


Abstract
In vitro and in vivo studies in human glioma models suggest that the antitenascin monoclonal antibody 81C6 labeled with the 7.2-h–half-life α-particle emitter 211At might be a valuable endoradiotherapeutic agent for the treatment of brain tumors. The purpose of this study was to develop methods for the production of high levels of 211At and the radiosynthesis of clinically useful amounts of 211At-labeled human/mouse chimeric 81C6 antibody. Methods: 211At was produced through the 209Bi(α, 2n)211At reaction using an internal target system and purified by a dry distillation process. Antibody labeling was accomplished by first synthesizing N-succinimidyl 3-[211At]astatobenzoate from the corresponding tri-n-butyl tin precursor and reacting it with the antibody in pH 8.5 borate buffer. Quality control procedures consisted of methanol precipitation, size-exclusion high-performance liquid chromatography (HPLC), and pyrogen and sterility assays, as well as determination of the immunoreactive fraction by a rapid procedure using a recombinant tenascin fragment coupled to magnetic beads. Results: A total of 16 antibody labeling runs were performed. Using beam currents of 50–60 μA α-particles and irradiation times of 1.5–4.5 h, the mean 211At production yield was 27.75 ± 2.59 MBq/μA.h, and the maximum level of 211At produced was 6.59 GBq after a 4-h irradiation at 55 μA. The decay-corrected distillation yield was 67% ± 16%. The yield for the coupling of the 211At-labeled active ester to the antibody was 76% ± 8%. The fraction of 211At activity that eluted with a retention time corresponding to intact IgG on HPLC was 96.0% ± 2.5%. All preparations had a pyrogen level of <0.125 EU/mL and were determined to be sterile. The mean immunoreactive fraction for these 16 preparations was 83.3% ± 5.3%. Radiolysis did not interfere with labeling chemistry or the quality of the labeled antibody product. Conclusion: These results show that it is feasible to produce clinically relevant activities of 211At-labeled antibodies and have permitted the initiation of a phase I trial of 211At-labeled chimeric 81C6 administered directly into the tumor resection cavities of brain tumor patients.
The potential utility of the α-particle–emitting radiohalogen 211At for targeted radiotherapy has been recognized for more than 25 y (1). 211At has a half-life of 7.2 h and α-particle emission is associated with each of its decays. In addition, as a consequence of its electron-capture decay branch, polonium K x-rays (77–92 keV) also are emitted, making it possible to conveniently image 211At using standard nuclear medicine planar and SPECT imaging equipment (2). The short range (55–70 μm) and high linear energy transfer of its α-particles offer the possibility of depositing highly focal and cytotoxic radiation on malignant cell populations while leaving neighboring normal tissues intact.
The relatively long half-life of 211At compared with other α-emitters of potential clinical relevance provides greater flexibility in terms of complexity of chemistry and nature of biologic carrier that can be investigated. This has motivated the development of a wide variety of strategies for selectively delivering 211At to tumors including colloids (3), drugs (4), melanin precursors (5), transporter substrates (6), thymidine uptake analogs (7), biotin analogs (8), bisphosphonate complexes (9), and monoclonal antibodies (mAbs) (10–12). With some of these compounds, the ability to selectively kill target cells with as few as 1–5 α-particle traversals per cell has been documented (13–15).
Although there is a compelling rationale for initiating human trials with some of these 211At-labeled compounds, patient studies have been impeded by the lack of methodologies for producing clinically relevant levels of 211At-labeled radiopharmaceuticals. There are 2 aspects to this problem. First, cyclotron targetry and 211At purification systems are needed to provide large quantities of 211At (>2 GBq) in chemical form appropriate for chemical manipulation. Second, labeling and purification procedures are required that are appropriate for high-level syntheses under conditions where radiolytic decomposition may play a role.
This article describes methods and procedures that were implemented for the production of clinical levels of 211At-labeled mAbs. These methodologies have permitted the initiation of the first clinical trial of a 211At-labeled endoradiotherapeutic agent, 211At-labeled human/mouse chimeric antitenascin 81C6 mAb (16). Doses of this α-particle–emitting mAb have been prepared with excellent quality control characteristics, and 12 glioma patients have received between 74 and 370 MBq 211At-labeled chimeric 81C6 administered directly into their tumor resection cavities.
Materials and Methods
Monoclonal Antibody
The human/mouse chimeric antitenascin mAb was generated by combining the variable region genes of murine 81C6 with human IgG2 constant region genes (17). Like its murine parent, chimeric 81C6 binds to fibronectin type III domains 6–12 of tenascin, a molecule that is overexpressed on human glioma cell lines and xenografts but not in normal brain tissue (18). The chimeric 81C6 hybridoma was grown in a CELLMAX 100 hollow fiber cartridge system (Cellco, Inc., Rancho Dominguez, CA). Cell culture media were separated from cells by centrifugation and passed through a 0.22-μm filter (Millipore, Bedford, MA). After adjusting the pH to 8.0 with a sterile 1.0 mol/L tris buffer, the chimeric mAb was purified by passage over a protein A-Sepharose column (Amersham Pharmacia Biotech, Piscataway, NJ) and dialyzed against 50 mmol/L tris-acetate buffer (pH 5.6). Chimeric 81C6 was further purified by passage over a 40-μm ABx column (JT Baker, Phillipsburg, NJ) using a 0%–100% gradient of 2 mol/L sodium acetate buffer (pH 6.0). Finally, tubes containing chimeric 81C6 were dialyzed against sterile 115 mmol/L phosphate buffer (pH 7.4), and the mAb was aliquoted into sterile, pyrogen-free vials through a 0.22-μm filter (Millipore). In accordance with the Food and Drug Administration (19), chimeric 81C6 was tested for bovine and murine viruses, mouse DNA, viral contaminants, unapparent viruses, mycoplasma, sterility, and pyrogens.
