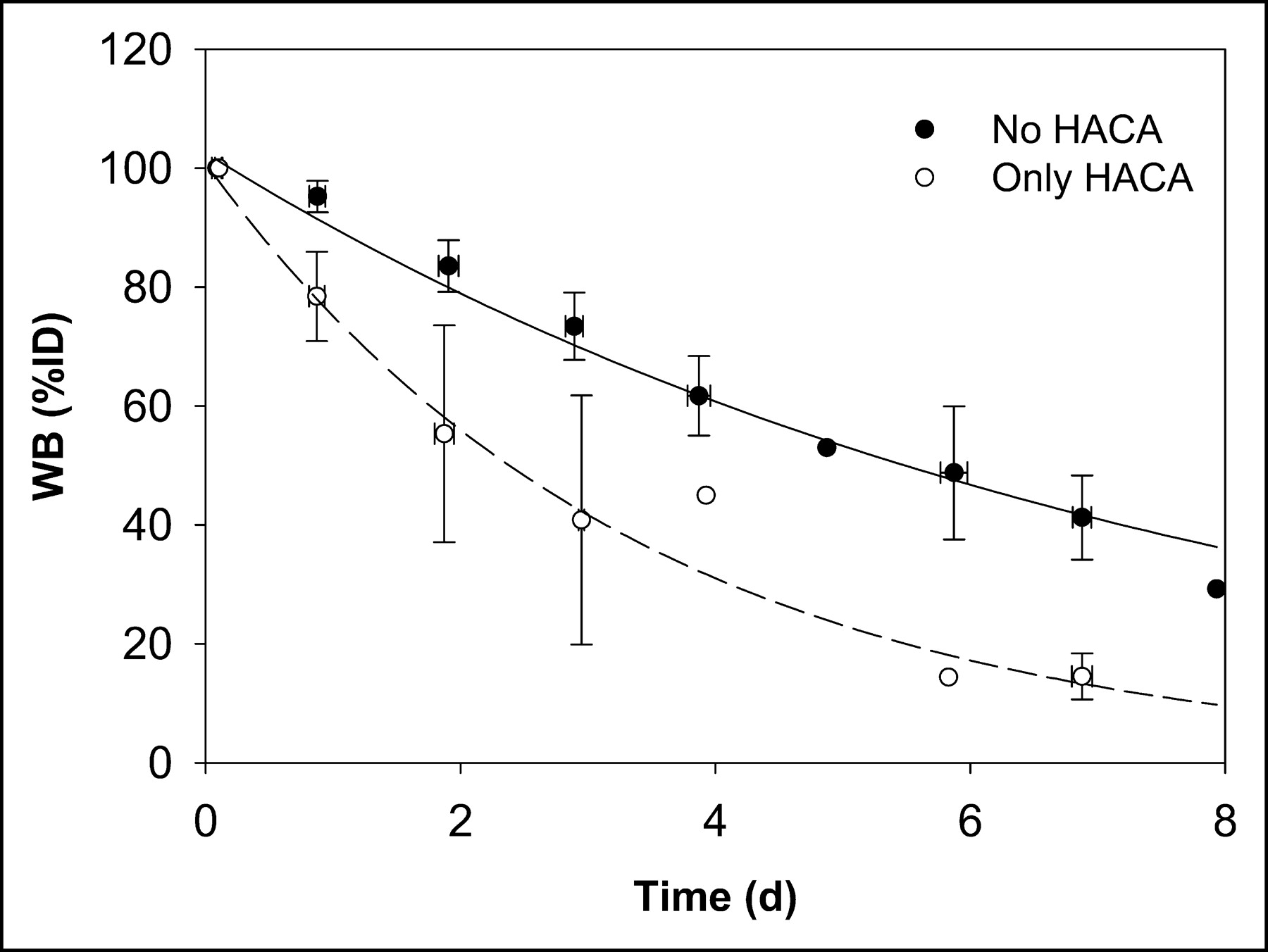
Whole-body clearance curves for all data excluding 2 administrations to 2 patients in whom HACA developed (no HACA) and for data on those administrations with altered kinetics due to HACA (only HACA).
In a similar manner, a mean serum clearance curve was generated by combining all data and fitting a biexponential function (not shown). Figure 4 shows biexponential serum clearance curves for all non-HACA administrations and for those (2 administrations) in which HACA developed.
