


Abstract
The high energy and short range of α-particles make them attractive for targeted radiotherapy. However, these properties can be problematic when the production of high activity levels of α-particle–emitting radiotherapeutics is required. For example, difficulties were encountered in the production of N-succinimidyl 3-[211At]-astatobenzoate (SAB), when 370-MBq doses of 211At-labeled antibody were required. The purpose of this study was to investigate a potential cause of this behavior—radiolytic degradation of the radiohalogenation precursor. Methods: Both N-succinimidyl 3-(tri-n-butylstannyl)benzoate (BuSTB) and N-succinimidyl 3-trimethylstannylbenzoate (MeSTB) were incubated with various 211At time–activity combinations such that the radiation dose received by the reaction medium ranged from about 0 to 20,000 Gy. Studies were performed using chloroform, methanol, and benzene as the solvent, and both at neutral pH and at a pH of ∼5.5, as used in SAB synthesis. The fraction of tin precursor remaining and the generation of unlabeled byproducts were determined from high-performance liquid chromatograms and then plotted against radiation dose. Results: Extensive radiolytic decomposition of BuSTB and MeSTB was observed in chloroform, with 50% degradation taking place even at doses below 500 Gy. Formation of a byproduct, most likely N-succinimidyl 3-chlorobenzoate, increased with radiation dose. A greater degree of stability was seen in both methanol and benzene, with more than 85% of the precursor remaining at 3,500 Gy. No cold byproducts were observed with either solvent. Conclusion: The nature of the solvent profoundly influences the ability to synthesize high activity levels of SAB and possibly other 211At-labeled radiopharmaceuticals.
Targeted radionuclide therapy is an emerging approach for cancer treatment that shows great promise because of its potential for delivering curative doses of radiation to malignant cell populations while sparing normal tissues. Although, in many cases, potentially useful molecular targets and labeled compounds have already been identified, putting these concepts into practice has frequently been impeded by methodologic limitations. The development of therapeutic radiopharmaceuticals provides additional challenges in comparison to diagnostic reagents in the realm of radiochemistry not only in terms of radiation safety but also with regard to the potentially deleterious effects of high radiation fields on labeling chemistry and shelf life. This is particularly important for α-particle–emitting radionuclides because they deposit large amounts of energy in a highly focal manner.
211At is one of the most attractive α-particle emitters for targeted radiotherapy (1). The α-particles emitted in the decay of this 7.2-h half-life radiohalogen have a range in tissue of 55–70 μm and a linear energy transfer (LET) of about 100 keV/μm. The most successful approach for labeling molecules with 211At has been electrophilic demetallation of stannyl or silyl precursors (2–6). This radiosynthetic strategy has been able to reliably provide a variety of 211At-labeled radiopharmaceuticals including peptides, proteins, and small molecules at activity levels of approximately 37 MBq.
Recently, we have shown it possible to produce significantly higher levels of N-succinimidyl 3-[211At]astatobenzoate (SAB) from an N-succinimidyl 3-(tri-n-butylstannyl)benzoate (BuSTB) precursor to meet the needs of a clinical study of an 211At-labeled monoclonal antibody (mAb) (7). However, difficulties have been encountered when 370-MBq doses of 211At-labeled mAb were required. Two problems surfaced. First, the yield of SAB decreased to 30%–40%. Second, after the reaction of SAB with the mAb, 40%–60% of the 211At was retained on the walls of the reaction vessel and the immunoreactivity of the labeled mAb was unacceptable.
Because the doses delivered during the radiolabeling procedures exceed 1,000 Gy (7), we hypothesized that with increasing dose escalation, α-particle irradiation could have deleterious effects on the synthesis of SAB, through degradation either of the tin precursor required for its synthesis or of the SAB product itself. To investigate this possibility, we performed the current study, in which the synthesis of SAB was evaluated as a function of radiation dose. We selected 3 solvents compatible with the conditions needed for electrophilic astatodestannylation, including chloroform and methanol, both of which have been used previously for SAB synthesis (5,7), and benzene, which has generally been considered nearly radiation inert (8). Our goal was not only to understand the phenomena responsible for the difficulties in SAB synthesis at high radiation doses but also to obtain some guidance as to solvent-related effects that may be relevant to the synthesis of other therapeutic radiopharmaceuticals.
MATERIALS AND METHODS
General
The experiments were performed with the 2 tin precursors that have already been used for SAB radiosynthesis. BuSTB and N-succinimidyl 3-(trimethylstannyl)benzoate (MeSTB) were prepared as described previously (9,10). Their purity was confirmed by thin-layer chromatography before each set of experiments. Although the clinical SAB production procedure uses 2 mg of BuSTB, the current studies were performed with a smaller amount of tin precursor (50 μg) in order to decrease the probability of direct interaction between the α-particles and the molecules of tin precursor itself. This change facilitated observing mainly the radiolysis effects due to the solvent, because almost all radiolytic decomposition of the tin precursor would be produced by radicals coming from the interaction between the α-particles and the solvent.
All solvents were of reagent grade or better and were used as purchased. High-performance liquid chromatography (HPLC) was performed on a System Gold (Beckman) equipped with a model 126 programmable solvent module, a model 168 diode array detector, a model 170 radioisotope detector, and a model 406 analog interface module. For reverse-phase chromatography, a 4.6 × 250 mm (10 μm) Xterra column (Waters) was used. Normal-phase HPLC was done on a 4.6 × 250 mm Partisil (10 μm) silica column (Alltech). The elution conditions for reverse-phase chromatography were as follows: Initially, at a flow rate of 1 mL/min, 48% of solvent B (95:5:0.1 acetonitrile:water:acetic acid) in solvent A (100:0.1 water:acetic acid) was held for 13 min, followed by a 48%–100% linear gradient of solvent B over 2 min, followed by 100% solvent B until the end of the HPLC run. The flow was increased from 1 to 1.5 mL/min between 15 and 15.5 min and held there until the end of the run. The ultraviolet signal at both 220 and 254 nm, as well as the radioactive signal, was monitored. Normal-phase chromatography was performed under isocratic conditions with a 70:30:0.12 hexane:ethyl acetate:acetic acid solvent at 1 mL/min.
Production of 211At
The 211At was produced at the Duke University Medical Center cyclotron as described previously (11) by bombarding natural bismuth metal targets with 28.0-MeV α-particles using the 209Bi(α, 2n)211At reaction. The MIT-1 internal target system (Cyclotron, Inc.), specifically designed for production of 211At, was used. The dry distillation system for isolation of the 211At from the bismuth cyclotron target was a variation of the equipment described previously (7).
Radiation Dose Calculations
Because of the short range of 211At α-particles compared with the dimensions of the reaction mixtures, we assumed that all α-particle decay energy was deposited in these solutions. In addition, we assumed that the reagents were uniformly distributed within the medium. The absorbed dose was calculated as follows (7):
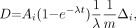 Eq. 1 where D is expressed in Gy and Ai is the initial activity in MBq, λ is the decay constant for 211At (s−1), t is the exposure time in seconds, m the mass of the solution in grams, and Δi is the mean energy emitted per nuclear transition. Dose contributions from α-particles and α-recoil nuclei were considered, in which case Δi = 1.09 × 10−3 Gy·g/MBq·s for 211At (12). To convert from volume to mass, densities of 1.49, 0.791, and 0.879 g/mL were used for chloroform, methanol, and benzene, respectively.