Production of 211At
211At was produced at the Duke University Medical Center (Durham, NC) cyclotron by bombarding natural bismuth metal targets with 28.0-MeV α-particles using the 209Bi(α, 2n)211At reaction. The MIT-1 internal target system (Cyclotron, Inc., Napa, CA), specifically designed for production of 211At, was used (20). The target plate was slightly modified from the previously described, reduced-size internal target. It consisted of a 2.1 × 10.2 × 0.6 cm (width × length × depth) aluminum plate with 2-mm-deep cooling fins machined into the back. The bismuth was deposited in a central depression of 1.5 × 10.2 cm, with a depth of 2 mm in the center and no depth at the edges. The target was mounted on the target ram using an aluminum clamping block.
The dry distillation system for isolation of the 211At from the bismuth cyclotron target, shown in Figure 1, is a variation of the equipment described previously (20). Modifications made to facilitate handling of higher 211At activity levels include trapping the 211At directly in 2-mL glass conical vials that are used as the reaction vessel for N-succinimidyl 3-[211At]astatobenzoate (SAB) synthesis and the use of a TCP-2 thermoelectric cold plate (Thermoelectric Unlimited, Inc., Vernon Hills, IL) to chill the collection traps in an aluminum cooling block to 0°C. In addition, an in-house–developed small-volume PIN photodiode-based miniature γ-detector (21) was coupled to a strip chart recorder and used to monitor the progress of the distillation. Each of the traps contained approximately 500 μL chloroform.
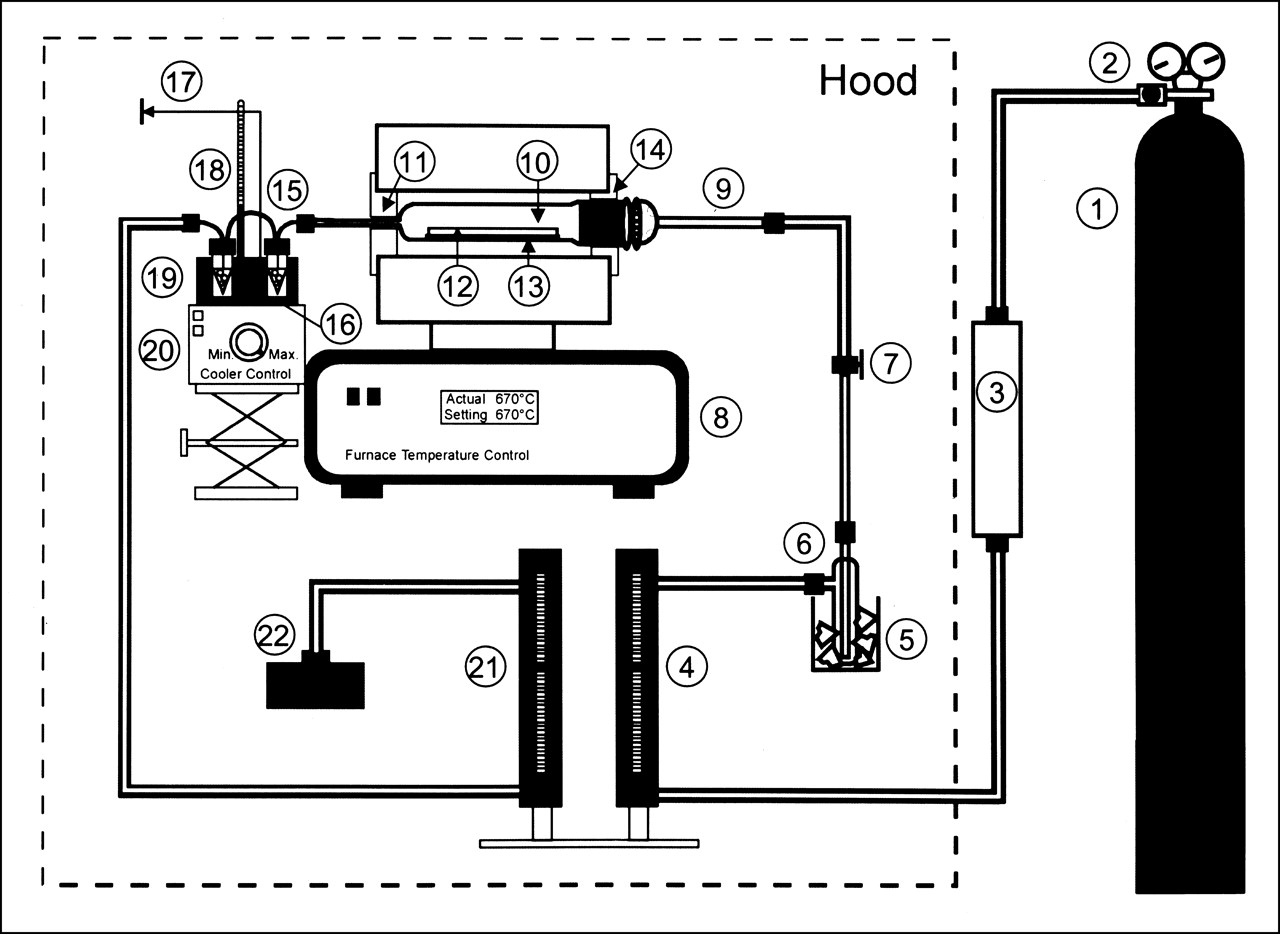
The distillation apparatus used for isolation of 211At from bismuth cyclotron target. (1) argon cylinder, (2) argon gas regulator, (3) gas filter, (4) inlet flow meter and valve, (5) Dewar containing dry ice, (6) ether trap, (7) Teflon valve, (8) tube furnace, (9) quartz inlet tube, (10) quartz still, (11) insulation ring, (12) aluminum-backed bismuth cyclotron target, (13) quartz support plate, (14) insulation ring, (15) glass still outlet tube, (16) radioactivity detector, (17) detector output to amplifier, counter, and strip chart recorder, (18) thermometer, (19) aluminum cooling block, (20) thermoelectric cooler, (21) outlet flow meter, and (22) charcoal filter trap.