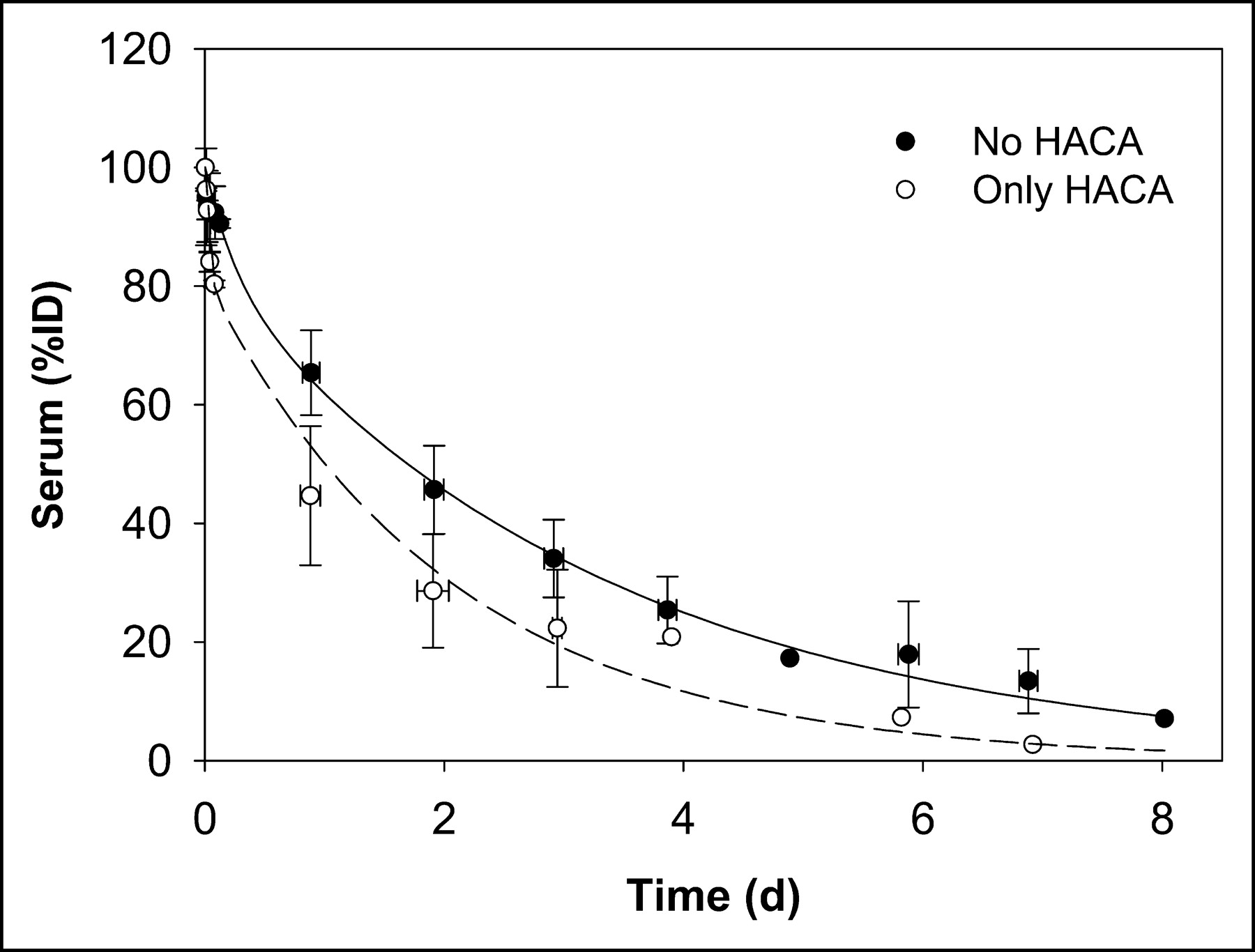
Serum clearance curves for all data excluding 2 administrations to 2 patients in whom HACA developed (no HACA) and for data on those administrations with altered kinetics due to HACA (only HACA).
