


Abstract
Accurate differential diagnosis of parkinsonism is of paramount therapeutic and prognostic importance. In addition, with the development of invasive therapies and novel disease-specific therapies, strategies for patient enrichment in trial populations are of growing importance. Imaging disease-specific patterns of regional glucose metabolism with PET and 18F-FDG allows for a highly accurate distinction between Parkinson disease (PD) and atypical parkinsonian syndromes, including multiple-system atrophy, progressive supranuclear palsy, and corticobasal degeneration. On the basis of a preliminary metaanalysis of currently available studies with inclusion of multiple disease groups, we estimated that the diagnostic sensitivity and specificity for visual PET readings supported by voxel-based statistical analyses for diagnosis of atypical parkinsonian syndromes are 91.4% and 90.6%, respectively. The diagnostic specificity of 18F-FDG PET for diagnosing multiple-system atrophy, progressive supranuclear palsy, and corticobasal degeneration was consistently shown to be high (>90%), whereas sensitivity was more variable (>75%). It is increasingly acknowledged that cognitive impairment represents a major challenge in PD, with mild cognitive impairment being a prodromal stage of PD with dementia (PDD). In line with clinical and neuropsychologic studies, recent PET studies demonstrated that posterior cortical dysfunction in nondemented PD patients precedes cognitive decline and the development of PDD by several years. Taken together, the current literature underscores the utility of 18F-FDG PET for diagnostic evaluation of parkinsonism and the promising role of 18F-FDG PET for assessment and risk stratification of cognitive impairment in PD.
- FDG PET
- Parkinson disease
- multiple-system atrophy
- progressive supranuclear palsy
- corticobasal degeneration
- mild cognitive impairment
Molecular neuroimaging using PET allows for quantitative visualization of functional and molecular processes in vivo. 18F-FDG is the most commonly used radiotracer for the assessment of regional cerebral glucose metabolism as a marker of neuronal function. By virtue of disclosing disease-specific alterations due to synaptic dysfunction, neuronal degeneration, and accompanying compensatory network changes, 18F-FDG PET has become an essential part in the diagnostic work-up of patients with neurodegenerative disorders, most notably dementia. Although disease-specific patterns of regional glucose metabolism in patients with parkinsonism have been known since the early days of PET imaging (1–3), the valuable role of 18F-FDG PET for differential diagnosis of parkinsonism has been increasingly acknowledged only in recent years. This interest was prompted by several promising studies from different research groups worldwide, particularly the group from the Feinstein Institute (Table 1).
Overview and Preliminary Metaanalysis of Available Studies on Differential Diagnosis of Parkinsonism by 18F-FDG PET, Including Multiple Disease Groups (Group of APSs as Target Condition; i.e., Positive Case)
Against this background, this clinically oriented review on the use of 18F-FDG PET in neurodegenerative parkinsonism provides the clinical practitioner with an update on the clinical demand and rationale for 18F-FDG PET imaging in parkinsonism, typical 18F-FDG PET patterns and their value for differential diagnosis of parkinsonism (including a preliminary metaanalysis of recent studies), and an outlook on the promising role of 18F-FDG PET for diagnostic assessment and risk stratification in cognitive impairment in Parkinson disease (PD).
CLINICAL DEMAND AND RATIONALE FOR 18F-FDG PET IMAGING IN PARKINSONISM
PD
The clinical diagnosis of PD, as the most common neurodegenerative cause of parkinsonism, relies on the presence of the cardinal motor manifestations: bradykinesia, resting tremor, and rigidity. A clear response to dopaminergic therapy and the presence of either olfactory loss or cardiac sympathetic denervation are supportive criteria (4). Moreover, diagnosis of clinically established PD requires absence of the features of atypical parkinsonian syndromes (APSs).
The histopathologic hallmark of PD is the so-called Lewy pathology (i.e., α-synuclein immunoreactive neuronal inclusions) (5). Intracerebral Lewy pathology starts at clearly defined induction sites and advances in a topographically predictable sequence affecting the brain stem, subcortical nuclei, and neocortex (6). Consequently, loss of nigrostriatal dopaminergic projection neurons of the substantia nigra pars compacta (5) is primarily responsible for the development of typical motor symptoms. Cortical spread of Lewy pathology in addition to concomitant, possibly synergistic pathologies (e.g., Alzheimer-type pathology) and distant effects via disruption of modulatory projections from noradrenergic, serotonergic, dopaminergic, and cholinergic brain stem and basal forebrain nuclei contributes to cognitive decline, ranging from PD with mild cognitive impairment (PD-MCI) to PD with dementia (7). Although controversial, the so-called 1-y rule (i.e., the diagnosis is PD with dementia (PDD) if the onset of dementia occurs more than 1 y after the onset of parkinsonism) (8) is used by clinicians to distinguish dementia with Lewy bodies (DLB) from PDD. The continuum of PD, PD-MCI, PDD, and DLB is often referred to as the spectrum of Lewy body disorders (9).
APSs
The APSs comprise multiple-system atrophy (MSA), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD), which are a heterogeneous group of neurodegenerative disorders characterized by levodopa-refractory parkinsonism and distinctive atypical clinical features (10). In cases of MSA, one may observe various combinations of progressive autonomic failure, parkinsonian, cerebellar features, and pyramidal features. MSA is classified as of the parkinsonian subtype (MSA-P) if parkinsonism is the most prominent feature and as of the cerebellar subtype (MSA-C) if cerebellar ataxia predominates (11). MSA is defined as a primary oligodendroglial α-synucleinopathy with consecutive neuronal degeneration (11). MSA pathology most severely affects the substantia nigra, caudate, putamen, cerebellar white matter, pontine nuclei, inferior olivary nucleus, and medullary tegmentum (12).