Procedures used for glassware preparation and target cleaning after the irradiation remained the same (20). The inlet and outlet argon flow rates were 12.7 and 11.6 mL/min, respectively. After insertion of the target in the apparatus, it took approximately 20 min to reach the desired distillation temperature of 670°C. The distillation was terminated when a plateau in activity, determined by the scintillation detector, had been reached. For these production runs, the distillation was terminated after 20–30 min heating at 670°C. After turning off the furnace and the argon gas flow, the Teflon tubing was removed from the traps. The activity level in the traps was measured using a CRC-7 dose calibrator (Capintec, Pittsburgh, PA) at the 133Xe setting. On the basis of previous comparisons with an efficiency-calibrated Ge(Li) detector, the displayed activity was multiplied by a factor of 2.3 to obtain the 211At activity.
Labeling Chimeric 81C6 mAb with 211At
SAB was prepared by adding the following reagents to the glass vial containing the 211At in chloroform: 2 mg N-succinimidyl 3-(tri-n-butylstannyl)benzoate in 20–25 mL chloroform, 3 μL tert-butylhydroperoxide, and 2 μL glacial acetic acid. The reaction mixture was shaken at room temperature for 30 min, evaporated to 30 μL, and purified using a Sep-Pak cartridge (Millipore) containing 650 mg of silica gel (22). The cartridge was sequentially eluted with 30 mL hexane, 30 mL 8% ethyl acetate in hexane, and 10 mL 30% ethyl acetate in hexane. The purified SAB was collected in the first 6 mL of the 30% ethyl acetate in hexane and was evaporated to dryness in a glass vial with a stream of argon.
The pH of a 10-mg aliquot of chimeric 81C6 (16.0–17.6 mg/mL) was adjusted to a final pH of 8.5–8.9 by the addition of saturated borate buffer in a 1:1 volume ratio. Using a 1-mL sterile syringe, the mAb was transferred to the vial containing SAB and incubated at room temperature for 15 min. The reaction was terminated by the addition of an equivalent volume (1.14–1.25 mL) of 0.2 mol/L glycine in borate buffer followed by an additional incubation of 3 min. The 211At-labeled chimeric 81C6 was purified by size-exclusion chromatography using a gas-sterilized, 1.5 × 10 cm borosilicate glass chromatography column (BioRad, Richmond, CA) loaded with Sepadex G-25 medium (Pharmacia, Piscataway, NJ). To minimize nonspecific binding of mAb, the column had been previously conditioned by the addition of 100 μL Albutein (25% human serum albumin, U.S. Pharmacopoeia [USP]; Bayer Corp., Elkhart, IN) followed by elution with 30 mL sterile 0.05 mol/L phosphate buffer. The 211At-labeled chimeric 81C6 was eluted from the column in 0.05 mol/L phosphate-buffered saline. Fractions containing the labeled mAb (3–5 mL) were pooled, sterile filtered through a 0.22-μm filter into a sterile, pyrogen-free glass vial, and stored at 4°C until injection. An aliquot (100–200 μL) was withdrawn for quality control determinations.
Immunoreactive Fraction
A magnetic-bead–based assay was developed to be able to rapidly evaluate the immunoreactivity of 211At-labeled chimeric 81C6 preparations before patient administration. Approximately 1 mL of 5-μm-diameter streptavidin-coated MPG magnetic beads (Controlled Pore Glass, Lincoln Park, NJ) were first incubated for 3 h at room temperature with 200 μg of biotinylated recombinant tenascin fragment 6–12 (positive) or tenascin fragment 6–11 (negative control) (23) and washed with binding buffer (115 mmol/L phosphate buffer, pH 7.4, containing 0.05% bovine serum albuminand 0.05% Brij 35). The beads were aliquoted at 12.5, 25, and 50 μL per 1.5 mL micro centrifuge tube, diluted to 0.5 mL with this buffer, and stored at 4°C until needed. To measure the immunoreactive fraction of 211At-labeled chimeric 81C6, 15–30 ng of labeled mAb were added in triplicate to the 3 concentrations of recombinant tenascin fragment 6–12 or tenascin fragment 6–11 coated beads and agitated for 30 min at room temperature. After separation of the beads from the supernatant using a magnetic separator, 211At activity levels in the fractions were counted in an LKB 1282 γ-counter (Wallac, Turku, Finland). Specific binding was calculated by subtracting nonspecific binding to the tenascin 6–11 coated beads from the binding to tenascin 6–12 coated beads. The immunoreactive fraction was calculated according to the method of Lindmo et al. (24).
Protein-Associated Activity
The standard method for measuring protein-associated radioactivity is to determine trichloroacetic acid precipitability; however, because [211At]astatide is nearly quantitatively precipitated by trichloroacetic acid, an alternative method was devised for use with 211At-labeled proteins (25). In this procedure, 10% bovine serum albumin was added to an aliquot of 211At-labeled chimeric 81C6 in triplicate in a microfuge tube, 500 mL methanol were added, and the mixture was incubated for 10 min at 4°C. After centrifugation, the precipitates and supernatants were counted in a γ-counter. Preparations with a methanol precipitability of <95% were considered unsuitable for human use.