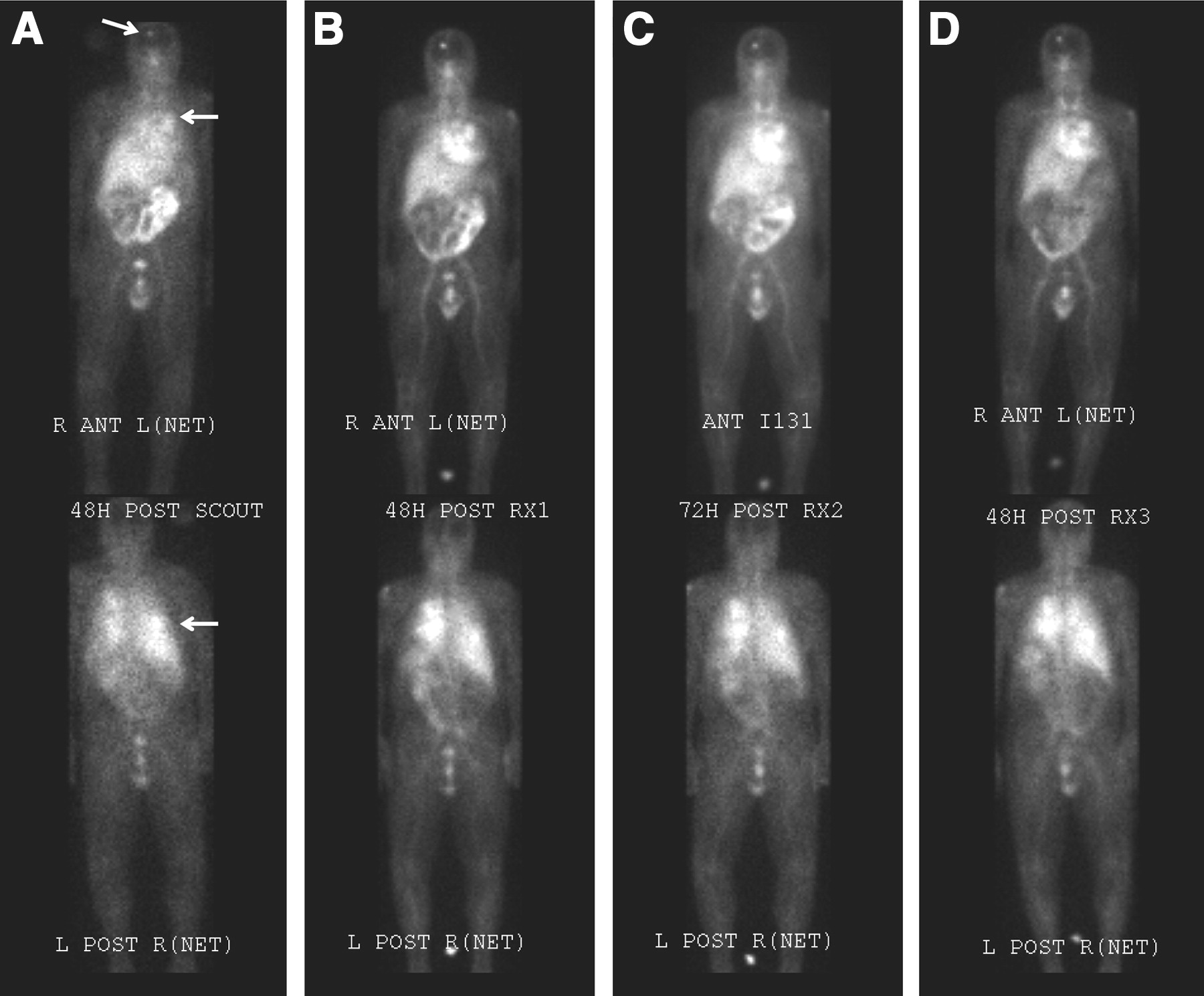
There was excellent targeting (Fig. 5) to all known lesions ≥2 cm in diameter, in all patients. These lesions included lung, liver, bone, and lymph node (including hilar and retroperitoneal adenopathy). The relative accumulation of radioactivity in tumor increased with time.

Anterior (top) and posterior (bottom) whole-body images acquired 2–3 d after the scout image (A) and after each therapeutic infusion (B-D) of 131I-cG250 (patient 3). Lesions are marked with arrows. ANT = anterior; POST = posterior.