The 4-repeat tauopathies PSP and CBD may be considered to represent different manifestations of a disease spectrum with several common clinical, pathologic, genetic, and biochemical features (13). In cases of PSP, recent Movement Disorder Society clinical diagnostic criteria propose 4 core functional domains—ocular motor dysfunction, postural instability, akinesia, and cognitive dysfunction—as characteristic clinical manifestations (14). Several clinical PSP presentations have been defined, with the combination of early-onset postural instability and falls with vertical ocular motor dysfunction representing the most frequent presentation (the so-called Richardson syndrome). Of particular relevance for the present context are also PSP presentations with prominent parkinsonism resembling PD (possibly including tremor and response to levodopa) and a corticobasal syndrome (14). PSP is histopathologically defined by intracerebral aggregation of the microtubule-associated protein tau, predominantly involving isoforms with 4 microtubule-binding repeats, in neurofibrillary tangles, oligodendrocytic coils, and, specifically, astrocytic tufts (14). Albeit heterogeneity in the distribution of tau pathology in PSP has to be recognized (reflecting clinical diversity), globus pallidus, subthalamic nucleus, and substantia nigra are consistently the regions most severely affected, whereas less tau immunoreactivity is detected in the posterior frontal cortex (15).
On the basis of autopsy-confirmed cases, 4 clinical phenotypes associated with CBD pathology have been proposed (16). The corticobasal syndrome phenotype, as the most frequent, prototypical variant, is characterized by an asymmetric presentation of limb rigidity or akinesia; limb dystonia or myoclonus; oro-buccal or limb apraxia; cortical sensory deficit; or alien limb phenomenon (16). Like PSP, CBD may also cause a frontal behavioral syndrome, primary progressive aphasia, and even a PSP-like syndrome. CBD is also characterized by widespread deposition of hyperphosphorylated 4-repeat tau in neurons and glia, the latter as astrocytic plaques. Strategic regions severely affected by CBD are the superior frontal gyrus, motor cortex, and substantia nigra (5).
Treatment and Prognosis
Clinicopathologic studies suggest that the clinical diagnosis of PD is inaccurate in about 20%–25% of patients (17,18). APSs are frequently misdiagnosed as PD. Even at advanced stages, when patients present with distinctive clinical features, a relevant fraction of patients with MSA and PSP (∼30%) and, particularly, CBD (≤74%) receive an incorrect diagnosis (19). The limited accuracy represents a major obstacle for treatment selection and reliable prognostic counseling: for PD, functional reconstitution of the dopaminergic nigrostriatal pathway is essential. Dopaminergic drugs alleviate both motor symptoms and (to some extent) nonmotor symptoms and improve quality of life (20). Trials investigating possible neuroprotective agents have supported, although not proven, disease-modifying effects of dopaminergics in PD (21). Given that common side effects (i.e., psychosis, orthostatic hypotension, and gastrointestinal disorders) may substantially compromise the clinical condition, reliable treatment-predictive biomarkers and diagnoses are desirable. Moreover, unnecessary treatment costs need to be avoided. For the management of advanced PD (when intractable levodopa-induced motor complications develop) invasive techniques, including deep-brain stimulation, stereotactic neurosurgery or other ablative treatments, and subcutaneous apomorphine therapy or intraduodenal levodopa infusion, are available (22,23). Experimental therapies have tried to restore striatal dopamine in PD patients by gene-based and cell-based approaches. Aside from ethical considerations and logistic challenges, data concerning the use of human fetal ventral mesencephalic allografts in PD are controversial (24,25).
The current focus of drug therapy for APSs is still primarily alleviation of symptoms, for which the therapy is often ineffective (10). Available data on interventional therapies in APSs are sparse: small case series highlight the risk of clinical worsening after deep-brain stimulation in histologically proven MSA patients and strongly discourage its use (26). In line with this finding, another case report revealed rapid post-thalamotomy deterioration in a patient who received a postmortem diagnosis of CBD (27). At present, no effective treatment exists to delay disease progression in APSs. In turn, disease-modifying trials with rasagiline and rifampicin for MSA or tideglusib and davunetide for PSP yielded negative results (10). Strategies to enhance trial success have been proposed, such as an enrichment design for novel disease-specific molecularly targeted therapeutics or identification of preclinical stages of parkinsonism, allowing for primary prevention and early disease-modifying trials. Most recently, several immunotherapeutic approaches to modify clearance, aggregation, and transport of α-synuclein (28) and tau (29) have been explored.
Survival is distinctly better in PD than in APSs: although some population-based studies did not find higher mortality (30), others convincingly demonstrated an increased age-adjusted mortality in PD (31). A higher age at onset, as in APSs, is also associated with a higher PD mortality (32), but median survival was still 10.3 y in a recent population-based cohort with a high average age of 70 y at diagnosis (30). However, development of PDD is associated with a strong increase in mortality (33). In fact, the mean time span between onset of dementia and death appears to be only about 2–4 y (33,34). Compared with PD, APSs are characterized by the early presence of additional debilitating symptoms and a more rapid progression to death (10). According to clinicopathologic studies, APSs share a comparably short survival of about 7–8 y from symptom onset, or less than 3–4 y from the typically somewhat delayed clinical diagnosis (32).
Taken together, accurate diagnosis of the underlying cause of parkinsonism is required for decisions on treatment strategies (including invasive techniques), inclusion in therapy trials (e.g., enrichment of patient populations for novel therapies), and prognostic statements.
DIFFERENTIAL DIAGNOSIS OF PARKINSONISM
In our opinion and as practiced by many centers, 18F-FDG PET may be used for differential diagnosis of parkinsonism when there remains substantial uncertainty about the underlying cause of parkinsonism after comprehensive evaluation by a movement disorder specialist and when the result of the examination is expected to alter patient management (e.g., ordering further diagnostic tests, medication, other treatments, and patient counseling). This view is strengthened by a larger prospective study supporting American Academy of Neurology level I evidence for 18F-FDG PET as a test for APS differential diagnosis (35,36) and the present metaanalysis.