Eq. 1 where D is expressed in Gy and Ai is the initial activity in MBq, λ is the decay constant for 211At (s−1), t is the exposure time in seconds, m the mass of the solution in grams, and Δi is the mean energy emitted per nuclear transition. Dose contributions from α-particles and α-recoil nuclei were considered, in which case Δi = 1.09 × 10−3 Gy·g/MBq·s for 211At (12). To convert from volume to mass, densities of 1.49, 0.791, and 0.879 g/mL were used for chloroform, methanol, and benzene, respectively.
Radiolysis Experiments
Most of these studies were performed at a pH of 5–5.5, the condition used for 211At labeling of biomolecules. In each case, the final volume was the same, and the reaction mixtures contained only the 211At activity, the tin precursor, and the acetic acid when acidic conditions were studied. A control vial with the same concentration of tin precursor, same solvent, same final volume, and same pH but without 211At, designated as the control, was also run with each experiment. All experiments were conducted at room temperature and, in order to have an oxygen concentration similar to that present during labeling procedures, under atmospheric conditions (no argon was bubbled).
To each half-dram flat-bottomed glass reaction vial were added 50 μg of the tin precursor (BuSTB or MeSTB) dissolved in 100 μL of the solvent to be tested (chloroform, methanol, or benzene) plus 20 μL of acetic acid, and the 211At in the same solvent in a volume such that the final reaction volume always was 520 μL. To study the effect of acetic acid, a component of our standard labeling reaction, we conducted separate experiments in which parallel reactions were performed on the same day, with and without acetic acid, at the concentration normally used for radioastatination (0.67 mol/L final concentration). In methanol, the approximate pH measured with pH paper was 5; measurement of pH in the aprotic solvents chloroform and benzene was not attempted. The 211At was used immediately after completion of the target distillation to minimize the impact of radiolysis while the 211At was in the solvent distillation trap. Depending on the experiment, activities ranging from about 10 to 250 MBq were used. The activity levels in the reaction vials were measured using a CRC-7 dose calibrator (Capintec) at the 133Xe setting. On the basis of previous calibration with a Ge(Li) detector (7), the displayed activity was multiplied by a factor of 2.3 to obtain the 211At activity.
Samples for HPLC analysis were taken at various times chosen on the basis of the doses to be studied in each experiment. The radiation dose deposited in the reaction vessel ranged from about 200 to 20,000 Gy with exposure periods ranging from minutes to about 24 h. In each case, an aliquot from the control (without 211At) was also obtained at the same exposure time for direct comparison. The activity, time, and volume were recorded each time aliquots were taken. The HPLC samples were aliquots of 50 μL except for exposure times of 24 h or more and when the initial activity was low; in those cases, aliquots of >50 μL were taken. Samples were injected on the HPLC column immediately after they were obtained.
HPLC analyses were performed using the normal-phase system for BuSTB and using the reverse-phase system for MeSTB. The BuSTB or MeSTB areas were integrated, and the results for the tin precursor remaining in each sample were expressed as percentage of the area of the control and plotted as a function of the radiation dose (Gy) deposited.
Synthesis of N-Succinimidyl 3-Chlorobenzoate (SClB) Standard
To aid in the analysis of some of the HPLC data, SClB was synthesized according to published methods (13,14). Briefly, 3-chlorobenzoic acid was reacted with N,N′-disuccinimidyl carbonate in 30% excess and pyridine in acetonitrile as solvent. The solvent was then evaporated in vacuo, the residue was redissolved in chloroform, and the organic phase was washed first with aqueous sodium bicarbonate solution and then with brine and finally dried with sodium sulfate.
RESULTS
To relate the results of these experiments to conditions potentially encountered in higher-dose 211At labeling, consider a situation in which a 370-MBq dose of 211At-labeled mAb is required. If one assumes that obtaining a SAB synthesis yield of 54% and a mAb coupling yield of 76% would be possible (7), then considering the synthesis and purification times involved, the starting activity of 211At would need to be more than 1,500 MBq. For a reaction volume of 520 μL and a reaction time of 20 min, the minimum dose that would be encountered when the reaction is run in chloroform would be about 2,500 Gy; thus, a dose of 3,000 Gy to the SAB chloroform reaction mixture is a reasonable benchmark for evaluative purposes. If one assumes that similar levels of radioactivity would be required if the reaction were run in other solvents, then the equivalent benchmark doses in methanol and benzene would be 4,700 and 4,250 Gy, respectively, because of the lower densities of these solvents.
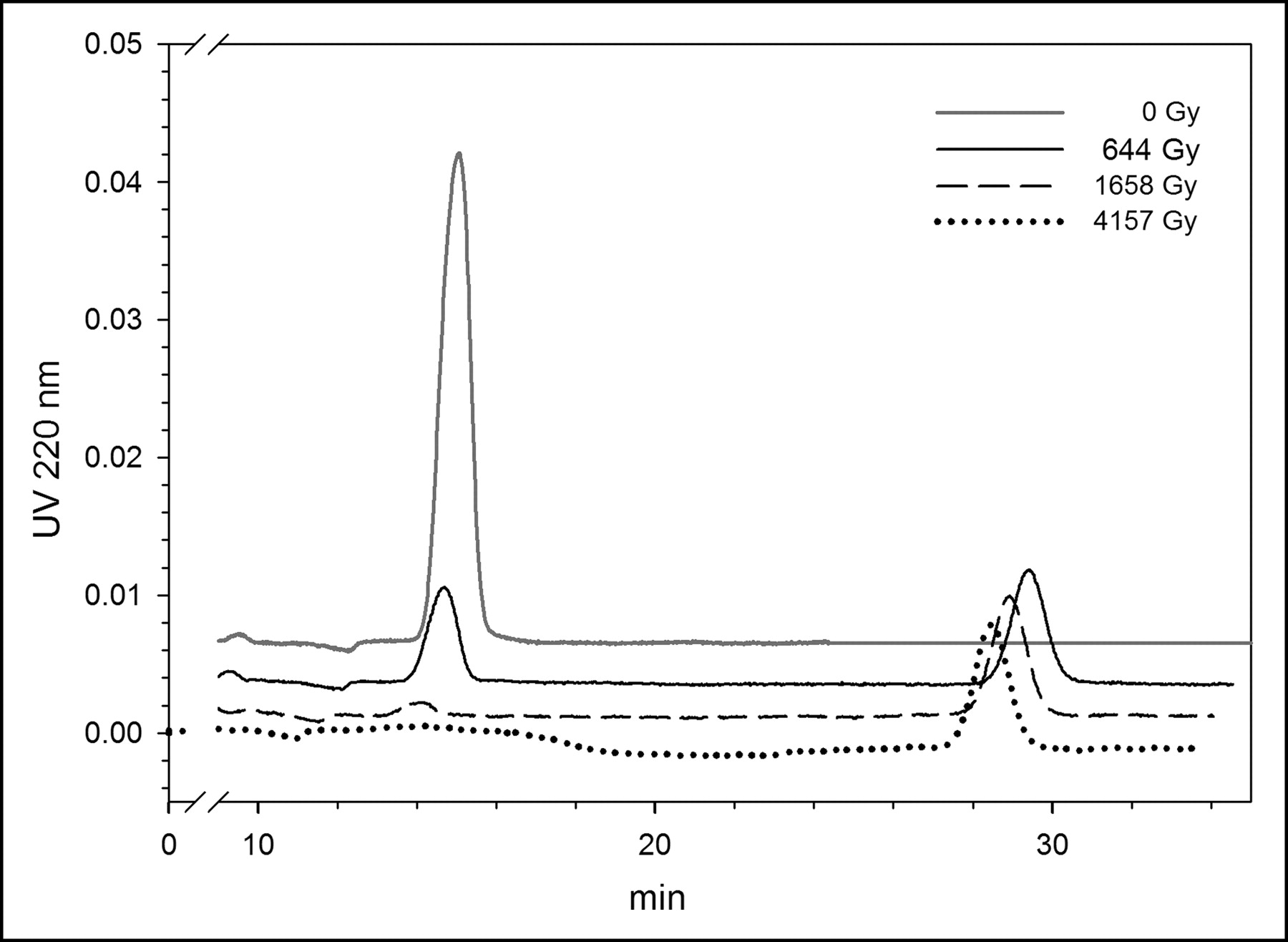
Our initial experiments were performed in chloroform because that was the solvent used in our clinical-level SAB production protocol. A series of typical normal-phase high-performance liquid chromatograms of BuSTB is shown in Figure 1. The tin precursor was exposed under acid conditions to 211At (59–252 MBq; 2.3–3.7 h) to create doses to the reaction mixture of 644, 1,658, and 4,157 Gy or, as a control, no 211At (0 Gy). In the chromatogram of the control sample, a single peak at 13.5–14.0 min was observed, the expected retention time of BuSTB in this HPLC system. However, as the radiation dose to the reaction mixture increased, the peak corresponding to BuSTB decreased and a second peak with a retention time of 28.8 min appeared. Although we have not yet confirmed its identity, we hypothesize that the byproduct peak might be SClB, produced through reaction of radiolytically generated chlorine species with the tin precursor. This possibility is consistent with the fact that the retention time of the byproduct on normal-phase HPLC was almost the same as that of an SClB standard.