The average absorbed dose to the liver per treatment course estimated from the analysis of scintigraphic images was 1.8 Gy (range, 0.9–3.2 Gy), and that to the index lesion was 8.3 Gy (range, 0.9–23 Gy). In general, the absorbed doses to liver and lesion were higher for patients receiving higher prescription doses (R2 = 0.7 and 0.5, respectively). On average, the estimated absorbed dose to liver was 2.2 times that delivered to the whole body, whereas that to the index lesion was 9.5 times greater. These data are summarized in Table 3.
Estimated Whole-Body, Tumor, and Liver Radiation-Absorbed Dose
Prescription Compliance
In general, compliance between the prescribed activity and that actually delivered was excellent, as shown by Table 1. There were 2 exceptions; in one case, the patient (patient 14) did not complete treatment because of infection. In the other case, the patient (patient 15) exhibited slower clearance than we had anticipated on the basis of the scout administration. This resulted in a gradual increase in the interfraction nadir of whole-body activity. The patient was scheduled to receive 5 fractions, but before he received his fifth fraction we made a projected dose calculation that suggested a significant overdose would be likely if the prescription was followed explicitly. We therefore decided to withhold the last scheduled treatment.
The relationship between prescribed and delivered absorbed dose to the whole body was close to linear. However, there was a systematic tendency to underestimate the delivered dose based on pretherapy pharmacokinetics. On average, the delivered dose was 10% higher than the prescribed value, but there was no correlation (R2 = 0.02) between the number of fractions and this ratio. Table 1 summarizes the prescribed and delivered administered activity and whole-body absorbed dose for each patient.
Toxicity
No significant nonhematopoietic toxicity was believed to be related to the study agent. Four adverse events of grade 3 severity (according to the Common Terminology Criteria for Adverse Events) occurred (fever [patient 3], loss of appetite [patient 7], septicemia [patient 14], and anemia [patient 12]), all of which were believed to be unrelated to radioimmunotherapy. Immediately after the second course of 131I-cG250 in patient 3, a grade 2 allergic reaction and dyspnea developed, considered possibly related to 131I-cG250, and a rash developed, considered probably related.
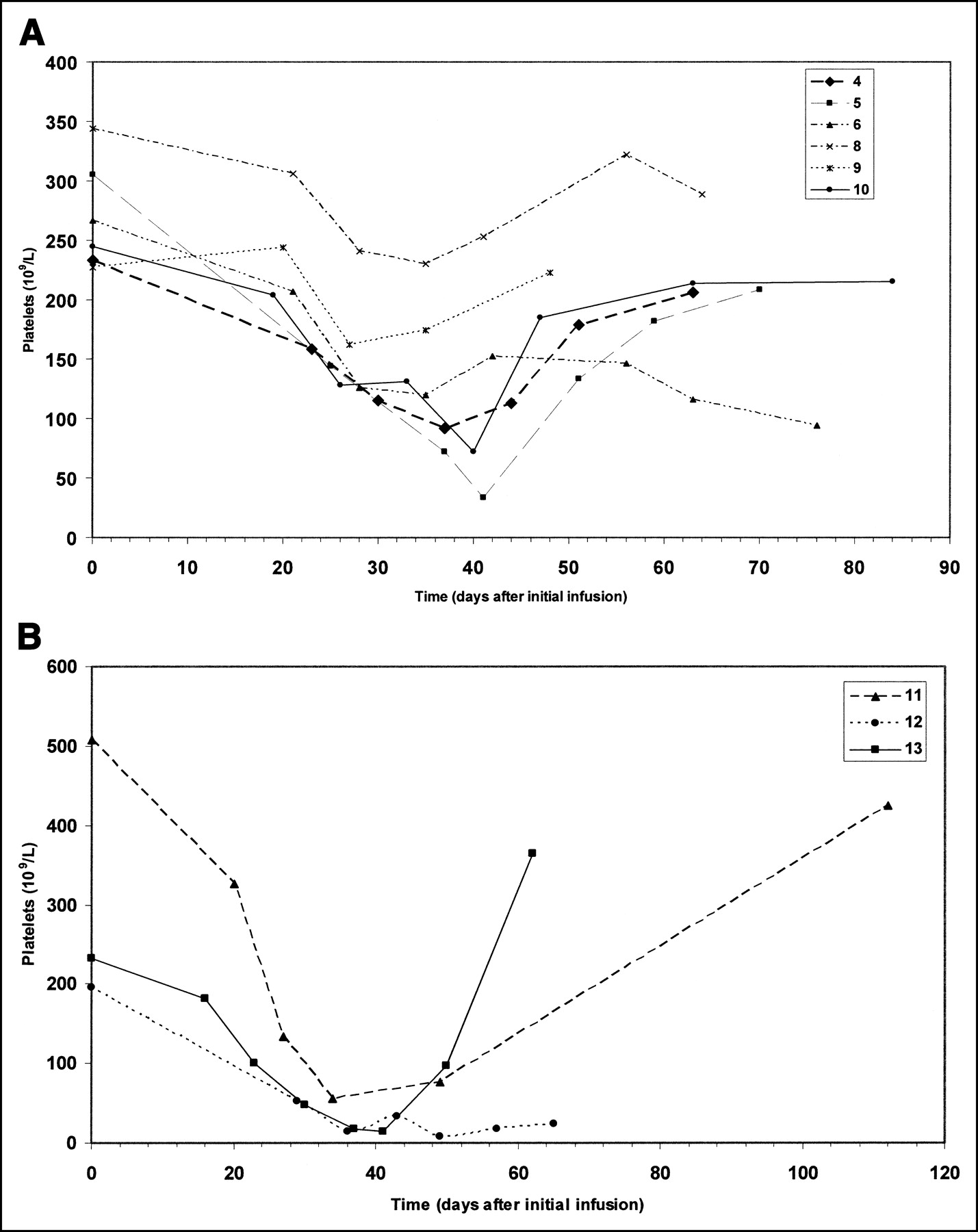