The preceding diagnostic work-up usually includes MRI, which may provide valuable diagnostic findings in secondary parkinsonism (e.g., vascular parkinsonism) and in APSs with relatively high specificity but low sensitivity (particularly in early disease stages) (37). In our experience, it is neither necessary nor effective in terms of costs and radiation burden to always verify beforehand a neurodegenerative cause of parkinsonism by imaging of nigrostriatal integrity (e.g., with 123I-FP-CIT [N-(3-fluoropropyl)-2β-carbon ethoxy-3β-(4-iodophenyl)nortropane] SPECT), because an 18F-FDG PET scan in patients with suspected APSs often provides typical findings that render additional imaging of nigrostriatal integrity dispensable. Technical aspects of 18F-FDG PET brain imaging (e.g., patient preparation, data acquisition, and image interpretation) have been presented in respective procedure guidelines (38,39).
Disease-Specific Patterns of Regional Glucose Metabolism
Disease-specific patterns of altered cerebral glucose metabolism used for differential diagnosis of PD and APS subgroups (35,40–42) are schematically illustrated in Figure 1. Furthermore, transaxial 18F-FDG PET images and outputs of currently used image analysis methods are depicted in Figure 2.
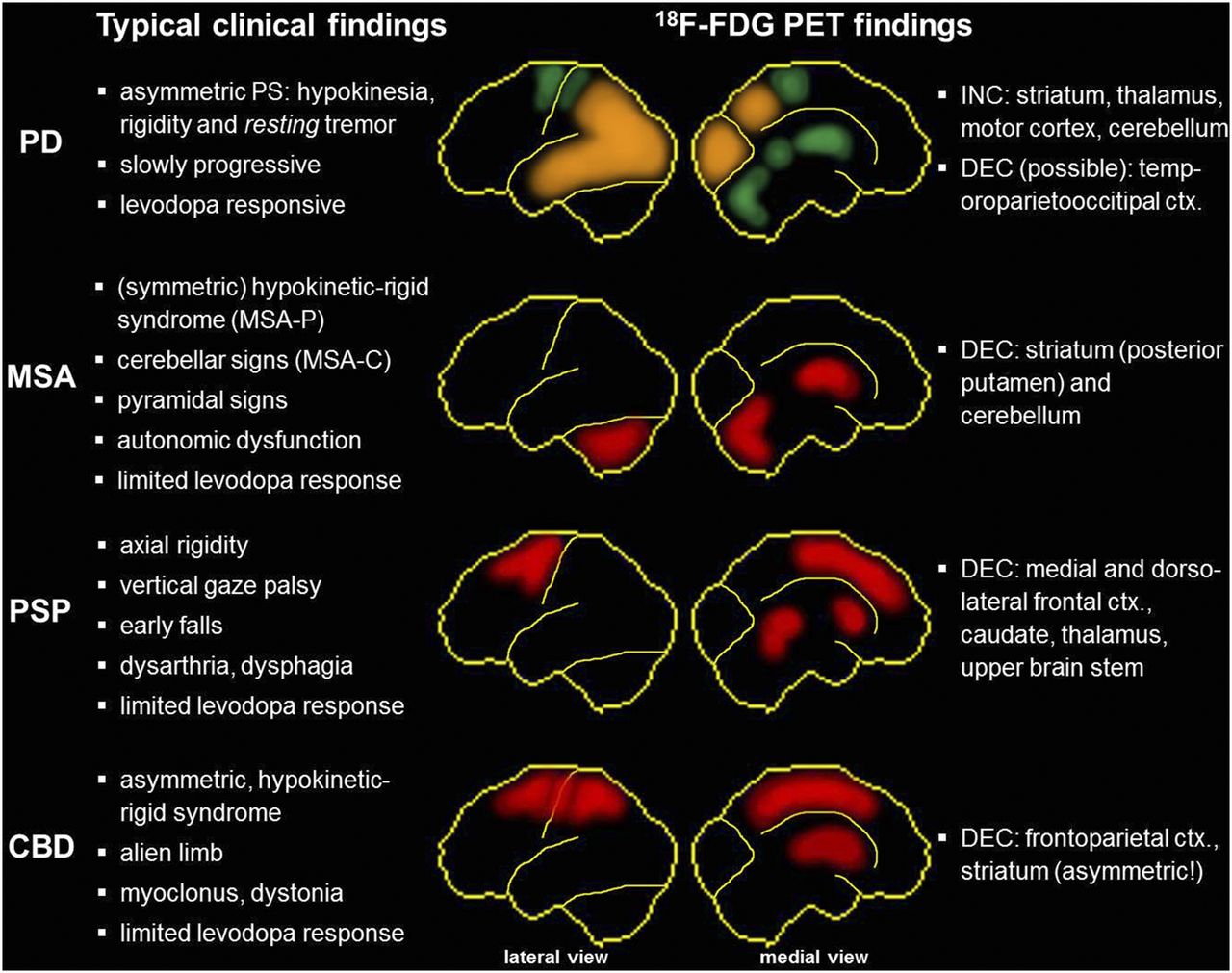
Summary and schematic illustration of typical clinical findings and disease-specific metabolic patterns yielded by 18F-FDG PET in parkinsonism. Green regions show relative hypermetabolism, red regions exhibit hypometabolism, and orange regions indicate possible metabolic decreases in PD (especially in patients with cognitive impairment). Shown are lateral (left) and medial (right) views of brain. Hypometabolism in CBD is usually markedly asymmetric, with lower metabolism contralateral to the clinically more affected body side. ctx = cortex; DEC = decrease; INC = increase. MSA-P and MSA-C = multiple-system atrophy with predominant parkinsonism and cerebellar symptoms, respectively; PS = parkinsonian syndrome.