High-Performance Liquid Chromatography
The molecular weight profile of the 211At-labeled chimeric 81C6 preparations was monitored by size-exclusion high-performance liquid chromatography (HPLC) using a 7.5 × 300 mm Toso Haas TSK-3000 SWXL column (Tosoh Biosep, Montgomeryville, PA) eluted with pH 6.7, 50 mmol/L phosphate buffer. A standard elution profile for unlabeled chimeric 81C6 was first established by monitoring OD at 280 nm and then a 1-MBq aliquot of the labeled mAb was run with its elution monitored by a model 270 flow-through γ-counter (Beckman, Fullerton, CA). Intact, monomeric 211At-labeled chimeric 81C6 was required to represent ≥90% of total radioactivity for the preparation to be deemed acceptable for human use.
Sterility and Pyrogen Testing
These assays were performed by the Duke University Medical Center Clinical Radiopharmacy. The standard USP sterility test was used; however, because this test involves a 14-d incubation period, the results of this test were not available before human administration of the labeled mAb. The level of pyrogens was determined before patient administration using the USP Limulus amebocyte lysate endotoxin test (Associates of Cape Cod, Woods Hole, MA).
Radiation Dose Calculations
Because of the potential for radiolysis to adversely affect labeling chemistry and the quality of the labeled mAb (26,27), the radiation dose deposited in solutions during the 211At distillation, SAB synthesis, and mAb coupling were calculated. Because of the short range of 211At α-particles, it was assumed that all of the α-particle decay energy was deposited in these solutions and that the reagents were uniformly distributed within the media. Dose contributions from α-particles and α-recoil nuclei were considered. The absorbed dose was calculated as follows:
 where Ai is the initial activity in Bq, λ is the decay constant for 211At, t is the exposure time, m is the mass of the solution, and Δi is the mean energy emitted per nuclear transition, equal to 1.11 × 10−10 Gy.kg/Bq.s for 211At (28). For solutions containing chloroform, a density of 1.49 g/mL was used to convert volume to mass. The correlation coefficient for the relationship between radiation absorbed dose and chemical yield or quality control parameters was calculated using the statistical function package of Microsoft Excel (Microsoft, Seattle, WA).
where Ai is the initial activity in Bq, λ is the decay constant for 211At, t is the exposure time, m is the mass of the solution, and Δi is the mean energy emitted per nuclear transition, equal to 1.11 × 10−10 Gy.kg/Bq.s for 211At (28). For solutions containing chloroform, a density of 1.49 g/mL was used to convert volume to mass. The correlation coefficient for the relationship between radiation absorbed dose and chemical yield or quality control parameters was calculated using the statistical function package of Microsoft Excel (Microsoft, Seattle, WA).
Results
A total of 16 batches of 211At-labeled chimeric 81C6 were prepared for clinical use, 12 of which were administered to glioma resection cavity patients. Three additional runs were successful; however, they were not administered because of clinical complications. The last run yielded a preparation with acceptable quality control characteristics; however, insufficient 211At-labeled mAb was available for injection because of the retention of high activity levels in the final reaction vessel.
These 211At production runs used beam currents of 50–60 μA α-particles and irradiation times of 1.5–4.5 h (Table 1) with a mean yield of 27.75 ± 2.59 MBq/μA.h. No significant differences were observed in 211At production as a function of beam current or irradiation time. The maximum level of 211At that was produced was 6.59 GBq after a 4-h irradiation at 55 μA. The decay-corrected distillation yield was 67% ± 16%.
Production and Distillation of 211At for Clinical Studies
Radiochemical purity of the preparations was monitored by counting an aliquot of the distilled 211At using a germanium detector coupled to a multichannel analyzer. Of particular concern is the presence of 210At, which can be produced through the 209Bi(α, 3n)210At reaction and emits 569.7-, 687.9-, and 897.8-keV γ-rays. No peaks corresponding to 210At were detected, and an upper limit of 210At contamination was set at <0.02% based on the detection sensitivity of the germanium detector.
Synthesis of SAB was begun between 5 and 40 min after the end of the distillation, depending on whether the 211At was produced during the day or, for the longer-duration runs, beginning at midnight. Yields for the synthesis of SAB are summarized in Table 2. SAB eluted in the 30% ethyl acetate in hexane fractions and accounted for 54% ± 10% of the activity added to the Sep Pak (Burdick and Jackson, Muskegan, MI). The mean fraction of activity eluted in the hexane and 8% ethyl acetate in hexane fractions, presumably representing an Ato species and an astatinated π-complex with the tin precursor aromatic ring (22), respectively, was 7% ± 3% and 27% ± 8%. The mean activity retained on the column, most likely in the form of [211At]astatide, was 12% ± 6%. The radiation dose deposited in the chloroform solution before initiation of the synthesis ranged from 125 to 3,943 Gy, and the dose delivered during the reaction ranged from 604 to 1,596 Gy. Because of the potential for radiolytic effects on the radioastatodestannylation reaction, the correlation between these radiation absorbed doses and SAB yield was investigated. As shown in Figure 2, there was no correlation between SAB yield and the radiation dose received by the reaction mixture. No correlations were found between the radiation dose delivered before and during the reaction and the fraction of activity either retained on the cartridge or eluted in the hexane, 8% ethyl acetate in hexane, or 30% ethyl acetate in hexane fractions.
