


Abstract
The biologic profiles of [99mTc]Demobesin 1 ([99mTc-N40–1,bzlg0,d-Phe6,Leu-NHEt13,des-Met14]BB(6–14)) and [111In]Z-070 were compared using various gastrin-releasing peptide receptor GRP-R)–expressing tissues of human and animal origin. Methods: The binding affinities of Demobesin 1, Z-070, and its metallated analogs were determined by receptor autoradiography on human cancer biopsy and mouse pancreas samples and by binding assays in rat AR4-2J and human PC-3 cell membranes. Biodistribution of [99mTc]Demobesin 1 and [111In]Z-070 was compared in nude mice bearing AR4-2J and PC-3 xenografts. Results: Demobesin 1, Z-070, and metallated Z-070 showed high affinity for the rat GRP-R in AR4-2J cell membranes (50% inhibitory concentration values = 0.17–0.45 nmol/L). In human PC-3 cell membranes, Demobesin 1 showed 11- to 15-fold higher affinity than the Z-070 peptides. These data were corroborated by results from human cancers and mouse pancreas. In AR4-2J and PC-3 tumor-bearing mice, [99mTc]Demobesin 1 and [111In]Z-070 displayed similar uptake in the rat tumor. However, in the human PC-3 xenografts, [99mTc]Demobesin 1 showed a 2- to 3-fold higher uptake than [111In]Z-070. Conclusion: Considerable differences between rat or mouse and human GRP-R–expressing tissues were found for the in vitro and in vivo characteristics of 2 radiolabeled bombesin analogs. This finding may have a significant impact in the selection of experimental tools in the development of bombesin analogs for GRP-R–targeting applications in humans.
- human gastrin-releasing peptide receptor
- rodent gastrin-releasing peptide receptor
- receptor targeting
- radiolabeled bombesins
Success in the area of somatostatin receptor–positive tumor targeting with diagnostic and therapeutic radionuclides (1,2) has stimulated research toward radionuclide targeting of alternative receptor systems overexpressed in tumors (2–4). Particular attention has been recently focused on the bombesin receptor (BB-R) family (5) comprising 3 members in humans: the neuromedin B receptor (NMB-R), the gastrin-releasing peptide receptor (GRP-R), and the orphan bombesin subtype-3 receptor (BB3-R) (6). These receptors can be pharmacologically distinguished by means of their different affinities for the mammalian peptides GRP (with high affinity for the GRP-R) and NMB (with high affinity for the NMB-R), whereas a native ligand for the BB3-R has not been identified yet (7–10). Expression of NMB-Rs and BB3-Rs has been documented in a few restricted tumor types (11,12). However, general interest has been primarily focused on GRP-Rs because of their massive expression in several frequently occurring human cancers (11–18), such as in primary prostatic invasive carcinoma with a >50% incidence of expression documented in androgen-insensitive bone metastases (13,14). In ∼60% of primary breast carcinoma cases, a high-density GRP-R expression is reported, reaching ∼100% in incidence in infiltrated lymph nodes (15–17). Recently, extremely high numbers of GRP-Rs have been detected in gastrointestinal stromal tumors as well (18).
As a consequence, several attempts have been made to develop radiolabeled bombesin analogs for GRP-R–targeted diagnostic imaging and radionuclide therapy of tumors (19). Functionalization of bombesin tetradecapeptide or BB(7–14) derivatives at the N-terminal with diethylenetriaminepenteacetic acid (DTPA) or 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) has enabled stable binding of radionuclides useful for SPECT, PET, or radionuclide therapy (19,20). On the other hand, the ideal nuclear properties, cost-effectivenes, and wide availability of 99mTc have intensified the search for useful 99mTc-labeled bombesins (19,21). Coupling of tetradentate N3S, P2S2, N4 chelator systems (affording 99mTcV(O)x-chelates, x = 1 or 2), or tricarbonyls combined with tridentate coligands (affording organometallic 99mTcI-chelates) have produced a few promising 99mTc-labeled bombesins (19,21).