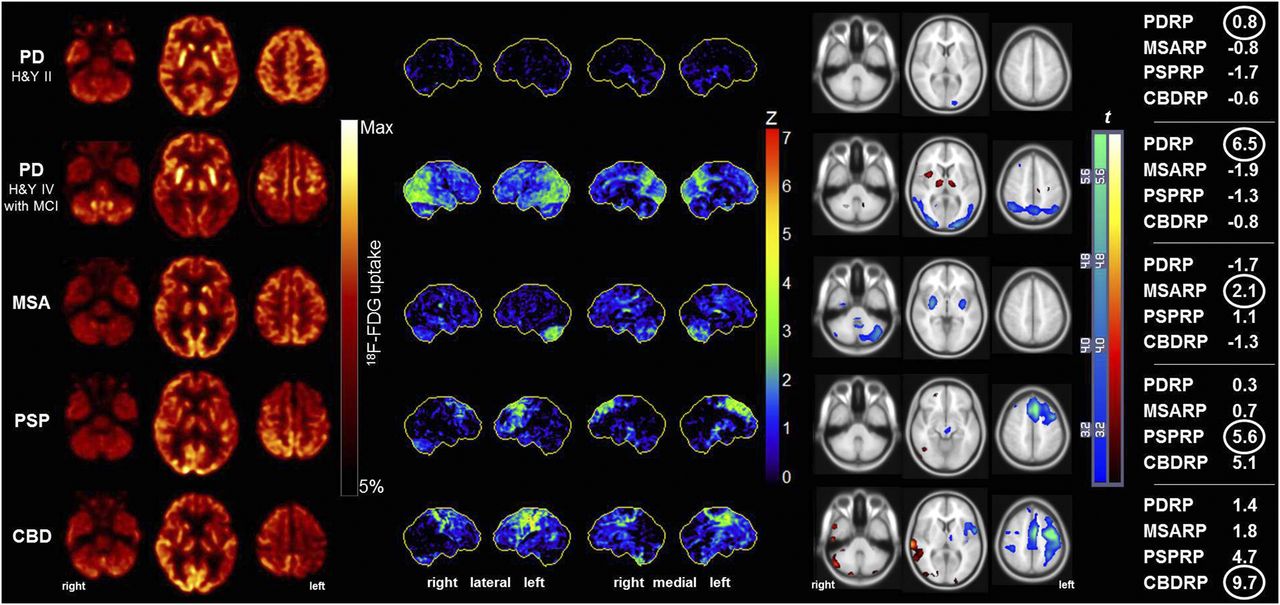
Typical 18F-FDG PET findings in individual patients with parkinsonism as depicted by the currently used image analysis methods. First panel shows spatially normalized transaxial 18F-FDG PET cross-sections at levels of cerebellum, basal ganglia, and dorsal frontoparietal cortex. Datasets were threshold-constrained for optimal display. Second panel shows individual results of voxelwise statistical analysis with NeuroSTAT/3D-SSP. Shown are lateral and medial views of brain. Metabolic deficits compared with age-matched control subjects are color-coded as z scores. Third panel shows individual results of voxelwise statistical analysis with SPM. Shown are t maps in comparison to age-matched control subjects overlaid on SPM MRI template. Metabolic decreases (blue color-scale) and increases (hot metal color-scale) were constrained to threshold at voxelwise P level of less than 0.01 and cluster extent of more than 50 voxels. Fourth panel shows z scores (relative to healthy controls) of metabolic covariance network expression for PD-related pattern, MSA-related pattern, PSP-related pattern, and CBD-related pattern (PDRP, MSARP, PSPRP, and CBDRP, respectively). Network showing highest expression (encircled) always matched reference diagnosis. Expression scores for PD-related cognitive pattern were 0.7 in patient with PD Hoehn & Yahr (H&Y) stage II without MCI and 2.6 in patient with PD H&Y IV with MCI.
Scans of PD patients often show no major abnormality at first glance. On closer inspection and especially on voxel-based statistical analyses, PD is characterized by a posterior temporoparietal, occipital, and sometimes frontal hypometabolism (especially in PD with cognitive impairment) and relative hypermetabolism of the putamen, pallidum, thalamus sensorimotor cortex, pons, and cerebellum.
Conversely, MSA patients show a marked hypometabolism of the (posterior) putamen, pons, and cerebellum, which may be more pronounced in the striatum or in the pons and cerebellum, depending on the predominant side of degeneration and, thus, clinical presentation (striatonigral/MSA-P and olivopontocerebellar/MSA-C, respectively) (43). An isolated cerebellar hypometabolism may also occur in other causes of cerebellar degeneration (e.g., paraneoplastic or spinocerebellar ataxia). Group analyses demonstrated also a frontal hypometabolism that may spread to parietal and temporal areas simultaneously with the onset of cognitive impairment (44). The latter finding, however, is less apparent in individual analyses and of little help for differential diagnosis by 18F-FDG PET.
In the case of PSP, regional hypometabolism is consistently noted in the medial, dorsal, and ventrolateral frontal areas (i.e., the anterior cingulate gyrus, supplementary motor area, precentral gyrus, and premotor–to–posterior prefrontal areas); caudate; thalamus; and upper brain stem. These findings have also been confirmed in a recent study with postmortem verification (45). The recently proposed Movement Disorder Society clinical diagnostic criteria for PSP set a framework for the diagnosis of several PSP predominance types (14), which can be expected to also differ on 18F-FDG PET. For example, the aforementioned functional domains have been linked to predominant bilateral regional hypometabolism of the anterior cingulate gyrus (vertical gaze palsy (46)), thalamus (repeated unprovoked falls (47)), midbrain (gait freezing (48)), and left medial and dorsolateral frontal lobe (nonfluent aphasia (49)).
Finally, CBD is characterized by a usually highly asymmetric hypometabolism of the frontoparietal areas, striatum, and thalamus contralateral to the most affected body side. Cortical hypometabolism may be pronounced in the parietal cortex and usually extends across the sensorimotor cortex into the cingulate gyrus and premotor–to–posterior prefrontal areas.
A corticobasal syndrome may also be caused by several diseases other than CBD, most notably Alzheimer disease and PSP. Consequently, the clinical diagnosis of CBD is notoriously inaccurate: only about 25%–50% of patients with a corticobasal syndrome are found to have CBD on postmortem examination, whereas only 25%–30% of patients with neuropathologically verified CBD present with a corticobasal syndrome (19,50). Likewise, imaging results in patients with clinically diagnosed PSP and CBD may be very similar. In direct comparison of clinically defined patient groups, PSP patients showed a lower metabolism in the midbrain, thalamus, and cingulate gyrus, whereas CBD patients revealed a more pronounced and asymmetric hypometabolism in the parietal and sensorimotor cortex and striatum (51–54). According to recent studies with support by postmortem data, parietal involvement and the asymmetry of the aforementioned frontoparietal hypometabolism appear to be most characteristic of CBD (45,55). However, asymmetric hypometabolism on 18F-FDG PET may also be encountered in PSP (Fig. 2), with an asymmetric PSP presentation being related to an asymmetric metabolism in the motor cortex, cingulate gyrus, and thalamus (51). Finally, a recent study suggested that more prominent temporoparietal than frontal hypometabolism points toward an underlying Alzheimer pathology in patients with corticobasal syndrome, as confirmed by amyloid PET and pathology in roughly two thirds of these cases (56).