Adverse events associated with any causal relationship to 131I-cG250 were generally of hematopoietic origin. No dose-limiting toxicity was believed to be related to the study agent at the 0.5-Gy dose level. In 1 patient of the 0.75-Gy cohort, grade 3 leukopenia (white blood cell count, 1.6 × 109/L) and grade 3 thrombocytopenia (platelets, 33 × 109/L) developed, considered possibly related to 131I-cG250 and qualifying as a dose-limiting toxicity. This patient was retreated after recovery from toxicity. Toxicity appeared to be cumulative in 2 of the 3 patients (patients 4 and 5) that were retreated at this dose level. Although nadirs were not lower after subsequent therapy, platelet counts after recovery from each therapy were always lower than at baseline. Three additional patients were studied in the 0.75-Gy cohort, and no additional dose-limiting toxicity was observed (Fig. 6A).

(A) Platelet counts over time in patients treated at the 75-cGy dose level. There was 1 case of reversible grade 4 thrombocytopenia. (B) Platelet counts over time in patients treated at the 100-cGy dose level. Grade 4 thrombocytopenia developed in two, one of whom died from progressive disease; the third patient had grade 3 thrombocytopenia. 11, 12, and 13 = patients 11, 12, and 13, respectively.
In the first patient at the 1.0-Gy dose, progressive disease developed after the last fraction of 131I-cG250. The patient subsequently experienced grade 4 anemia (hemoglobin, 4.6 g/dL), believed to be unrelated to the study agent (the patient’s baseline hemoglobin was 7.5 g/dL). The platelet count nadir was 49 × 109/L (grade 3).
Two additional patients in the 1.0-Gy cohort, patients 12 and 13, also had grades 3 and 4 toxicity after therapy (Fig. 6B). Patient 12 experienced grade 3 and eventually grade 4 thrombocytopenia (platelets, 8 × 109/L) and grade 3 anemia (hemoglobin, 7.7 × 109/L), requiring transfusions on several occasions, and grade 3 neutropenia (absolute neutrophil count, 0.7 × 109/L), requiring granulocyte-colony stimulating factor. The patient experienced neurologic deficits and hemiplegia. MRI revealed a cerebrovascular hemorrhage in an area of previously unknown brain metastases. The patient died 83 d after entry into the study.
Patient 13 had grade 4 neutropenia (absolute neutrophil count, 0.3 × 109/L) and grade 3 thrombocytopenia (platelets, 17 × 109/L). She subsequently recovered completely and is currently alive.
Because 2 of 3 patients at the 1.0-Gy dose level had agent-related grade 3 or greater toxicity, additional patients were prescribed 0.75 Gy. These patients had no grade 3 or greater hematopoietic toxicity.