Preclinical evaluation of these compounds has been performed in a variety of in vitro and in vivo models, comprising GRP-R–expressing cancer cells of either animal or human origin, animal or human tissue samples, and rodents bearing experimental tumors of either animal or human source. Although this practice, first followed in the somatostatin/somatostatin receptor system, unveiled no significant differences between human and animal somatostatin receptor–positive models, in the case of bombesin/GRP-R, the situation seems to be different. In fact, several studies have shown that bombesin-like peptides may have different structure–function relationships in different species, with a given compound functioning as an agonist in one species and as an antagonist in another (8–10,22). Despite the extensive information available on the effects of the bombesin peptide family in various species, relatively little is known about their action in humans (9,10). Recently, significant pharmacologic differences have been reported between rat and human BB3-Rs despite their high degree of sequence similarity (∼80% identity) (23). Similarly, despite a high interspecies homology (>90%) between the mouse, rat, and human GRP-R (24), differences in the binding characteristics or function of GRP-R–affine bombesin analogs have been reported between animals and humans (10,25–27). Therefore, to design more effective bombesin-based radiopharmaceuticals, it is very important to understand the pharmacologic and cellular basis of actions of the bombesin-like peptides and their receptors in humans and not rely solely on data derived from nonhuman models.
In this study, 2 radiolabeled bombesin analogs were evaluated for their potential use for GRP-R targeting of tumors in humans using mouse, rat, and human origin in vitro and in vivo models. The 111In-compound, based on a DOTA-modified agonist, is compared with the antagonist-based [99mTc]Demobesin 1 ([99mTc-N40–1,bzlg0,d-Phe6,Leu-NHEt13, des-Met14]BB(6–14)), showing high affinity and selectivity for the human GRP-R and persistent uptake in human xenografts in mice (28). Several interspecies discrepancies in the performance of the 2 compounds are pinpointed and discussed herein, revealing the importance of selecting appropriate experimental tools in the evaluation of new compounds intended for use in humans.
MATERIALS AND METHODS
Synthesis of Bioconjugates
Synthesis of Demobesin 1 was performed in solution as described previously (28).
Z-070 ([DOTA0–1,PEG40,d-Tyr6,β-Ala11,Thi13,Nle14]BB(6–14)) was synthesized on the solid support. Briefly, the peptide was assembled on a Rink amide 4-methylbenzydrylamine resin (NovaBiochem AG) according to classical Fmoc (9-fluorenylmethyloxycarbonyl) chemistry on a semiautomatic peptide synthesizer provided by Rink Combichem Technologies. Trityl and t-butyl were used as protecting groups of His and d-Tyr, respectively, and t-butoxycarbonyl was used for Trp. Fmoc-15-amino-4,7,10,13-tetraoxapentadecanoic acid (Fmoc-PEG4-OH; Quanta BioDesign Ltd.) and 1,4,7,10-tetraazacyclododecane-1,4,7-tris(acetic acid-t-butyl ester)-10-acetic acid (DOTA-tris(t-butyl ester; Macrocyclics) were consecutively coupled following the same method with 4 and 4.8 eq. O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (Fluka Chemie GmbH) to the peptide ratio used, respectively. Cleavage from the resin and deprotection as well as product purification and identification were performed according to a previously described protocol (29). Mass spectrometry of the product was performed on a Finnigan SSQ 7000 spectrometer, and a matrix-assisted laser desorption ionization was performed on a Voyager sSTR instrument (Applied Biosystems).
MW (molecular weight): 1771.86; MS-ES(−): 1809.7 [M+K-H]−, 904.4 [M+K-2H]2−; MS-ES(+): 1811.7 [M+K+H]+, 906.9 [M+K+2H]2+; MALDI, m/z (%): 1772.7 (100, [M+H]+), 1794.7 (8, [M+Na]+), 1826.6 (2, [M+K]+); HPLC tR (high-performance liquid chromatography; tR = retention time) (MN, CC 250/4 Nucleosil 120/3 C18 column, 80% B to 40% B from 0 to 30 min, 40% B to 0% B from 31 to 34 min, and 0% B to 80% B from 35 to 40 min, where A = MeCN and B = 0.1% TFA (trifluoroacetic acid), at 1 mL/min flow rate and UV [ultraviolet] detection at 254 and 280 nm): 18.5 min. Purity: >98%. (MS-ES is electrospray mass spectrometry; MS MALDI is matrix-assisted laser desorption ionization mass spectrometry.)
Peptide Radiolabeling
Labeling of Demobesin 1 with 99mTc was performed according to a published protocol (28) using the eluate of a commercial 99Mo/99mTc generator (Mallinckrodt Medical B.V.) as a 99mTcO4− source.
For labeling Z-070 with 111In, 111InCl3 (Mallinckrodt Medical B.V.) was provided in 0.01N–0.2N HCl, and 37 MBq were used. Labeling was conducted by adding 2 μL of a 10−3 mol/L stock solution of Z-070 in 0.1% acetic acid/ethanol (8:2, v/v) and 50 μL of 2 mmol/L sodium ascorbate/20 mmol/L sodium acetate, pH 4.6, in a polypropylene centrifuge tube and heating the reaction mixture at 90°C for 40 min (30,31).