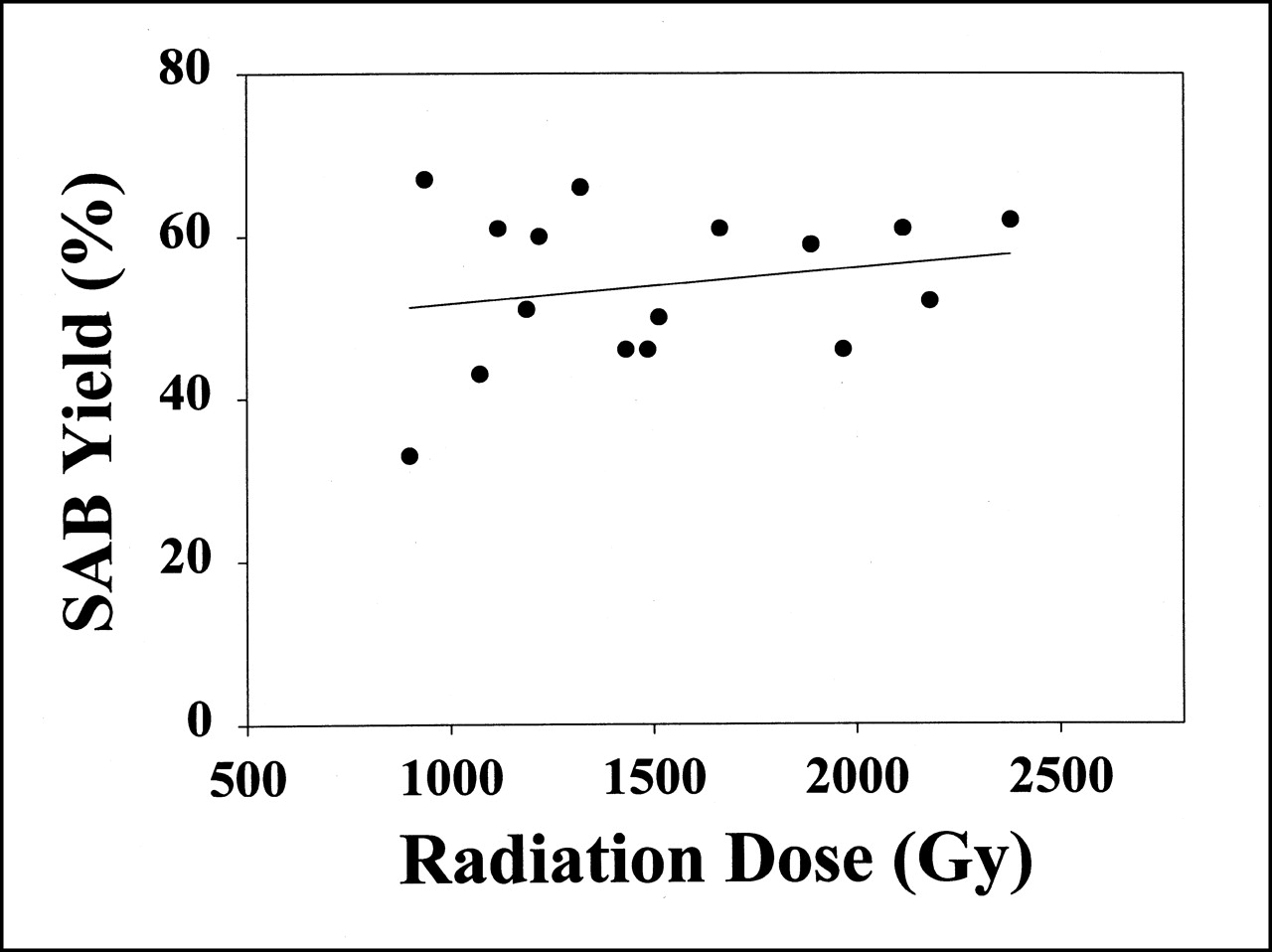
Plot of yield of SAB as function of radiation dose deposited in destannylation reaction mixture. No significant correlation was observed (r2 = 0.05).
Synthesis of [211At]SAB and 211At-Labeled Chimeric 81C6
The radiolabeling yield for the coupling of SAB to chimeric 81C6 was 76% ± 8% after a 20-min reaction at room temperature (Table 2). Because of the 10-mg mAb protein dose and the activity levels, which were required for this clinical protocol, no attempt was made to maximize specific activity. The maximum specific activity for 211At-labeled chimeric 81C6 was 52 MBq/mg, which resulted in about 1 of 3,000 mAb being labeled. The radiation dose to the reaction medium ranged from 399 to 1,196 Gy, and there was no significant correlation between radiation dose and coupling yield.