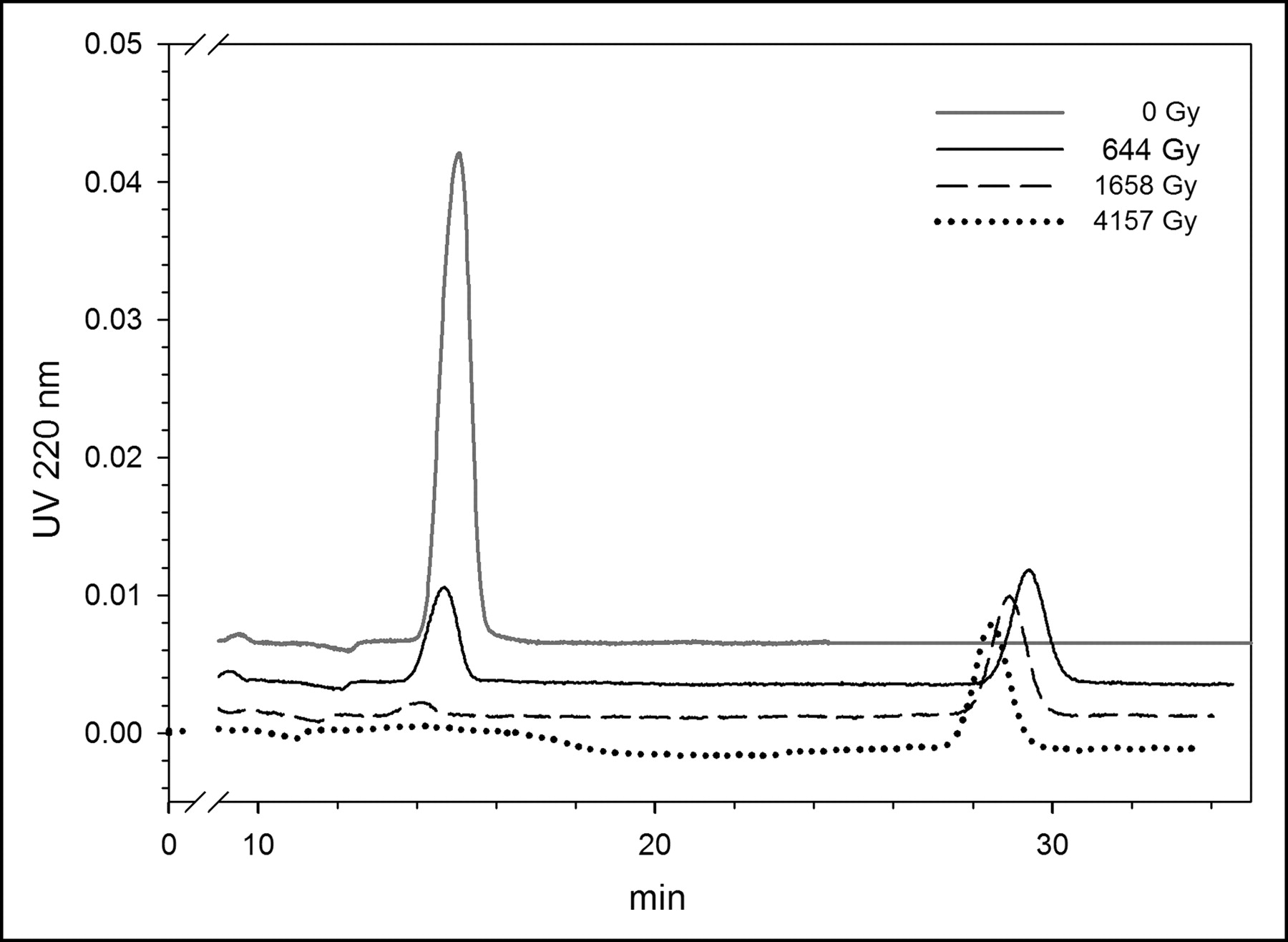
Normal-phase HPLC profile of BuSTB radiohalogenation precursor after exposure to 211At at doses of 644, 1,658, and 4,157 Gy, as well as control (0 Gy).