Alternatively (29), 10 μg (5.64 nmol) Z-070 were dissolved in sodium acetate buffer (300 μL, 0.4 mol/L, pH 5) and, after addition of 111InCl3 (111–185 MBq [3–5 mCi]), the mixture was heated at 95°C for 25 min. For use in the internalization experiments, a 1.5 molar excess of InCl3·5H2O (Fluka Chemie GmbH) was added and the solution was heated again at 95°C for 25 min. The radiolabeled product was purified over a SepPak C18 cartridge (Waters Corp.) to afford [111In]Z-070 with a specific activity of ≥32.56 GBq/μmol (≥ 880 mCi/μmol). For injection in rats, the addition of InCl3·5H2O was omitted and a radioligand solution in 0.9% NaCl with 0.1% bovine serum albumin (BSA) was used.
Quality Control
Labeling yields were determined using instant thin-layer chromatography silica gel (ITLC-SG; Gelman). In the case of [111In]Z-070, strips were developed with 0.1 mol/L sodium citrate in the presence of 4 mmol/L DTPA, pH 4.6. The labeled peptide remained close to the origin, whereas [111In]DTPA migrated with the solvent front (30,31). For detection of reduced hydrolyzed 99mTc in the [99mTc]Demobesin 1 labeling mixture, strips were developed with 1 mol/L ammonium acetate/MeOH (1:1, v/v), as previously described (28).
Radiochemical purity was further studied by HPLC on a Waters XTerra RP-8 cartridge column, 5 μm, 150 × 4.6 mm. The flow rate was 1 mL/min with a linear gradient starting from 15% solvent A (acetonitrile) and 85% solvent B (0.1% TFA in water) and reaching 75% solvent A and 25% solvent B in 60 min ([111In-DOTA0]Z-070) or 30 min ([99mTc]Demobesin 1).
Radioiodination of [Tyr4]BB was conducted according to a published protocol (7).
Metallation of Bioconjugates
[InIII]Z-070.
A solution of Z-070 (0.5 μmol) in 500 μL 0.4 mol/L sodium acetate buffer (pH 5) was incubated for 20–25 min at 95°C with 1.5 μmol InCl3·5H2O in 0.04 mol/L HCl. After cooling, the mixture was purified over a SepPak C18 cartridge. [InIII]Z-070 in water was collected after methanol evaporation (29).
Analytic HPLC purity: ≥98%; MW: 1886; MS MALDI, m/z (%): 1885.2 (100, [M+H]+), 1907.9 (10, [M+Na]+).
[YIII]Z-070 and [LuIII]Z-070.
The products were similarly prepared using Y(NO3)3·5H2O or Lu(NO3)3·5H2O, respectively, as the metal source.
Analytic HPLC purity: ≥98%; [YIII]Z-070 MW: 1860.77; MS MALDI, m/z (%): 1858.7 (100, [M+H]+), 1880.7 (10, [M+Na]+); [LuIII]Z-070 MW: 1946.83; MS MALDI, m/z (%): 1944.7 (100, [M+H]+), 1967.7 (100, [M+Na]+).
[ReV]Demobesin 1.
The isostructural trans-[ReV(O)2(N4)]+1 species (32–34) was accessible by reaction of Demobesin 1 with an excess of the [(n-C4H9)4N][ReVOCl4] precursor (35) in MeOH/H2O medium in the presence of Hünig’s base (N-ethyldiisopropylamine). [ReV]Demobesin 1 was collected by HPLC purification.
Analytic HPLC purity: ≥98%; [ReV]Demobesin 1 MW: 1565.8; MS MALDI, m/z (%): 1567.3 ([M+H]+).
Cell Culture
Human androgen-independent prostate cancer PC-3 cells (American Type Culture Collection, LGC Promochem) were cultured in Dulbecco’s modified Eagle medium GLUTAMAX-I (Invitrogen Life Technologies) supplemented by 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were kept in a controlled humidified atmosphere containing 5% CO2 at 37°C. Passages were performed weekly using a 0.05% trypsin/0.02% EDTA (w/v) solution. Rat acinar pancreatic carcinoma AR4-2J cells (kindly offered by Dr. Ralf Kleene, Philipps University, Marburg, Germany) were grown confluent in F-12K nutrient mixture supplemented by 10% (v/v) fetal bovine serum, 1 mmol/L glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin in humidified air containing 5% CO2 at 37°C. Subculturing was performed using a 0.05% trypsin/0.02% ethylenediaminetetraacetic acid (EDTA) (w/v) solution.