The maximum tolerated dose was therefore considered to be 0.75 Gy.
DISCUSSION
Most clinical trials with radioimmunotherapy have been performed using a single large dose of radiolabeled antibody. A notable exception has been the fractionated radioimmunotherapy performed at this center with 213Bi-huM195 (21); a fractionated schema was used in this study because the small number of antigenic sites necessitated a small mass amount of antibody per infusion, thus limiting the amount of radioactivity that could be administered at a given time. There have been no phase 1 studies using a fractionated radioimmunotherapy schema based on delivering an escalating radiation-absorbed dose to critical normal organs.
The current study was based on a radiation-absorbed dose schema for several reasons. First, we have shown that radiation-absorbed dose is a better predictor of toxicity than is administered activity (17). Second, given the variation in antibody pharmacokinetics between patients, it would have been problematic to calculate the amount of radioactivity necessary in the fractionation schedule. Third, marrow sparing was one of the hypotheses favoring fractionation (22).
It was possible to predict clearance of 131I-cG250 on the basis of the clearance characteristics of an initial scout administration and to make adjustments in real time based on clearance measurements. This has important implications for future trials using absorbed dose schemata. Clearance changes in patients in whom an antiantibody response developed demonstrated that this trial design is applicable only to nonimmunogenic agents, for which pharmacokinetics remain relatively constant with repeated administration. Basing serum and whole-body clearance on measurements taken over a week tended to result in an overestimate of the activity required to deliver a specified whole-body absorbed dose. However, this overestimation may be corrected for by adjusting the number of infusions or activity per infusion based on real-time monitoring of clearance (as was done for patient 15).
Tumor targeting was noted in all patients with clear cell renal cancer, demonstrating no need for immunohistochemistry-based preselection of these patients. Tumor targeting of 131I-cG250 did not decrease with multiple infusions, suggesting no saturation of tumor receptor sites. Images in some patients showed uptake in testes and small bowel. Immunohistochemical analysis has not shown any reactivity of G250 with normal testicular or bowel tissue; hence, we are unable to explain the transient uptake of activity in these organs. However, the antigen is carbonic anhydrase IX, which may be transiently expressed at low levels (i.e., undetectable by immunohistochemical analysis) in these tissues.
Fractionated radioimmunotherapy with 131I-cG250 is safe. No nonhematologic toxicity was found in this study. Dose-limiting toxicity was hematopoietic, with the maximum tolerated dose being 0.75 Gy to the whole body. This dose is comparable to estimates of maximum tolerated whole-body radiation-absorbed dose obtained from other studies, both in lymphoma and in solid tumor. The use of fractionated radioimmunotherapy therefore did not result in any obvious marrow-sparing effect.
Hepatic function was unaltered, similar to the observations of Steffens et al. (16), presumably because of the protective effect of the scout dose. The radiation-absorbed dose to the liver ranged from 0.9 to 3.2 Gy, increased with dose level, and was relatively unchanged in proportion to the whole-body absorbed dose (2.2 ± 0.4 per gray in whole body), confirming our earlier observations with murine G250 (9). Tumor radiation-absorbed doses were estimated using an index lesion in each patient. Although most patients had more than one lesion, absorbed dose to all lesions could not be determined for various reasons, including overlapping of normal structures (e.g., heart for thoracic vertebral lesions). The total radiation-absorbed dose to tumor did not exceed 23 Gy per treatment course, and the mean radiation-absorbed dose ratio for tumor to whole body was 9.5 (SD, 6.4). No significant changes in tumor targeting or residence time were seen with successive treatments.
No major clinical responses were noted. All patients had progressive disease at the time of entry into the protocol. Four of the 15 patients treated were eligible for retreatment because their disease had stabilized. Two of these patients received 3 cycles; their disease was stable for 7 and 13 mo. Two other patients are still alive more than 3 y after treatment, suggesting that fractionated radioimmunotherapy may result in stabilization of previously progressive disease. However, until these observations can be verified with a trial specifically designed to establish disease stabilization as a valid endpoint in patients with progressive disease, they must remain anecdotal. The design suggested by Fazzari et al. (23) and Scher and Heller (24) may permit timely observations about therapeutic potential with a minimum of patients.