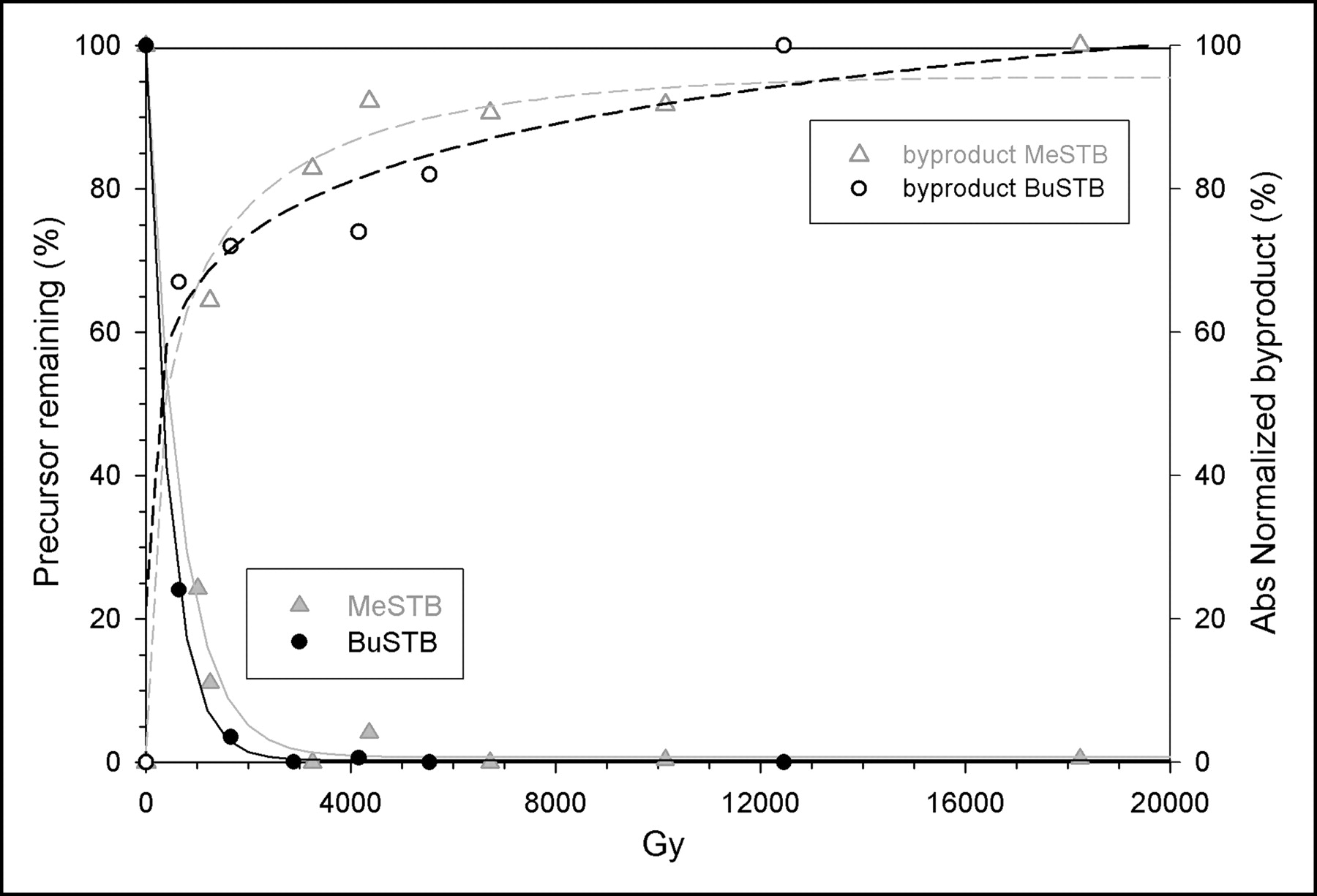
The percentage of BuSTB or MeSTB added to the reaction mixture that remained intact as a function of radiation dose was determined by comparison of the integrated area at each dose level to a control sample obtained the same day. As shown in Figure 2, extensive decomposition of both tin precursors was observed with increasing dose in chloroform. The data for BuSTB were fit to an exponential curve with a good correlation (r2 = 0.999), and this equation shows that a 50% loss of BuSTB occurs with a radiation dose of only 314 Gy. This is equivalent to the dose delivered during a SAB synthesis in chloroform by an initial 211At activity of 189 MBq. The degradation of the MeSTB precursor likewise increased exponentially with radiation dose and declined to 50% of initial levels at 450 Gy. At doses of 3,000 Gy, only 0.4% and 1.6% of BuSTB and MeSTB, respectively, would remain.
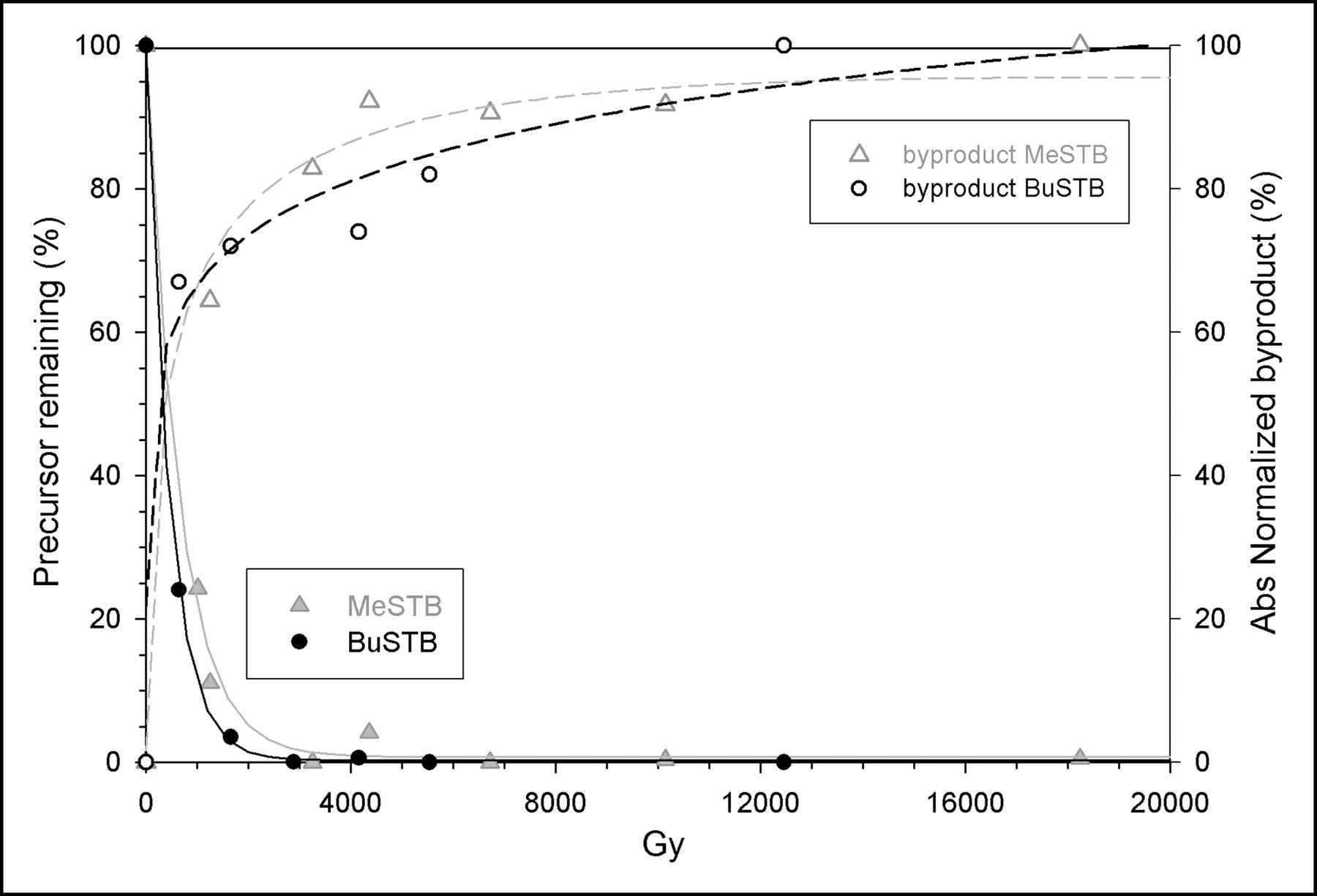
Radiolytic decomposition of BuSTB and MeSTB in chloroform and generation of a cold byproduct as a function of radiation dose.
The area under the byproduct peak also was determined and expressed as a percentage of its maximum value. The production of the byproduct peak increased with increasing radiation dose for both BuSTB and MeSTB and reached levels greater than 50% of maximum at a dose of less than 1,000 Gy. The dose at which the curve for precursor loss crosses the curve for generation of byproduct was calculated to be 274 and 422 Gy for BuSTB and MeSTB, respectively.