Receptor Autoradiography Studies
Competition binding studies for Demobesin 1, Z-070, [InIII]Z-070, and [LuIII]Z-070 were performed on cryostat sections of human prostate cancer expressing human GRP-Rs and on cryostat sections of mouse pancreas expressing mouse GRP-Rs, using [125I-d-Tyr4]BB as GRP-R–preferring ligand, as described in detail previously (13,14,25).
Competition Binding Assays on Cell Membranes
Bioconjugate affinities were tested by competition binding assays in cell membranes harvested either from PC-3 cells used as the human GRP-R source (36) or from AR4-2J cells serving as the source of rat GRP-R (37). Experiments were conducted as previously described using [125I-Tyr4]BB as the radioligand and [Tyr4]BB as the reference compound (28).
Internalization
For internalization studies, the PC-3 and AR4-2J cell lines were used and experiments were performed as previously described (29). A high excess BZH2 ([DOTA0–1,GABA0,d-Tyr6,β-Ala11,Thi13Nle14]BB(6–14)) (GABA = γ-aminobutyric acid; 0.58 μmol/L concentration) was used to determine nonspecific internalization.
Tissue Distribution
Lewis Rats.
Male Lewis rats were implanted subcutaneously with 10 × 106 AR4-2J cells freshly expanded in sterilized phosphate-buffered saline (PBS). Fourteen days after inoculation, rats were injected in the back leg vein with 200 μL [111In-DOTA0]Z-070 solution in 0.1% BSA in physiologic saline (∼407 kBq [∼11 μCi], 0.1 μg peptide). A separate group of 4 animals was injected with 225 μL of radiopeptide additionally containing 50 μg BZH2 (blocked animals). Rats were sacrificed by CO2 asphyxiation under isoflurane anesthesia at 4 and 24 h after injection. Organs of interest were collected, rinsed of excess blood, blotted, weighed, and counted in a γ-counter. The percentage injected dose per gram (%ID/g) of each tissue was calculated using appropriate standard solutions.
Nude Mice.
Female Swiss nu/nu mice (Charles River Laboratories) of 8 wk of age were inoculated in the left flank with a 150-μL suspension of 1.5–2 × 107 PC-3 cells in PBS buffer. Two weeks later, the same animals subcutaneously received a 150-μL suspension of 0.8–1 × 107 AR4-2J cells in PBS buffer in their right flank. Masses that were well palpable were grown 12 d later in both inoculation sites and biodistribution was conducted. Each mouse received a 100-μL bolus containing 185 kBq (5 μCi) [99mTc]Demobesin 1 (∼5 pmol total N4 peptide) and 74 kBq (2 μCi) [111In-DOTA0]Z-070 (∼4 pmol total DOTA peptide) in PBS through the tail vein. Animals were sacrificed in groups of 4 at 1, 4, or 24 h after injection by cardiac puncture under light ether anesthesia. Two separate groups of mice intravenously received 100 μg (first group) or 250 μg (second group) [Tyr4]BB along with the radiopeptides to determine the nonspecific accumulation at the target sites (blocked animals). These animals were sacrificed at 1 h after injection. Biodistribution was completed as described with radioactivity measurements conducted in 2 energy windows (120–160 keV for 99mTc and 140–270 keV for 111In).
All animal experiments were performed in compliance with national and European guidelines for animal treatment, and protocols were approved by either the Bundesamt für Veterinärwesen (approval no. 789) or the Veterinary Commission, Prefecture of Athens, Greece.
RESULTS
Peptide Conjugates
Synthesis and characterization of Demobesin 1 have been described elsewhere (28). Z-070 was synthesized using Fmoc strategy, affording an overall yield of approximately 23% based on the removal of the first Fmoc group.
Radiotracers
As previously reported, formation of [99mTc]Demobesin 1 was straightforward after incubation of Demobesin 1 with 99mTcO4−, SnCl2 at alkaline pH in the presence of citrate anions (28). Incorporation of 111In by the DOTA unit of Z-070 was complete after heating the 111InCl3 and Z-070 mixture under acidic conditions. Formation of desired radiopeptides was confirmed by comparison of radiolabeled products with nonradioactive metallated authentic samples by HPLC coupled to parallel photometric and radiometric detection modes (Table 1). Labeling yields >98% were verified by combined ITLC and RP-HPLC methods. Typical radiochromatograms of the 111In- and 99mTc-labeling reaction mixtures are shown in Figure 1.