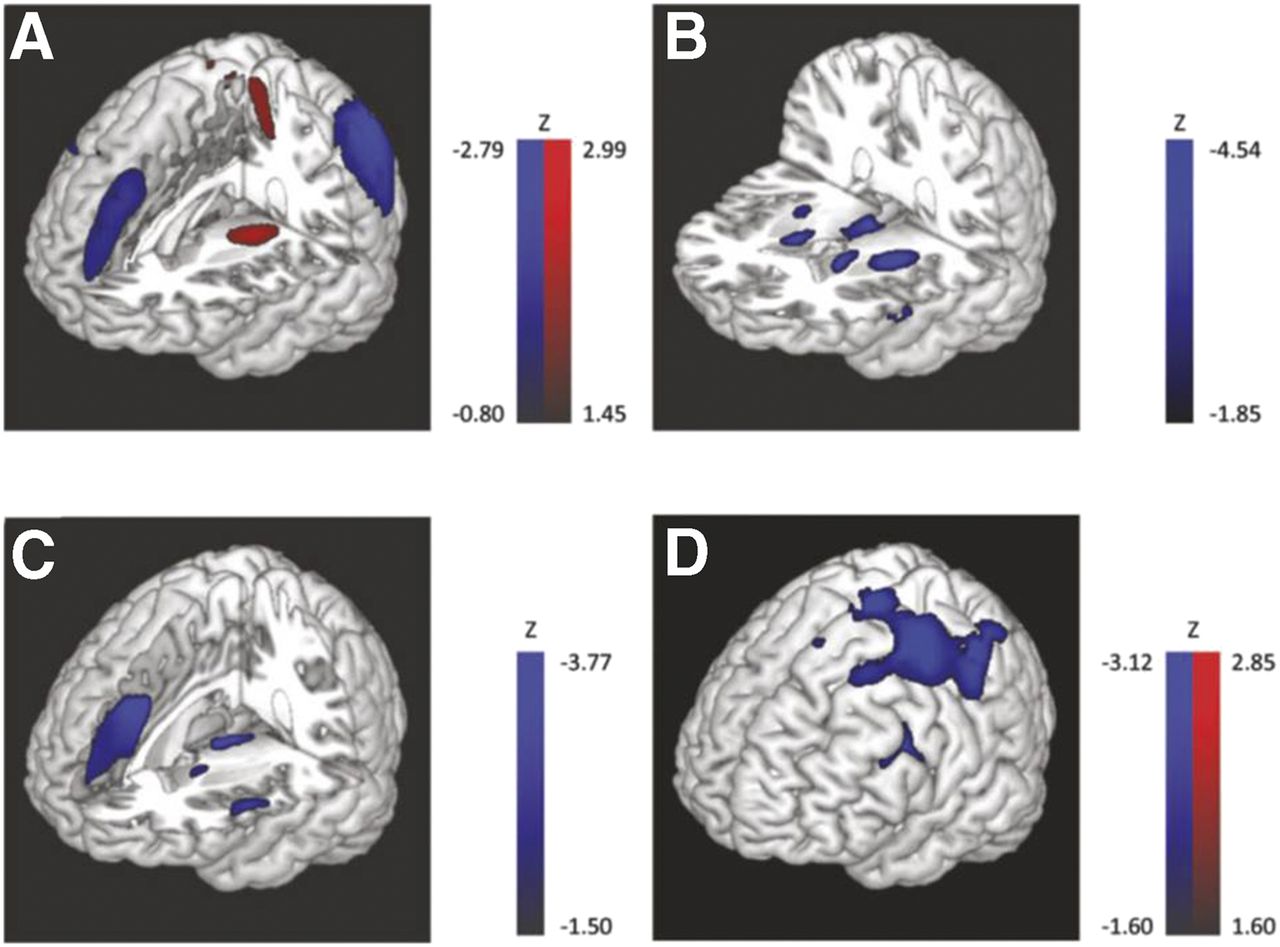
The aforementioned results were gained mostly from categoric comparisons. However, they fit the results gained from spatial covariance analyses, which were used to detect abnormal metabolic networks in PD, MSA, PSP, and CBD. These disease-related patterns (i.e., PD-, MSA-, PSP-, and CBD-related patterns) (Fig. 3) were demonstrated to be highly reproducible, to correlate with disease severity and duration (albeit to a variable extent in PD, MSA, and PSP), and to allow for prospective discrimination between cohorts (55,57–60). Furthermore, these patterns were confirmed by several independent groups relying on different patient populations and equipment (61–63). Expression of the PD-related pattern inversely correlates with striatal dopaminergic integrity (as assessed by 6-18F-fluoro-l-dopa PET) (64) and is already significantly increased in the ipsilateral (presymptomatic) hemisphere of patients with hemiparkinsonism (65). Likewise, recent studies demonstrated that expression of the PD-related pattern is also increased in the rapid-eye-movement sleep behavior disorder (66,67), being a significant predictor of phenoconversion to PD or DLB (assessed with perfusion SPECT (66)). In parallel to the respective metabolic changes (i.e., decrease in putamen/pallidum, sensorimotor cortex, and cerebellum; increase in precuneus) and symptom improvement, expression of the PD-related pattern was also found to decline with levodopa administration and deep-brain stimulation of the subthalamic nucleus in PD (68). Thus, covariance patterns of cerebral glucose metabolism represent interesting, observer-independent biomarkers for early diagnosis and therapy monitoring.