Radiolytic decomposition of both tin precursors was considerably less in methanol than in chloroform (Fig. 3). There was a trend for a higher rate of decomposition at doses less than about 2,000 Gy than at higher doses. At the higher-activity-SAB benchmark equivalent dose of about 4,700 Gy, more than 90% of both BuSTB and MeSTB remained intact, and even at doses more than 3 times this level, less than 20% of the precursor was degraded. No cold byproducts were observed even when the methanol-based reaction mixture was exposed to doses as high as 20,000 Gy.
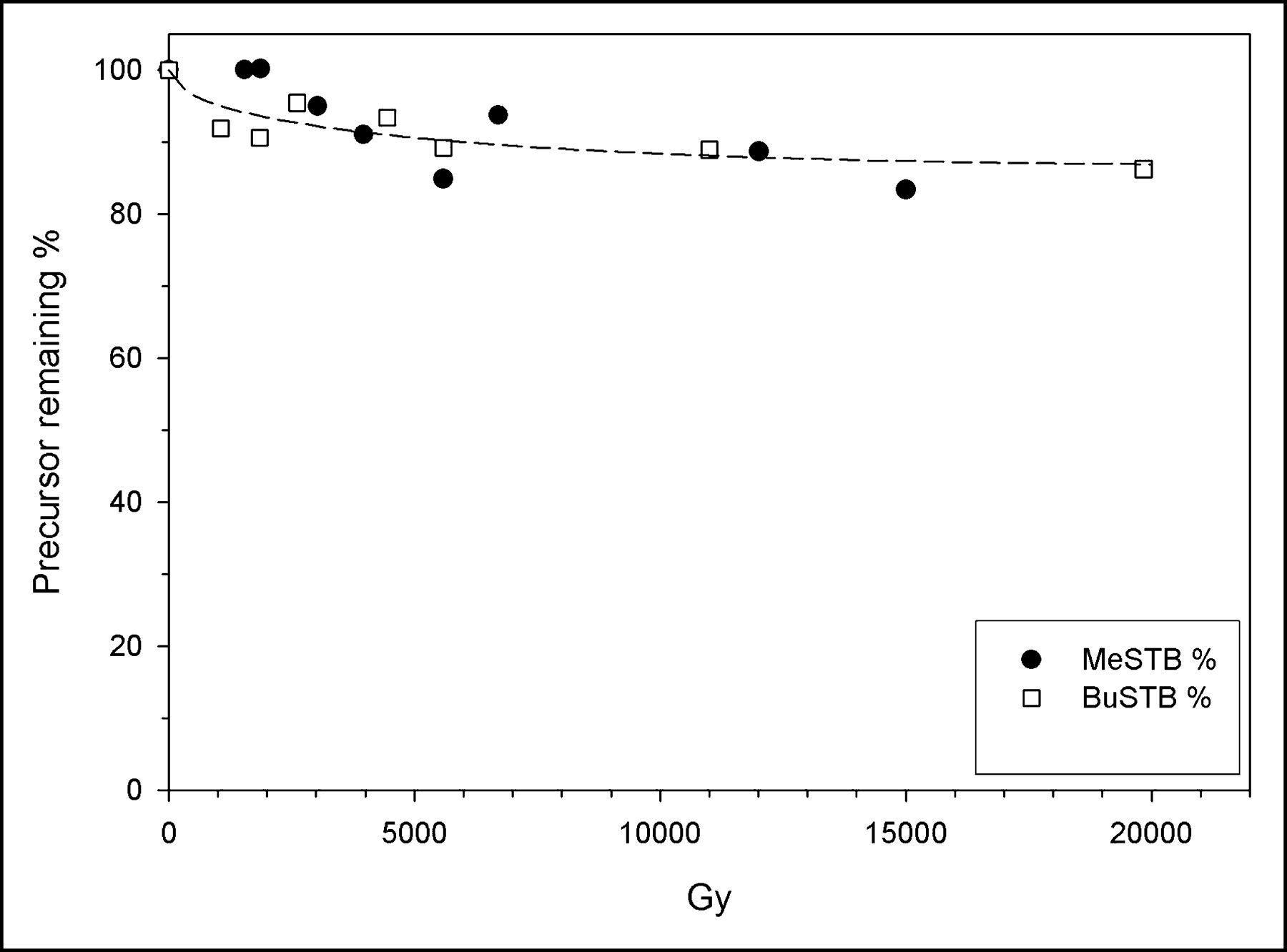
Radiolytic decomposition of BuSTB and MeSTB in methanol under acidic conditions as a function of radiation dose.
When the effect of α-particle irradiation on the integrity of BuSTB and MeSTB in benzene was evaluated under acidic conditions, the degree of degradation of the tin precursors was even lower than that observed in methanol (Fig. 4). Loss of precursor followed a linear function, with a decomposition rate of 0.8% ± 0.2% per 1,000 Gy for BuSTB and 0.7% ± 0.2% per 1,000 Gy for MeSTB. Thus, at the higher-activity-SAB benchmark equivalent dose of about 4,250 Gy, more than 95% of both precursors would remain available for astatodestannylation reactions. No cold byproducts were observed in benzene at any dose level.

Radiolytic decomposition of BuSTB and MeSTB in benzene under acidic conditions as a function of radiation dose.
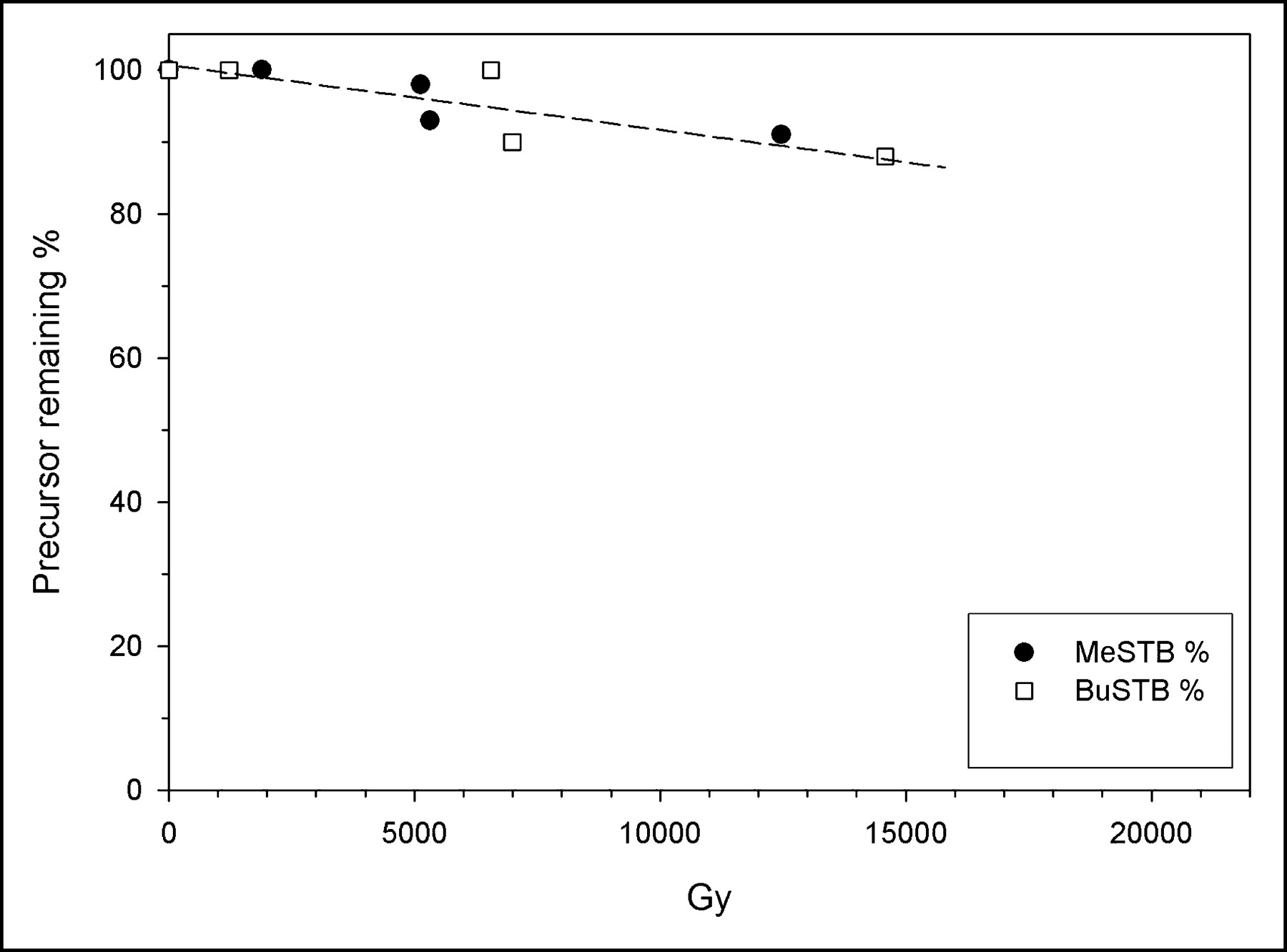
The effect of pH on decomposition of the tin precursors as a function of α-particle dose also was investigated. Paired experiments were performed on the same day at acidic and neutral pH; however, exact matching of radiation doses was difficult because of radionuclide decay and slight variations in the time required to analyze the paired samples with HPLC. Nonetheless, differences in the radiation doses received by the acidic and neutral samples used for direct comparison ranged from only 5% to 10%. In general, the results obtained at neutral and acidic pH were quite similar in either chloroform or benzene. In contrast, as summarized in Table 1, degradation of both tin precursors in methanol was greater at neutral pH. For example, the percentage of BuSTB remaining after exposure to 211At under acidic (4,890 Gy) and neutral (4,630 Gy) conditions was 97.9% ± 2.0% and 80.2% ± 1.6%, respectively.
Radiolytic Degradation of Tin Precursors in Methanol in Presence and Absence of Acetic Acid
DISCUSSION
Molecular radiotherapy has emerged as one of the most promising growth areas of nuclear medicine. The success of this approach depends on a wide variety of factors, not the least of which is identifying a radionuclide with properties well matched to the characteristics of the tumor and the vehicle that will be used to achieve tumor-selective targeting (1). For certain clinical applications—including compartmentally spread neoplasms, minimum residual disease settings, and targeting of tumor vasculature—that would benefit from treatment with short-range emissions, α-particle emitters offer significant advantages (15,16).