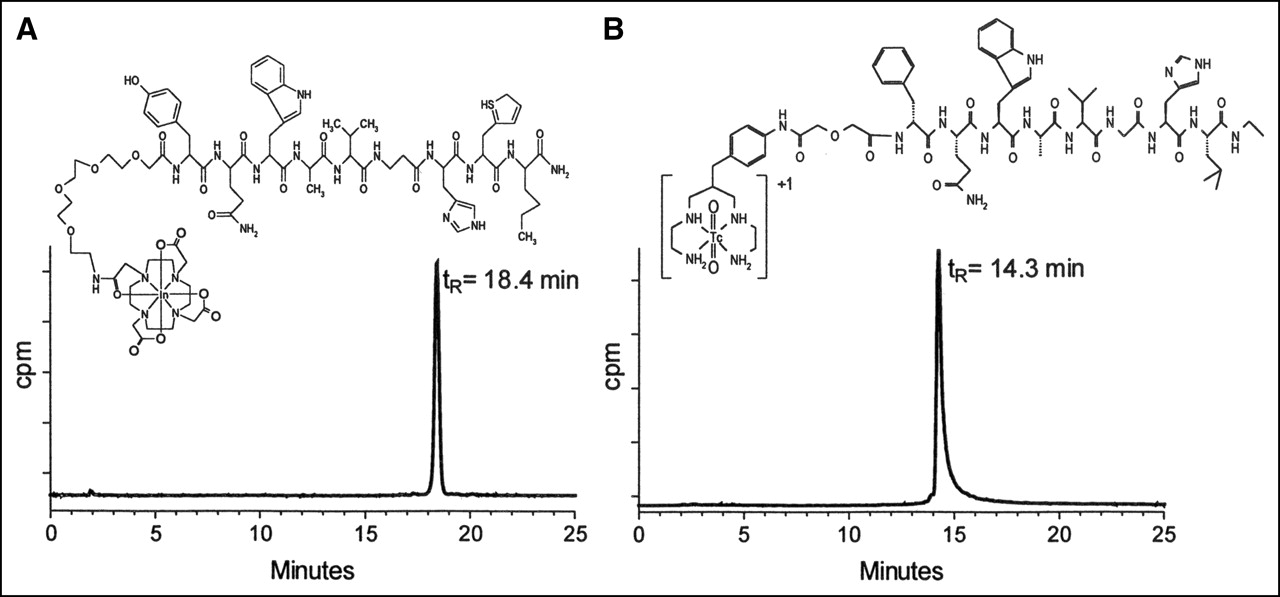
RP-HPLC analysis of [111In]Z-070 (A) and [99mTc]Demobesin 1 (B).
Comparative HPLC Profiles of Peptide Conjugates and Their Metal Complexes
Receptor-Binding Studies
The 50% inhibitory concentration (IC50) values determined during competition binding assays in AR4-2J cell membranes for Demobesin 1, Z-070, and its InIII- and YIII-metallated analogs were within a range of 0.17–0.45 nmol/L (Table 2), revealing that the rat GRP-R is not able to distinguish Demobesin 1 from the Z-070 peptides. In contrast, in PC-3 cell membranes expressing the human GRP-R, Demobesin 1 displayed IC50 values 11- to 14-fold lower (0.35 ± 0.33 nmol/L) than Z-070 and its metallated analogs (3.87–5.14 nmol/L) (Table 2).
Binding Affinity (IC50 in nmol/L [mean ± SEM]) to Human GRP-R and Mouse or Rat GRP-R of Bombesin Analogs
Receptor Autoradiography Studies
During competition binding assays on cryostat sections of mouse pancreas, the mouse GRP-R showed very high affinity for Z-070, [InIII]Z-070, and [LuIII]Z-070 whereas, during assays on cryostat sections of GRP-R–expressing human tumors, the human GRP-R showed a >13-fold lower affinity for these 3 compounds. Conversely, both the human and the mouse GRP-R showed a high affinity for Demobesin 1 (Table 2).