Disease-related metabolic covariance patterns: PD-related (A), MSA-related (B), PSP-related (C), and CBD-related (D). Metabolic increases and decreases are color-coded in blue and red, respectively; voxel loadings are z-scored. CBD-related pattern is usually markedly asymmetric (i.e., higher contralateral to clinically more affected body side). (Reprinted with permission of (58).)
Diagnostic Value: Preliminary Metaanalysis
Several larger, in part prospective studies investigated the applicability of 18F-FDG PET for the differential diagnosis of parkinsonism. Given the paucity of disease-specific treatments for APS subtypes, distinguishing PD from APSs (as an umbrella diagnosis) is of major therapeutic and prognostic relevance. To better characterize the value of 18F-FDG PET for diagnosis of APSs (as the target condition—i.e., positive case—as opposed to PD) and to compare the analysis methods used so far, we conducted a preliminary metaanalysis, which is described in the supplemental materials (available at http://jnm.snmjournals.org).
Table 1 summarizes the results of currently available studies and the resulting metaanalyses. PET image analyses can be divided into 2 major approaches (Fig. 2). (A review of image analysis methods was recently published by Meles et al. (69).) Five studies (35,40,41,70,71) used observer-dependent visual reads supported by voxel-based statistical analyses in comparison to healthy control subjects (done by statistical parametric mapping [SPM; http://www.fil.ion.ucl.ac.uk/spm/] or NeuroSTAT/3D-SSP [https://neurostat.neuro.utah.edu/]) (72). Three studies used observer-independent automated statistical classifications of patients with parkinsonism that applied logistic regression based on the expression of metabolic covariance patterns (42,73) or a relevance vector machine in combination with bootstrap resampling for nonhierarchic multiclass classification (74).
Most studies found 18F-FDG PET to be highly accurate (>90%) at distinguishing between APSs and PD. Figure 4 gives the summary receiver-operating-characteristic curves for the 2 analysis approaches used. The estimated pooled sensitivity of observer-dependent visual reads supported by voxel-based statistical analyses was significantly higher than the pooled sensitivity of observer-independent automated classifications (91.4% vs. 76.5%; P < 0.01), whereas the pooled specificity was slightly, though not significantly, higher for the automated analyses (94.7% vs. 90.6%; Table 1).

Summary receiver-operating-characteristic curves for differentiation between PD and APSs by 18F-FDG PET (APSs as target condition; i.e., positive case). Green and red lines represent summary receiver-operating-characteristic curves of observer-dependent visual reads supported by, respectively, voxel-based statistical analyses (methods 1.1 and 1.2 in Table 1; area under curve, 0.96 [95% confidence interval, 0.94–0.98]) and observer-independent automated classifications (methods 2.1 and 2.2 in Table 1; area under curve, 0.94 [0.90–0.97]).
Various factors might influence the diagnostic performance of 18F-FDG PET. However, current studies support the robustness of the method: exploring the effect of disease duration on classification accuracy, Eckert et al. (40) showed that agreement with the final clinical diagnosis was slightly higher for patients with early-stage PD (disease duration < 5 y) than for those with late-stage PD (98% vs. 92%) when using SPM-supported readings, but not for visual readings alone (88% vs. 97%). In the studies by Hellwig et al. (35) and Tripathi et al. (73), no difference was observed for distinguishing between PD and APSs when stratifying by disease duration (area under the receiver-operating-characteristic curve ≥ 0.93), whereas in the study by Tang et al. (42), diagnostic sensitivity was somewhat higher in patients with a longer disease duration (≥2 y) than in those with a shorter disease duration (APSs, 87% vs. 72%; PD, 88% vs. 77%).
Interestingly, in the latter study, diagnostic performance increased with longer clinical follow-up (≥2 y), suggesting that the apparent lower performance of imaging classification in patients with short follow-up (<2 y) is probably caused by initial clinical uncertainty, not incorrect imaging diagnosis (42).
In the study by Hellwig et al. (35), 11 of 34 patients belonging to the spectrum of Lewy body disorders received a final diagnosis of PDD or DLB after follow-up. Exclusion of these patients did not affect the diagnostic performance of 18F-FDG PET (area under the receiver-operating-characteristic curve, 0.93 vs. 0.94 before vs. after exclusion of PDD/DLB).
Given the aforementioned effects of treatment on regional metabolism and metabolic network expression, it may be advisable to scan patients in the off-medication state. However, a study comparing the diagnostic performance of the PD-related pattern in distinguishing between healthy controls and PD patients in the off-medication and on-medication states showed no major impact (62), and the effect of medication on regional metabolism in patients with APSs is unknown. The studies summarized in Table 1 were done under both conditions, without apparent impact.
The notably higher sensitivity of observer-dependent visual reads supported by voxel-based statistical analyses may represent an advantage over observer-independent automated analyses. In turn, observer dependence may be a disadvantage if expertise is limited. However, Eckert et al. (40) demonstrated that nonexpert investigators using SPM-supported reads performed better (92% accuracy) than experts using visual reads alone (85%), whereas in the study by Tripathi et al. (71), 2 experts performed comparably with (91%) and without (92%) using SPM-supported reads. Since NeuroSTAT/3D-SSP does not cover the striatum, SPM might be preferable, especially if reads are performed by nonexperts. The current automated method based on disease-specific network expression is limited with regard to cases classified as “indeterminate parkinsonism,” which accounted for 14% and 19% of patients in 2 studies (42,73). This issue seems to limit sensitivity for early APSs (73) and may also become more relevant if patients with severe structural abnormalities or alternative diagnosis are included.
Aside from multiclass relevance vector machine analysis (74), the specificity of the PET diagnoses for the APS subgroups MSA, PSP, and CBD usually exceeded 90% (as requested for a confirmatory test), whereas sensitivity was 77%–96% for MSA, 74%–100% for PSP, and 75%–91% for CBD (35,40–42,70,71,73). Such subgroup classifications will be important for patient enrichment in future trials with disease- or pathology-specific treatments. However, given the clinical and imaging ambiguity in the case of PSP and CBD, it may be advisable to use a combined PSP/CBD tauopathy category for PET readings, which reaches a sensitivity and specificity of 87% and 100%, respectively (35).