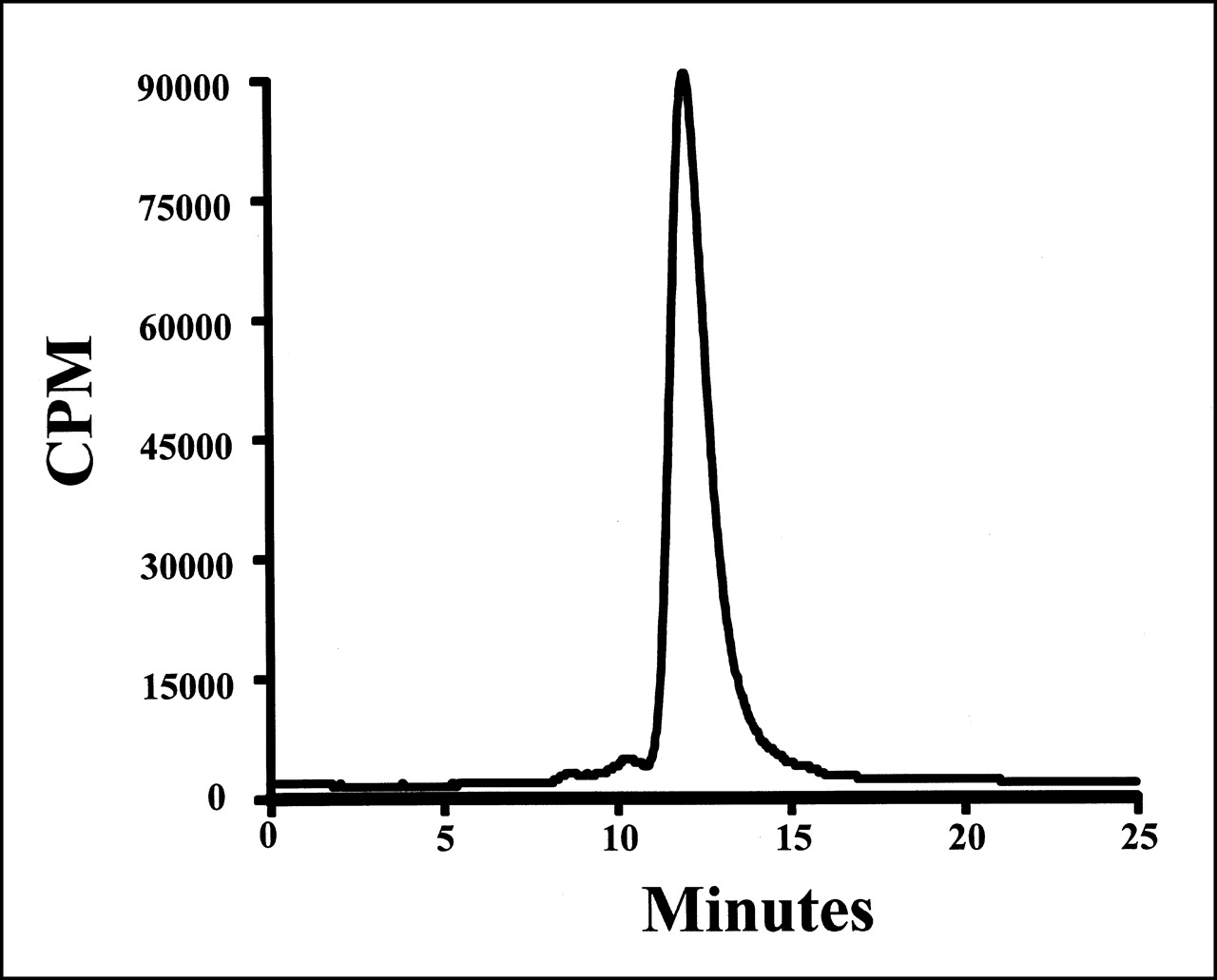
The fraction of the 211At activity in the clinical preparations that was protein associated, determined by methanol precipitation, was 98.5% ± 1.0%. The fraction of 211At activity that eluted with a retention time corresponding to intact IgG was 96.0% ± 2.5%, with the remainder present as either aggregates (3.1% ± 2.5%) or low-molecular- weight impurities (0.9% ± 1.2%). The size-exclusion HPLC profile for preparation 2 is illustrated in Figure 3. All preparations had a pyrogen level of <0.125 EU/mL and were determined to be sterile.

Size-exclusion HPLC profile of 211At-labeled chimeric 81C6 mAb (211At-labeling run 2).
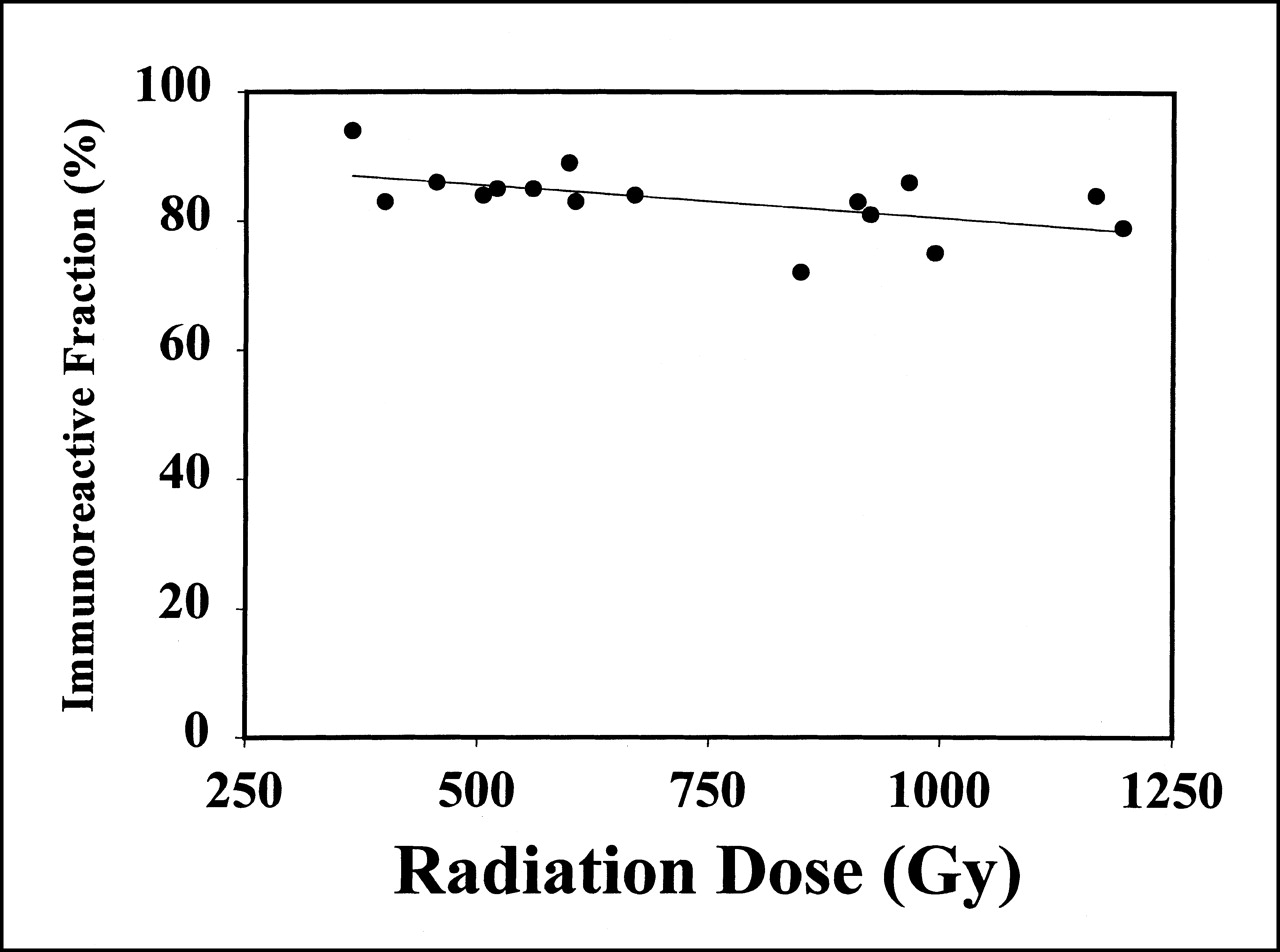
To determine the immunoreactive fraction of 211At-labeled chimeric 81C6 before patient administration, magnetic beads coated with either tenascin 6–12 (positive) or tenascin 6–11 (control) fragments were used. After correcting for nonspecific binding, the mean immunoreactive fraction for these 16 preparations was 83.3% ± 5.3%. In Figure 4, the immunoreactive fraction is plotted as a function of the radiation dose received by the mAb during radiolabeling and purification. Regression analysis indicated that these data were best fit by the following equation: immunoreactive fraction = 90.8% − 0.010 × radiation dose (Gy). However, this slight decrease in immunoreactivity with increasing radiation dose was not a significant effect (r2 = 0.29).

Plot of immunoreactive fraction of 211At-labeled chimeric 81C6 antibody as function of radiation dose received by antibody. No significant correlation was observed (r2 = 0.29).
Discussion
The short path length and high linear energy transfer of α-particles are potentially valuable characteristics for the treatment of certain types of tumors because of the possibility of delivering highly cytotoxic radiation doses to malignant cell populations while sparing adjacent normal tissues. A wide variety of compounds have been labeled with the α-particle emitter 211At and, in some cases, encouraging results have been obtained in cell culture and animal models of human cancers (29). Among the carrier systems that have been investigated for achieving the selective delivery of 211At to tumors, mAbs and their fragments have received the most attention. 211At-labeled mAbs and mAb fragments have been previously reported that are reactive with many types of malignancies, including renal cell carcinoma (10), glioma and melanoma (10), osteosarcoma (12), lymphoma (30), and ovarian carcinoma (31).