Although the therapeutic potential of α-particle–emitting radionuclides such as 211At has been appreciated for more than 25 y (6), their clinical application has faced many practical impediments (17). During a clinical trial of a 211At-labeled mAb (7), difficulties in the production of higher doses of the 211At-labeled radiopharmaceutical demonstrated the need for further adaptation of radiosynthetic strategies to compensate for high-radiation-field conditions. Because of the short range and high energy of the α-particles emitted after the decay of 211At and the short physical half-life, the radiation dose delivered to the reaction mixture can exceed 1,000 Gy even at initial activity levels of 740 MBq (7). An additional factor is that unlike synthetic procedures with radiometals, which take place under aqueous conditions, most 211At labeling is done in organic solvents (6). From a radiation chemistry perspective, this is an important distinction because aqueous and organic solvents differ in the nature and number of radicals that are generated after exposure of the solvent to radiation (18). In addition, the potential deleterious effects of radiolysis are quite variable, including generation of oxidizing and reducing species as well as of molecules that can form byproducts with either an essential precursor or the radionuclide itself.
The goal of the current study was to investigate solvent-related radiolytic effects on the radiosynthesis of SAB. This 211At-labeled molecule was selected not only because of the practical need to efficiently produce it at high levels for clinical studies but also because of its potential utility for labeling other proteins and peptides. In addition, these results should be pertinent to the production of chemically analogous 211At-astatobenzoate conjugates from their corresponding trialkylstannyl precursors, a strategy that has been used for labeling peptides, bisphosphonates, and biotin (19–21).
Developing radiopharmaceutical chemistry strategies for α-particle emitters such as 211At that will be suitable for use at high radiation doses presents challenges additional to those generally encountered with β-emitters such as 131I and 90Y. In addition to the higher radiation dose and dose rate to the labeling reaction mixture from short-half-life α-emitters, critical differences in radiolysis mechanisms may be present with α-particle emitters. For example, by varying the dose rate, Bibler (22) showed that 2 mechanisms are involved: a dose rate–dependent homogeneous mechanism, by which reactions occur between radicals distributed homogeneously throughout the solution, and a spur mechanism, which is not dependent on dose rate or moderated by radical scavengers. As might be expected because of their more densely ionizing nature, spur overlap becomes more important for high-LET radiation such as α-particles (23). The extent to which spur overlap occurred in our studies was not determined. Such calculations require the use of stochastic Monte Carlo and independent-reaction-time simulation techniques (23), which would be difficult to apply to these experimental conditions. Thus, extrapolation from studies reported in the radiolysis literature will be difficult, because most of those publications deal with conditions vastly different from those necessary for high-radiation-level 211At labeling.
The most important conclusion from the current study is that chloroform, the solvent used to prepare SAB for clinical 211At-labeled mAb production, is not suitable for use at high radiation doses. We observed not only extensive radiolytic decomposition of the tin precursor but also radiolytically induced generation of a nonradioactive byproduct, putatively SClB. These phenomena could explain the lower SAB yields, mAb coupling efficiencies, and immunoreactivities that were observed at higher doses (7).