Internalization Experiments
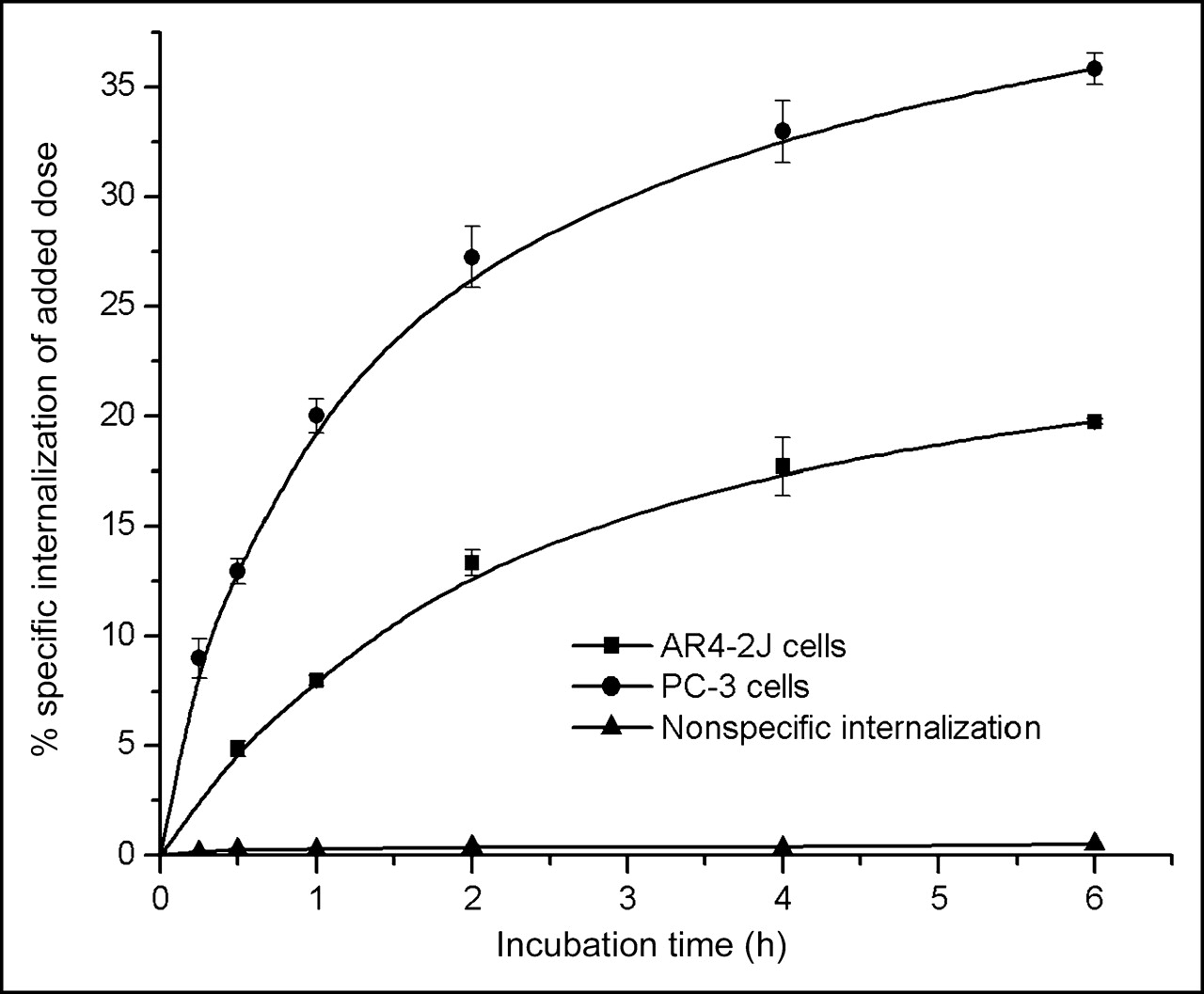
As presented in Figure 2, [111In]Z-070 internalized rapidly in both cell lines without reaching a plateau up to 6 h of incubation at 37°C. Internalization was strongly reduced in the presence of excess BZH2. The surface-bound peptide corresponded to <7.5% of added activity (data not shown). Internalization was much higher in PC-3 than in AR4-2J cells.

Comparison of [111In]Z-070 internalization rate in rat acinar pancreatic tumor AR4-2J cells and in human prostate adenocarcinoma PC-3 cells. Data were acquired from 2 independent experiments in triplicates and are expressed as specific internalization per million cells.
Animal Studies
AR4-2J Tumor-Bearing Rats.