The goal of the current study was to develop methodologies capable of producing clinically relevant levels of 211At-labeled mAbs. A recent report (32) described procedures for labeling mAbs with another promising α-emitter, 46-min–half-life 213Bi, which have permitted the initiation of a clinical trial of a 213Bi-labeled anti-CD33 humanized mAb in patients with leukemia (33). Although many of the considerations for labeling mAbs with 211At and 213Bi are similar, there are some differences. 213Bi is available from an 225Ac/213Bi generator, whereas 211At is cyclotron produced. Because we currently use a cyclotron primarily dedicated to clinical PET, scheduling clinical production runs can be complicated. In contrast, the longer half-life of 211At offers greater flexibility in compensating for unavoidable changes in patient treatments. For example, a 30-min delay in therapy administration time would reduce available 211At by <5% compared with 36% for 213Bi. The longer half-life of 211At also provides more time for radionuclide purification and mAb labeling, and permits the performance of all quality control procedures, except sterility testing, before administration of the labeled mAb to patients.
Using the internal target system designed specifically for 211At production, it was possible to produce up to 6.59 GBq of 211At in a 4-h irradiation, a level of 211At that appears to be higher than ever produced previously. In a review of 211At production methodologies, the maximum total yield of 211At that was reported was 5,000 MBq after an 8- to 10-h bombardment with 29-MeV α-particles (34). The amount of 211At produced in the current study was more than sufficient for producing clinically useful levels of 211At-labeled mAbs and, depending on the labeling chemistry and dosimetry, should be sufficient to produce other 211At-labeled radiopharmaceuticals as well. No significant difference in 211At production rate was observed over the range of beam currents and irradiation times that were used, suggesting that further upscaling of 211At production should be possible.
The yield for the distillation of 211At from the cyclotron target was nearly the same as that which we obtained previously at lower activity levels using a similar distillation system (21) and which other investigators have reported for other 211At dry-distillation approaches (30).
Because α-particle decay results in the deposition of high radiation doses in relatively small volumes, radiolysis is a potential confounding factor in the preparation of therapeutic levels of α-particle–emitting radiopharmaceuticals. The high ionization density of α-particles has been reported to compromise the immunoreactivity of a 211At-labeled antiosteosarcoma mAb at doses above 1,000 Gy (27). Use of ascorbic acid as a radioprotectant was required to maximize the recovery of 213Bi-labeled HuM195 mAb (32). These observations led us to investigate the potential effects of radiation dose on 3 aspects of the 211At-labeled mAb production process.