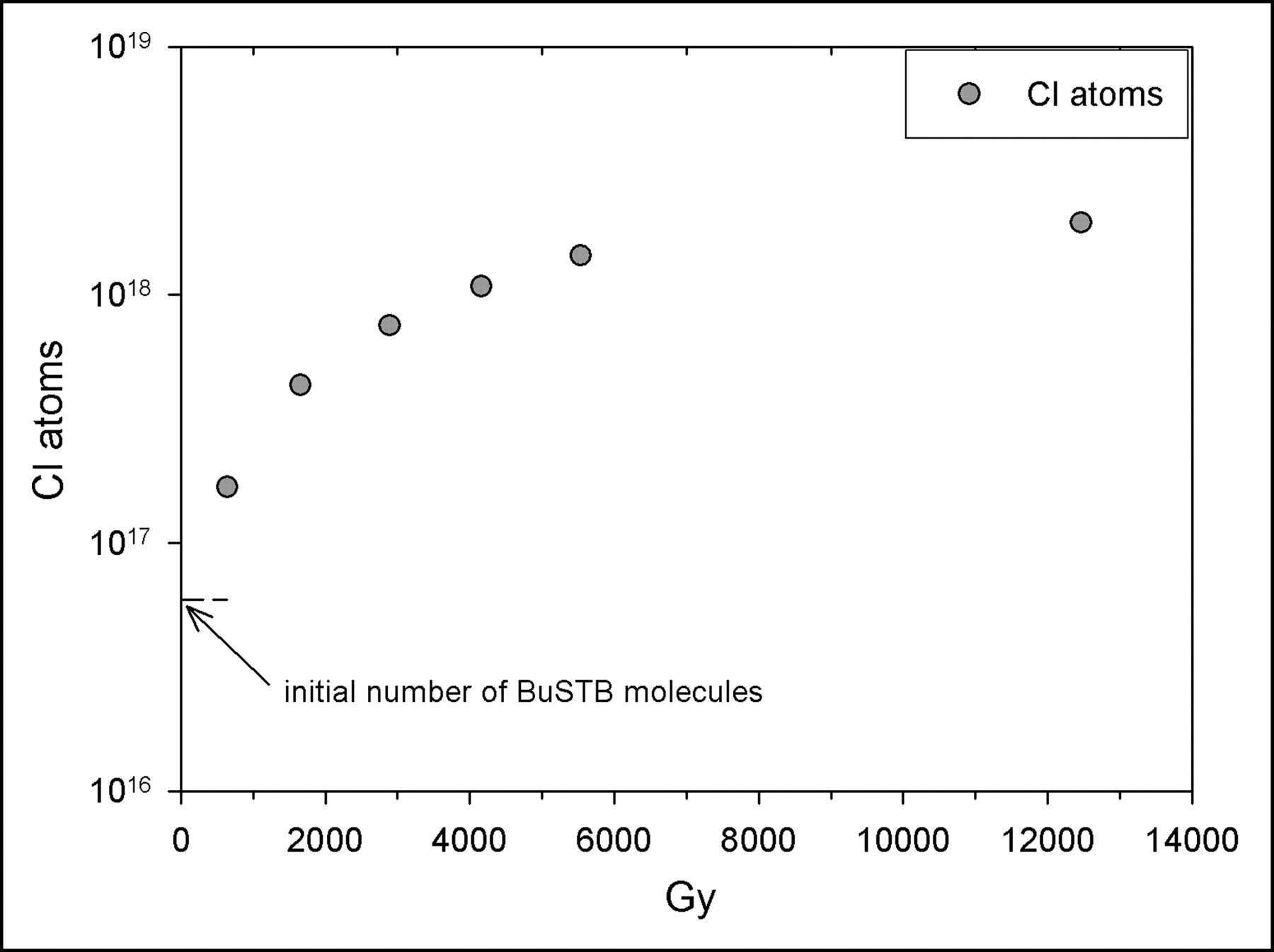
The problems encountered with chloroform at high radiation doses are consistent with the known sensitivity of aliphatic halides to radiation. Chloroform is known to undergo many reactions and has a complex radiation chemistry resulting in the generation of a variety of free radicals, including CCl3 and Cl, as well as stable products (18). The reaction between the very reactive Cl atom and BuSTB, leading to the hypothesized SClB, is likely. Using a G(Cl) of 5.4 (18), the number of Cl atoms was calculated at the dose levels used in our experiments. As shown in Figure 5, the initial number of Cl atoms produced was higher than the initial number of BuSTB molecules (50 μg are equivalent to 5.9 × 1016 molecules) even at the lowest dose level studied (644 Gy). At a dose of 3,000 Gy, the number of Cl atoms was more than an order of magnitude higher than the number of BuSTB molecules.

Calculated number of Cl atom radicals produced in chloroform by radiolysis as a function of radiation dose.
Because of the high rate of production and chemical properties of Cl, the probability is high that this radical could account for the presence of the byproduct and also explain the extensive radiolytic decomposition of BuSTB. These processes could be described with a simple consecutive reaction mechanism:
 Eq. 2 where Z is the product of decomposition of the byproduct. Even though a constantly increasing amount of the byproduct versus dose was observed, one cannot assume that the byproduct will be completely stable in this highly reactive environment. Also, this simplified kinetic description is valid only if Cl is the most important radical for the radiolytic decomposition of BuSTB. The validity is supported by the fact that the probability of interaction between the α-particles and BuSTB is much lower than with solvent molecules because of the small amount of BuSTB used (98 nmol). Also, although 211At reacts with BuSTB, the number of 211At atoms was orders of magnitude lower than the number of BuSTB molecules, and this reaction will not affect the macroscopic BuSTB concentration within the sensitivity limits of the ultraviolet HPLC detector used to monitor the reaction.
Eq. 2 where Z is the product of decomposition of the byproduct. Even though a constantly increasing amount of the byproduct versus dose was observed, one cannot assume that the byproduct will be completely stable in this highly reactive environment. Also, this simplified kinetic description is valid only if Cl is the most important radical for the radiolytic decomposition of BuSTB. The validity is supported by the fact that the probability of interaction between the α-particles and BuSTB is much lower than with solvent molecules because of the small amount of BuSTB used (98 nmol). Also, although 211At reacts with BuSTB, the number of 211At atoms was orders of magnitude lower than the number of BuSTB molecules, and this reaction will not affect the macroscopic BuSTB concentration within the sensitivity limits of the ultraviolet HPLC detector used to monitor the reaction.
If the experimental data are expressed as the molarity of BuSTB and are plotted as a function of time in seconds, an exponential radiolytic decomposition rate following first-order kinetics is apparent (Fig. 6). Taking this behavior into account, the resulting kinetics equations describing the proposed reaction mechanism (1) will be (24):
 Eq. 3
Eq. 3
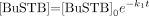 Eq. 4
Eq. 4
 Eq. 5
Eq. 5
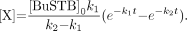 Eq. 6
Eq. 6
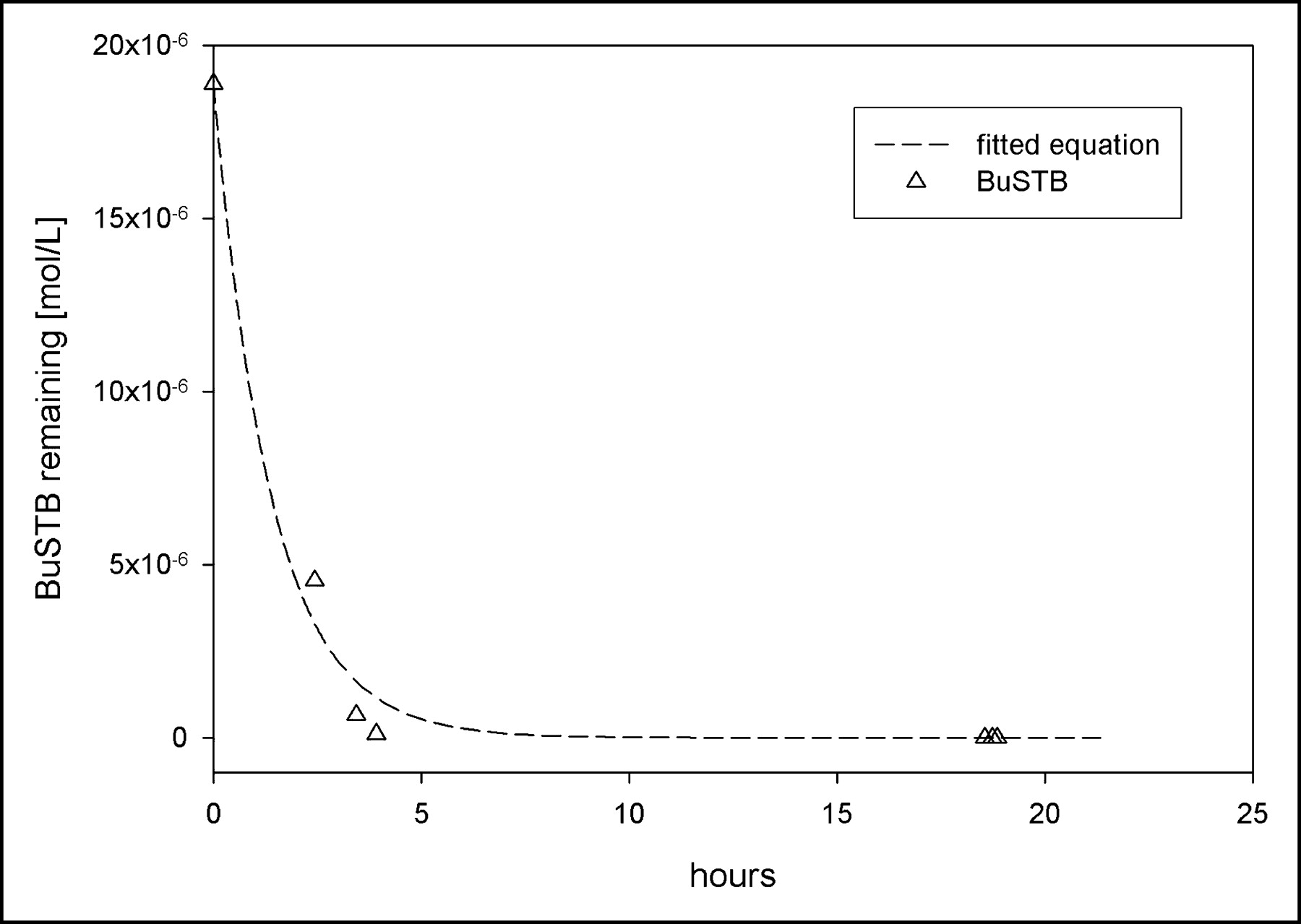
Kinetics of radiolytic decomposition of BuSTB in chloroform: experimental data and fitted first-order equation [BuSTB] = [BuSTB]0e−k1t.