Finally, a recent study investigated the prognostic value of 18F-FDG PET in parkinsonism (32). Using a follow-up of up to 6 y (median survival duration: PD, not reached; APSs, 4.1 y after PET), risk stratification concerning overall survival yielded by PET was at least as good as that provided by the clinical diagnosis filed 1 y after PET (age-adjusted hazard ratios relative to PD for PET: PSP/CBD, 5.2, and MSA, 5.6; for clinical diagnosis: PSP/CBD, 4.5, and MSA, not significant). That study highlighted for the first time the prognostic relevance of 18F-FDG PET, which is of particular importance not only for the patients but also for their relatives and caregivers.
OUTLOOK
Cognitive Impairment in PD
The onset of cognitive impairment in PD, by the definition given earlier, follows the onset of motor symptoms (8), and both the diagnosis of PDD and the diagnosis of PD-MCI require an established PD diagnosis (75,76). Cognitive impairment is frequent in PD, with up to 80% of all patients developing dementia over the long term (77,78). The putative prodromal stage of PDD—PD-MCI (requiring functional independence, in contrast to PDD, in which activities of daily living are impaired)—is common in nondemented patients with PD (cross-sectional prevalence of 27% (79)). In a population-based study, 20% of incident PD cases had PD-MCI, and another 20% developed PD-MCI over the 5-y study period. PD-MCI was a major risk factor for PDD, with an annual conversion rate of 13% (79), which is similar to the rate of conversion for MCI (without PD) to Alzheimer dementia (80). However, a significant proportion of patients who had PD-MCI also reverted to normal cognition in that study (25% over 5 y (79)).
The spatial distribution of Lewy-related pathology contributes to the type of predominant cognitive deficit in a given patient, and the most affected cognitive domains in PD are the executive, visuospatial, attention, and memory functions (76,81). The diagnosis of PD-MCI requires objective cognitive impairment, at least on a scale of global cognitive abilities (level 1 (76)). However, greater diagnostic certainty and sensitivity is achieved with at least 2 neuropsychologic tests probing each of the 4 cognitive domains mentioned above plus the language domain (level 2 (76)). Although the current diagnostic criteria for PD-MCI recommend formal neuropsychologic testing across a range of cognitive domains, it might be especially efficient to focus on visuospatial abilities or semantic fluency, as there is emerging evidence of a dual cognitive syndrome in PD (82,83): a frontostriatal syndrome and a posterior cortical syndrome. These syndromes are presumably independent but frequently occur together. The cognitive deficits associated with the frontostriatal syndrome typically involve phonemic verbal fluency and other executive functions, such as flexibility or planning abilities. By contrast, figure drawing, but also semantic verbal fluency or episodic memory, are involved in the posterior cortical syndrome. Importantly, the risk for developing dementia is not associated with the frontostriatal syndrome but is significantly increased in the presence of the posterior cortical syndrome (83), thereby having special prognostic value. Whereas disturbance of the dopaminergic transmitter system (from degeneration of the substantia nigra pars compacta) presumably underlies the frontostriatal syndrome, altered cholinergic transmission (from degeneration of the nucleus basalis of Meynert) is primarily associated with the posterior cortical syndrome (81). Consistently, a recent metaanalysis provided strong evidence for the efficacy of acetylcholinesterase inhibitors (donepezil, rivastigmine) in the treatment of cognitive (and psychiatric) symptoms in PDD/DLB (84).
Because of the high prevalence and clinical importance of cognitive impairment in PD, valid biomarkers for risk stratification are wanted.
18F-FDG PET Imaging of Cognitive Impairment in PD
Interestingly, cortical hypometabolism, particularly in the posterior temporoparietal and occipital areas, is frequently observed in nondemented PD patients (Figs. 1 and 2). These changes often match the changes seen in PDD and also DLB, thus raising the question of whether they predict cognitive decline in PD.
Both PDD and DLB are characterized by a widespread lateral frontal and temporoparietal hypometabolism in addition to an occipital hypometabolism, which distinguishes PDD and DLB from Alzheimer dementia (85,86). In turn, metabolic differences between PDD and DLB, if any, appear to be subtle (87). The widespread involvement of cortical and subcortical areas in the spectrum of Lewy body disease was also demonstrated by studies showing an extensive positive correlation between results on the mini-mental status examination and regional glucose metabolism in the lateral parietal, occipital, temporal, and frontal association cortices; anterior cingulate; precuneus; and caudate nucleus across the spectrum of PD to PDD (88,89).
Consequently, spatial covariance analyses were found to be sensitive enough to identify a PD-related cognitive pattern even in nondemented patients. The pattern is characterized by metabolic decreases in the rostral supplementary motor area, dorsal premotor area, prefrontal cortex, precuneus, and parietal cortex and increases in the cerebellar vermis and dentate nucleus (90). A PD-related cognitive pattern was found to be reproducible across institutions and patient populations (minor differences, such as regarding the caudate nucleus and anterior cingulate, may stem from different tests), to correlate with executive and memory performance, and to be increased in PD-MCI compared with cognitively unimpaired PD patients (90–92). Despite its spatial overlap with the PD-related pattern, PD-related cognitive pattern is orthogonal to the PD-related pattern and does not correlate with the severity of motor impairment (90,93). Expression of the PD-related cognitive pattern seems to correlate with nigrostriatal dopaminergic dysfunction of the caudate nucleus (64,94) and with levodopa-induced improvement in verbal learning in a subgroup of PD patients without dementia (95). Thus, assessing expression of PD-related cognitive pattern may be a valuable tool for objectifying and monitoring cognitive dysfunction in PD. Future studies need to explore the predictive value of PD-related cognitive pattern for cognitive decline in PD-MCI in comparison to more specific posterior cortical measures, given the particular prognostic value of the posterior cortical syndrome.