One possibility is that radiolysis during the distillation of 211At from the cyclotron target could interfere with subsequent chemical reactions. In our procedure, the 211At is trapped in chloroform under an argon atmosphere. Even in the absence of oxygen, the radiation chemistry of chloroform is complex, resulting in the generation a variety of free radicals including . CCl, . CCHCl2, and . CCCl3, as well as HCl and multiple chloroalkanes (35,36). Traces of oxygen or water lead to many more products including peroxides, and increase the number of radicals that are generated. Potential effects of these products include the reaction of free radicals with 211At and the generation of higher oxidation state astatinated species. The extent to which these processes take place while the 211At is in the chloroform trap is unknown at this time. However, because of the fact that 211At is present in extremely low levels, the number of free radicals that is generated far exceeds the number of astatine atoms (36). For this reason, we investigated whether the radiation dose deposited in the chloroform solution before the astatodestannylation reaction had any influence on SAB yield, and no significant effect was observed.
α-Particle irradiation could have deleterious effects in the SAB synthesis step by degrading the tin precursor required for its synthesis, as well as the 211At-labeled acylation agent itself. In a preliminary study, we observed only minor degradation of SAB at doses up to 40,000 Gy; however, only about 40% of the N-succinimidyl 3-(tri-n-butylstannyl) benzoate remained intact at 22,000 Gy (26). For this reason, a high level of tin precursor was used in the labeling reaction to avoid decreased SAB yields at high radiation doses. No decrease in SAB yield at higher radiation doses was observed in the current study; however, it should be noted that the maximum dose delivered during SAB synthesis was approximately 1,600 Gy, where only about 10% degradation of tin precursor would be predicted.
From the perspective of clinical application, the most important potential effect of α-particle mediated radiolysis, in addition to the reproducibility of the production process, is on the properties of immunoconjugate that is produced. One problem that could occur is the reaction of radiolytically generated chloride species with the tin precursor, followed by coupling of the chlorinated N-succinimidyl ester with the mAb, compromising conjugation efficiency and immunoreactivity. Direct effects of radiation on mAb structure also are possible. Radiation doses as low as 300 Gy can cause protein fragmentation in the presence of oxygen, whereas protein aggregation is the primary effect observed under anaerobic conditions (37,38). Radiation has also been reported to shift the protein isoelectric point to more acidic pH and cause conformational changes that can interfere with biologic function (39,40).
It is therefore not surprising that exposure of a mAb and a F(ab′)2 fragment to 211At at doses of >1,000 Gy resulted in a significant reduction in immunoreactivity, with a 50% decrease in antigen binding seen at approximately 2,500 Gy (27). In the current study, the maximum radiation dose received by chimeric 81C6 antitenascin mAb was 1,200 Gy. Plotting the immunoreactive fraction versus the radiation dose received during the mAb labeling and purification yielded a line with a negative slope; however, the slight decrease in immunoreactivity at higher doses was not significant. In addition, size-exclusion HPLC failed to reveal dose-related increases in 211At-labeled mAb aggregation or mAb fragments of lower molecular weight. It should be noted that these results were obtained without the addition of a radioprotective agent. We anticipate that the preparation of higher activity levels of 211At-labeled mAbs will require the addition of a radioprotector such as ascorbic acid, which was used to enhance the recovery of 213Bi-labeled mAbs (32).
Conclusion
Using an internal cyclotron target system, it has been possible to produce sufficient quantities of 211At to synthesize clinically useful levels of 211At-labeled radiopharmaceuticals. Methods have been developed for the preparation and rapid evaluation of 211At-labeled chimeric antitenascin mAb 81C6 with excellent preservation of immunoreactivity. This has permitted the initiation of a phase I trial involving direct administration of the labeled mAb into surgically created brain tumor resection cavities (16). Our results to date are consistent with excellent in vivo stability of this 211At-labeled construct, and encouraging responses have been observed. We are currently attempting to further optimize these procedures and apply them to other mAbs of potential utility for α-particle radioimmunotherapy.
Acknowledgments
The authors acknowledge the contributions of Drs. Roy Larsen and Bent Schoultz to the initial development phase of the production and labeling methods described in this paper. The assistance of Dr. Bruce Wieland and other members of the staff at the Duke University Medical Center cyclotron with 211At production is gratefully acknowledged. This work was supported in part by National Institutes of Health grants CA42324 and NS20023 and U.S. Department of Energy grant DOE-DE-FG-05–95ER62021.
Footnotes
Received Oct. 26, 2000; revision accepted Feb. 20, 2001.
For correspondence or reprints contact: Michael R. Zalutsky, PhD, Department of Radiology, Duke University Medical Center, Box 3808, Durham, NC 27710.
References
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- An Improved 211At-Labeled Agent for PSMA-Targeted {alpha}-Therapy
- Clinical Experience with {alpha}-Particle Emitting 211At: Treatment of Recurrent Brain Tumor Patients with 211At-Labeled Chimeric Antitenascin Monoclonal Antibody 81C6
- CD74: A New Candidate Target for the Immunotherapy of B-Cell Neoplasms
- Targeted cancer therapy with a novel low-dose rate {alpha}-emitting radioimmunoconjugate
- Radiopharmaceutical Chemistry of Targeted Radiotherapeutics, Part 3: {alpha}-Particle-Induced Radiolytic Effects on the Chemical Behavior of 211At
- First Clinical Experience with {alpha}-Emitting Radium-223 in the Treatment of Skeletal Metastases
- Radiopharmaceutical Chemistry of Targeted Radiotherapeutics, Part 1: Effects of Solvent on the Degradation of Radiohalogenation Precursors by 211At {alpha}-Particles
- Therapy of Small Subcutaneous B-Lymphoma Xenografts with Antibodies Conjugated to Radionuclides Emitting Low-Energy Electrons
- Pharmacokinetics, Dosimetry, and Toxicity of the Targetable Atomic Generator, 225Ac-HuM195, in Nonhuman Primates
- Microdosimetric Analysis of {alpha}-Particle-Emitting Targeted Radiotherapeutics Using Histological Images
- Overcoming the Obstacles to Clinical Evaluation of 211At-Labeled Radiopharmaceuticals