Results from biodistribution of [111In]Z-070 in Lewis rats bearing AR4-2J tumors are presented in Table 3 as %ID/g. [111In-DOTA0]Z-070 displayed rapid clearance from blood, muscle, and GRP-R–negative tissues, except for the kidneys, the main organ of radiopeptide excretion. [111In-DOTA0]Z-070 showed an uptake of 1.55 ± 0.25 %ID/g in the AR4-2J tumor and 4.29 ± 0.61 %ID/g in the GRP-R–positive pancreas at 4 h after injection. Coinjection of excess BZH2 caused a 91% reduction of tumor uptake with a reduction similar to that evident in the pancreas (97.5%), bowel (84%), stomach, and adrenal (66%) values. As a result, high tumor-to-background ratios were attained by the tracer (Table 3).
Biodistribution Analysis (%ID/g ± SD, n = 4) and Tissue Ratios of [111In]Z-070 in AR4-2J Tumor-Bearing Lewis Rats
Nude Mice.
Biodistribution results for [111In]Z-070 and [99mTc]Demobesin 1 in female nude mice bearing both human PC-3 and rat AR4-2J tumors are included in Tables 4 and 5, respectively. Data expressed as %ID/g for the 1-, 4-, and 24-h time intervals show a very rapid blood clearance for both compounds. The bulk of radioactivity was sequestered mainly via the kidneys and the urinary system with [99mTc]Demobesin 1 showing also a higher degree of hepatobiliary excretion. Intestinal activity of both [99mTc]Demobesin 1 and [111In]Z-070 could be partly blocked by coinjection of excess [Tyr4]BB, implying, at least in part, a receptor-mediated process. Thus, for [111In]Z-070, coinjection of 100 μg [Tyr4]BB was sufficient to cause an ∼82% drop in the bowel values. In the case of [99mTc]Demobesin 1, this amount reduced intestinal activity only by 35%, whereas coinjection of 250 μg [Tyr4]BB resulted in a total 54% drop of intestinal accumulation. These findings imply that the higher intestinal accumulation of [99mTc]Demobesin 1 could be partially a result of its stronger binding to the natural bombesin-binding sites in the intestinal wall (38). [99mTc]Demobesin 1 showed a higher uptake than [111In]Z-070 in mouse pancreas. Coinjection of 100 μg [Tyr4]BB reduced pancreatic uptake by ∼90%. Again, a higher amount of [Tyr4]BB (250 μg) further reduced pancreatic uptake.
Biodistribution of [111In]Z-070 in Double (Rat AR4-2J and Human PC-3) Tumor-Bearing Athymic Mice
Biodistribution of [99mTc]Demobesin 1 in Double (Rat AR4-2J and Human PC-3) Tumor-Bearing Athymic Mice
In the AR4-2J tumor, expressing the rat GRP-R, [99mTc]Demobesin 1—despite its minimal internalization (28)—showed slightly higher tumor values (4.4 ± 0.9 %ID/g at 1 h after injection) during the initial time intervals than the rapidly internalizing [111In]Z-070 (3.2 ± 0.7 %ID/g at 1 h after injection). At 24 h after injection, [111In]Z-070 showed a 2.5-fold higher accumulation in the rat implant. In the human PC-3 xenografts, [99mTc]Demobesin 1 displayed a clearly higher uptake than the 111In peptide at all time points. Uptake in both tumors could be blocked in a dose-dependent manner by coinjection of [Tyr4]BB, suggesting a receptor-specific process. Again, [Tyr4]BB was less effective in competing with [99mTc]Demobesin 1 than with [111In]Z-070 for the bombesin-binding sites in the human xenografts.
DISCUSSION
A great number of bombesin analogs labeled with metallic radionuclides, including 99mTc, have been proposed recently for targeted diagnostic imaging and radionuclide therapy of GRP-R–expressing tumors. Although the feasibility of this approach has been established in a few patient studies (39,40), a systematic comparison of data extracted from preclinical testing or estimation of their clinical relevance is not possible due to the heterogeneity of biologic models used (19,28,29). Thus, GRP-R–positive cell lines of rat, mouse, and human sources have been used alternatively for in vitro tests and for developing experimental tumors on rats or mice. On the other hand, recent studies have demonstrated pharmacologic differences of bombesin receptors from different species despite their high sequence homology (8–10,23–27). Furthermore, the function of most bombesin-like peptides in the human is not well established yet, despite the wealth of data compiled from studies in animals. As a consequence, it is becoming more evident that—unlike in the case of radiolabeled somatostatin analogs—validation of radiolabeled bombesin-based peptides should rather rely on experimental tools expressing the human GRP-R as best matching human conditions. Systematic correlation of data derived from similar—if not identical—experimental models expressing the human GRP-Rs is expected to rapidly boost research toward more effective bombesin-like radiopharmaceuticals for the diagnosis and treatment of cancer.