The results of several longitudinal studies investigating cognitive decline and regional metabolism in PD have recently become available: In a seminal work, Bohnen et al. (96) demonstrated that conversion from PD to PDD was heralded by significant hypometabolism in the posterior cingulate, occipital cortex (Brodmann area 18/19), and caudate nucleus. Hypometabolism in the primary visual cortex (Brodmann area 17) was also observed in cognitively stable PD patients. Metabolic decline was widespread in converters on 2-y follow-up PET, involving the association cortices, posterior cingulate, hippocampus, and thalamus. Subsequent studies on nondemented PD patients confirmed these results, showing that bilateral hypometabolism in the precuneus and lateral posterior temporoparietal and occipital regions at baseline predicted cognitive decline in the following 3 y (97–99). These findings agree with earlier cross-sectional studies (87,100).
PET studies on PD-MCI patients support the notion that PD-MCI represents a prodromal stage of PDD, especially in those with posterior cortical hypometabolism. Patients with MCI typically exhibit decreased temporoparietal, occipital, precuneal, and frontal metabolism when compared with healthy controls and, to a lesser extent, PD patients without MCI (88,91,98,101,102). These changes were more pronounced in multidomain than single-domain MCI (91,102). Recent studies using the current diagnostic criteria for PD-MCI demonstrated that the overall pattern of hypometabolism gradually develops from smaller clusters of mainly parietal and occipital (and sometimes frontal) hypometabolism in cognitively normal PD patients, to more widespread parietal, occipital, frontal, and (to a lesser extent) posterior temporal clusters in PD-MCI, and then to extensive parietal, occipital, frontal, and temporal hypometabolism in PDD (88,98,103). Interestingly, hypometabolism on 18F-FDG PET preceded spatially matching atrophic changes on MRI (103). Across the entire group of patients with PD, PD-MCI, or PDD, regional glucose metabolism correlated with memory and visuospatial functions in the posterior temporal and parietal regions and also with attentional, executive, and language functions in the frontal regions (88,98). Several additional clinical observations support the strong negative prognostic value of posterior cortical hypometabolism. For example, recent longitudinal studies demonstrated that visual hallucinations and hyposmia, which coincided with a marked temporoparietal and occipital hypometabolism in PD (with or without MCI), were associated with an increased conversion to PDD (104,105). Likewise, PD patients with the rapid-eye-movement sleep behavior disorder showed lower cognitive performance and a higher likelihood of MCI and posterior cortical hypometabolism than did PD patient without this disorder (106). Finally, the aforementioned long-term follow-up study (32) found that patients with posterior cortical hypometabolism exhibited twice the mortality risk of PD patients without such a pattern.
Taken together, these findings indicate that posterior cortical hypometabolism has an importance of which the nuclear medicine practitioner should be aware. Although it is probably premature to propose clinical use of posterior cortical hypometabolism as a predictor of cognitive decline in PD, this finding may prompt further examinations and special consideration under specific circumstances (e.g., counseling of patients with PD-MCI or before initiating invasive therapies such as deep-brain stimulation).
CONCLUSION
18F-FDG PET allows for accurate differentiation between PD and APSs, which is of paramount therapeutic and prognostic importance. Furthermore, 18F-FDG PET provides a highly specific differential diagnosis between the APS subtypes MSA, PSP, and CBD. However, given the limited accuracy of the clinical diagnosis as the reference standard, future studies with postmortem verification are needed to validate the diagnostic imaging patterns, particularly in tauopathies.
Current 18F-FDG PET studies strongly support the concept that PD-MCI represents a prodromal stage of PDD. These studies underline that posterior cortical hypometabolism in nondemented PD patients is not only a diagnostically useful epiphenomenon but also a negative prognostic marker. Future prospective studies are needed to confirm the prognostic value of 18F-FDG PET.
Acknowledgments
Regarding Figure 2, we thank Drs. Yilong Ma and David Eidelberg of the Center for Neurosciences, Feinstein Institute for Medical Research, Manhasset, NY, for providing the analyses of disease-specific metabolic network expression.
Footnotes
Published online Sep. 14, 2017.
Learning Objectives: On successful completion of this activity, participants should be able to (1) describe the clinical demand and rationale for differential diagnosis in parkinsonism, (2) recognize and differentiate the disease-specific patterns of regional glucose metabolism associated with Parkinson disease and the different atypical parkinsonian syndromes, and (3) identify regional metabolic changes that predict cognitive decline in Parkinson disease.
Financial Disclosure: Dr. Meyer receives support from GE and Piramal for a research study outside the present topic. The authors of this article have indicated no other relevant relationships that could be perceived as a real or apparent conflict of interest.
CME Credit: SNMMI is accredited by the Accreditation Council for Continuing Medical Education (ACCME) to sponsor continuing education for physicians. SNMMI designates each JNM continuing education article for a maximum of 2.0 AMA PRA category 1 credits. Physicians should claim only credit commensurate with the extent of their participation in the activity. For CE credit, SAM, and other credit types, participants can access this activity through the SNMMI website (http://www.snmmilearningcenter.org) through December 2020.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- 1.↵
- 2.
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.
- 53.
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.
- 59.
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- Received for publication May 31, 2017.
- Accepted for publication August 10, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- [18F]Florzolotau PET for the Differential Diagnosis of Parkinsonism in Patients with Suspected 4-Repeat Tauopathies
- Atypical Parkinsonian Syndromes: Structural, Functional, and Molecular Imaging Features
- SNMMI Procedure Standard/EANM Practice Guideline for Brain [18F]FDG PET Imaging, Version 2.0
- Added value of FDG-PET for detection of progressive supranuclear palsy
- In vivo assessment of astrocyte reactivity in patients with progressive supranuclear palsy
- Molecular Imaging Findings on Acute and Long-Term Effects of COVID-19 on the Brain: A Systematic Review
- 18F-FDG PET Imaging in Neurodegenerative Dementing Disorders: Insights into Subtype Classification, Emerging Disease Categories, and Mixed Dementia with Copathologies
- Synaptic density and neuronal metabolic function measured by PET in the unilateral 6-OHDA rat model of Parkinsons disease
- Limits for Reduction of Acquisition Time and Administered Activity in 18F-FDG PET Studies of Alzheimer Dementia and Frontotemporal Dementia
- Principal Components Analysis of Brain Metabolism Predicts Development of Alzheimer Dementia