Studies by other groups, notably DeNardo et al., have demonstrated a marrow-sparing effect in clinical fractionated radioimmunotherapy trials (22). These studies used a fractionation schema based on amounts of radioactivity rather than a schema based on radiation-absorbed dose, and each fraction was less than the maximum tolerated dose. In theory, fractionated radioimmunotherapy may have an advantage over single-dose radioimmunotherapy because it addresses the issue of dosimetric heterogeneity in tumors (11). Our preliminary observations suggested that the intratumor distribution of radioactivity may vary from fraction to fraction. We are now estimating absorbed dose distributions within tumors.
131I may not be the optimal nuclide for successful nonmyeloablative radioimmunotherapy with cG250. Radiometals labeled to cG250 appear to have better tumor retention (25,26) and potentially higher tumor-to-nontumor ratios than does iodine. The therapeutic index of radiometals may be sufficient to permit nonmyeloablative therapy with chimeric G250. We have chelated cG250 with dodecanetetraacetic acid, a macrocycle capable of stably attaching radiometals (including 90Y and 177Lu) to cG250, and are initiating clinical trials with these agents in patients with advanced clear cell renal cancer.
CONCLUSION
Fractionated radioimmunotherapy with 131I-labeled cG250 was safe and feasible, with excellent correlation between predicted and actual clearance of radiolabeled antibody fractions from serum and whole body. Tumor targeting was consistent within and between treatment courses, with tumor radiation-absorbed doses not changed between successive treatment courses. The fractionation schema used was largely nonimmunogenic. The maximum tolerated dose of 0.75 Gy is comparable to that with other 131I-labeled antibodies, suggesting that dose-limiting toxicity is not significantly affected by fractionation. Nonmyeloablative fractionated radioimmunotherapy with 131I-cG250 did not result in any major clinical responses in metastatic clear cell renal cancer. Further trials with nuclides that increase the therapeutic ratio will help determine the feasibility of nonmyeloablative radioimmunotherapy in renal cancer and other solid tumors.
Footnotes
Received Dec. 22, 2003; revision accepted Mar. 19, 2004.
For correspondence or reprints contact: Chaitanya R. Divgi, MD, Nuclear Medicine Service, Memorial Sloan-Kettering Cancer Center, 1275 York Ave., New York, NY 10021.
E-mail: divgic{at}mskcc.org
REFERENCES





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- In Vivo Imaging of the Programmed Death Ligand 1 by 18F PET
- Tyrosine Kinase Inhibitor Sorafenib Decreases 111In-Girentuximab Uptake in Patients with Clear Cell Renal Cell Carcinoma
- Correlation of In Vivo and In Vitro Measures of Carbonic Anhydrase IX Antigen Expression in Renal Masses Using Antibody 124I-cG250
- Carbonic Anhydrase IX Is Not an Independent Predictor of Outcome for Patients With Clear Cell Renal Cell Carcinoma
- Phase I Evaluation of J591 as a Vascular Targeting Agent in Progressive Solid Tumors
- A pilot study of monoclonal antibody cG250 and low dose subcutaneous IL-2 in patients with advanced renal cell carcinoma
- A phase I multiple dose, dose escalation study of cG250 monoclonal antibody in patients with advanced renal cell carcinoma
- Treatment of Metastatic Renal Cell Carcinoma With Autologous T-Lymphocytes Genetically Retargeted Against Carbonic Anhydrase IX: First Clinical Experience
- Radioimmunotherapy with [131I]cG250 in Patients with Metastasized Renal Cell Cancer: Dosimetric Analysis and Immunologic Response
- Enhanced Efficacy of Radioimmunotherapy with 90Y-CHX-A''-DTPA-hu3S193 by Inhibition of Epidermal Growth Factor Receptor (EGFR) Signaling with EGFR Tyrosine Kinase Inhibitor AG1478
- Radioimmunotherapy of Prostate Cancer Using 90Y- and 177Lu-Labeled J591 Monoclonal Antibodies: Effect of Multiple Treatments on Myelotoxicity
- Lack of Efficacy of Two Consecutive Treatments of Radioimmunotherapy With 131I-cG250 in Patients With Metastasized Clear Cell Renal Cell Carcinoma
- Radioimmunotherapy of Prostate Cancer: Does Tumor Size Matter?
- Radioimmunotherapy for Colorectal Cancer
- Current Status of Therapy of Solid Tumors