In the present study, we were able to identify differences in the characteristics of 2 radiolabeled bombesin peptides, [111In]Z-070 and [99mTc]Demobesin 1, for the rodent or human GRP-R. These radiopeptides—although displaying similar profiles in rat or mouse GRP-R–expressing cells, tissues, and tumor-bearing animals—could be clearly distinguished in conditions using tissues that express the human GRP-R.
Thus, Z-070 and its metallated analogs showed high affinity for the rat GRP-R found in AR4-2J membranes. Concordant with this finding are the subnanomolar IC50 values of Z-070–related peptides for the mouse GRP-R determined by receptor autoradiography in mouse pancreas. By changing from rodent to human GRP-R–positive models—that is, to PC-3 cell membranes and human prostate cancer cryostat sections—the affinities of Z-070 and its metallated analogs dropped by 13-fold. In contrast, no significant differences were revealed in the affinity of Demobesin 1 versus the human or the mouse GRP-R.
It is interesting to observe how affinity differences affect individual tumor uptake in mice bearing both rat (AR4-2J) and human (PC-3) xenografts after taking into account other important parameters as well, such as internalization rates. In the AR4-2J tumors, [99mTc]Demobesin 1 and [111In]Z-070 showed similar uptake, as a result of their equally high affinities for the rat GRP-R and regardless of the high internalization capacity of the 111In tracer in AR4-2J cells. However, in the human PC-3 xenograft, [99mTc]Demobesin 1 showed an ∼2- to 3-fold higher uptake than that of [111In]Z-070 at all time intervals. This finding can be attributed to the higher affinity of Demobesin 1 for the human GRP-R in comparison with [111In]Z-070. In general, the tumor uptake of the 2 compounds seems to be very much related to their respective affinities for the rat or human GRP-R, with internalization playing a minor role. The animal experiments could confirm the capability of the human GRP-R—in contrast to the rodent GRP-R—to distinguish between [99mTc]Demobesin 1 and [111In]Z-070 also in vivo. Further studies seem to be warranted to adopt reliable and universally accepted criteria for the preclinical validation of GRP-R–targeting drugs.
CONCLUSION
The present study has demonstrated differences in the characteristics of radiolabeled bombesins for the rat or mouse and human GRP-R, both in vitro and in vivo. It emphasizes the importance of carefully selecting experimental tools in the validation of new bombesin-based radiopharmaceuticals for use in patients because of interspecies differences in the structure and, most importantly, in the pharmacology of human and animal GRP-Rs.
Acknowledgments
This work has been assisted by the European Union through COST Action B12: “Radiotracers for In Vivo Assessment of Biological Function”, WG-3: “Radiolabeled Biologically Active Peptides”. Financial support was provided by the Swiss National Science Foundation (grant 3100A0-100390) and by Biomedica Life Sciences, S.A.
Footnotes
Received Nov. 4, 2004; revision accepted Jan. 23, 2005.
For correspondence or reprints contact: Theodosia Maina, PhD, Institute of Radioisotopes–Radiodiagnostic Products, National Center for Scientific Research Demokritos, 153 10 Ag. Paraskevi Attikis, Athens, Greece.
E-mail: mainathe{at}rrp.demokritos.gr
REFERENCES
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Theranostic Perspectives in Prostate Cancer with the Gastrin-Releasing Peptide Receptor Antagonist NeoBOMB1: Preclinical and First Clinical Results
- 99mTc Radiotracers Based on Human GRP(18-27): Synthesis and Comparative Evaluation
- PET of Tumors Expressing Gastrin-Releasing Peptide Receptor with an 18F-Labeled Bombesin Analog
- Alpha- versus Beta-Particle Radiopeptide Therapy in a Human Prostate Cancer Model (213Bi-DOTA-PESIN and 213Bi-AMBA versus177Lu-DOTA-PESIN)
- Of Mice and Humans: Are They the Same?--Implications in Cancer Translational Research
- Evaluation of a 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid-Conjugated Bombesin-Based Radioantagonist for the Labeling with Single-Photon Emission Computed Tomography, Positron Emission Tomography, and Therapeutic Radionuclides
- [64Cu-NOTA-8-Aoc-BBN(7-14)NH2] targeting vector for positron-emission tomography imaging of gastrin-releasing peptide receptor-expressing tissues
- 18F-Labeled Bombesin Analogs for Targeting GRP Receptor-Expressing Prostate Cancer