To find the value for the kinetic constant k1, we fit the experimental data to an exponential function y=y0e−k1t and calculated the parameters of the equation through a nonlinear regression algorithm. Because we are interested in a time span on the order of the anticipated reaction time, and because the concentration of remaining BuSTB is negligible thereafter, only the data before 4 h were considered in calculating the regression. A value of 0.0002 mol/L/s was obtained for k1, corresponding to a high rate of radiolytic decomposition as was observed experimentally. The calculated regression curve agreed well with the experimental data even after 4 h (Fig. 6). If k1 ≫ k2, then this equation can also describe the evolution of the byproduct, with its concentration [X] proportional to the difference (e−k1t−e−k2t); [X] therefore would be expected to rise rapidly as was observed experimentally (Fig. 2). This kinetic description must be considered only a practical approximation; although Cl is present at a high concentration, it probably is not the only radical contributing to the radiolytic degradation of BuSTB.
Although having different conditions from our study, a study of the effect of γ-rays at doses up to 10 kGy/h on aqueous-chlorinated organic mixtures noted severe radiolytic dechlorination; chloroform underwent nearly total radiolytic dechlorination at doses ≥ 3,000 Gy (25,26). Little information is available on the α-particle radiolysis of chlorinated organics; however, inorganic chlorine salts in water have been investigated. In this case, the formation of oxidizing chlorinated species ClO−, ClO2−, and ClO3− in similar yield was noted at doses ≤ 500 kGy (27). The main difference between γ- and α-induced radiolysis was a higher production of ClO− and ClO2− with α-particles, presumed to be related to the high primary yield of Cl (28).
These studies make clear that a solvent other than chloroform that is compatible with electrophilic astatodestannylation should be used for higher-level SAB synthesis. Of the 3 solvents that were evaluated, benzene offered the lowest degree of radiolytic decomposition for both tin precursors, which is consistent with the fact that benzene is generally considered nearly radiation inert because of its stability. This contention is based on studies with low-LET radiation. Recent studies of the heavy ion radiolysis of liquid benzene with 1H, 4He, and 12C at doses of 100–500 Gy and 60Co γ-ray irradiation at 16 Gy/min did show that radical production was relatively low (0.72 radicals/100 eV); however, phenyl radicals did react with benzene, which ultimately could lead to polymer formation (29). Of potential relevance to 211At labeling is the observation that iodine was an efficient scavenger of these phenyl radicals, producing iodobenzene. As will be discussed in a future publication, reaction of astatine with benzene, possibly by an analogous mechanism, precludes the use of this solvent for high-activity-level astatodestannylation reactions.
Both BuSTB and MeSTB at high radiation doses were also stable in methanol, a solvent that has been used to label the para analog of SAB, albeit at considerably lower 211At activity concentrations (5). The radiolysis of methanol has been shown to yield hydrogen, carbon monoxide, formaldehyde, and glycol as the major products (30). Of relevance to the current study is the observation that increasing the LET of the radiation decreased the yield of radicals that can be scavenged (H, OH, ·CH3, and ·CH2OH) and increased the yield of molecular products. The addition of acid increased G(H2) and G(CH2O), and in the presence of oxygen, formaldehyde was the major stable product. The increased radical scavenging due to intraradical reactions at acidic pH is consistent with the lower decomposition of the tin precursors observed in the presence of acetic acid. In addition, the increased intraradical reactions inside the spur that should take place with increasing radiation dose could also explain the decreasing rate of radiolytic decomposition observed for BuSTB and MeSTB at higher dose rates.
CONCLUSION
The effect of high radiation fields on the labeling chemistry of α-particle–emitting radiotherapeutics such as those labeled with 211At has largely been unexplored. In this study, we have shown that the nature of the organic solvent used for synthesizing SAB from a tin precursor has a profound effect on the radiation sensitivity of this reaction. In chloroform, we observed not only rapid degradation of the tin precursor but also concomitant formation of a stable byproduct that could interfere with subsequent use of SAB for labeling biomolecules. Chloroform has frequently been used in the preparation of SAB for labeling mAbs (7,31–33) and other molecules (4), as well as in the synthesis of other 211At-labeled compounds such as 5-211At-astato-2′-deoxyuridine (3). Radiolytic factors would likely interfere with attempts to scale up these procedures for clinical use. Our results presented here, and our forthcoming description of SAB radiosynthesis at high doses, indicate that methanol is a much better choice than chloroform or benzene as a solvent for the synthesis of high activity levels of 211At-labeled radiopharmaceuticals.
Acknowledgments
The authors thank Kevin Alston for excellent technical assistance. This work was supported in part by U.S. Department of Energy grant DE-FG02-05ER63963; National Institutes of Health grants CA42324, CA91927, and NS20023; and a grant from the Pediatric Brain Tumor Foundation.
Footnotes
Received Aug. 31, 2004; revision accepted Dec. 8, 2004.
For correspondence or reprints contact: Michael R. Zalutsky, PhD, Department of Radiology, Duke University Medical Center, Box 3808, Durham, NC 27710.
E-mail: zalut001{at}mc.duke.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Direct Procedure for the Production of 211At-Labeled Antibodies with an {varepsilon}-Lysyl-3-(Trimethylstannyl)Benzamide Immunoconjugate
- Radiopharmaceutical Chemistry of Targeted Radiotherapeutics, Part 3: {alpha}-Particle-Induced Radiolytic Effects on the Chemical Behavior of 211At
- Potential and Pitfalls of Therapy with {alpha}-Particles