


Abstract
This investigation aimed to validate 5-[76Br]bromo-2′-fluoro-2′-deoxyuridine (BFU) as a proliferation marker using PET. Methods: Five megabecquerels 76Br-BFU were injected into the tail vein of Sprague-Dawley rats. At 6 or 16 h after injection, the rats were killed and the radioactivity concentration was measured in 6 different organs and blood. The fraction of radioactivity incorporated into DNA was determined for the spleen and small intestine. In parallel experiments, the animals were pretreated with hydroxyurea. In a few experiments, the urinary excretion of radioactivity was measured from administration of 76Br-BFU until 6 h. A sample of urine was analyzed with HPLC. In separate experiments, rats were given different doses of cimetidine, and the organ uptake and the fraction of radioactivity in DNA were determined at 24 h. Results: The highest organ uptake of radioactivity was found in the spleen, followed by the small intestine. Approximately 90% of the radioactivity in these organs was incorporated into DNA, and inhibition by hydroxyurea was pronounced. Intact tracer constituted more than 95% of the radioactivity in urine. With cimetidine, the uptake of radioactivity increased approximately 2–5 times at different doses, whereas the urine radioactivity decreased markedly. Conclusion: 76Br-BFU was predominantly incorporated into DNA after administration in vivo in rats. If cimetidine was given in combination with the tracer, an increased contrast of radioactivity concentration between organs of high proliferation and organs of low proliferation was observed. The investigation suggested that 76Br-BFU has good potential as a PET tracer for the assessment of proliferation in vivo.
In clinical practice, PET has proven to be an important tool for the diagnosis and grading of malignancy. Several publications have shown high uptake of 18F-fluorodeoxyglucose and 11C-methionine in tumors, often with high contrast to surrounding normal tissues (1–5). Although a correlation between uptake of these tracers and malignancy has been shown in comparisons of the same type of tumor, use of tracer uptake as an indicator of proliferation potential is generally not possible.
Several attempts have been made to develop methods for the assessment of proliferation potential, which is a key element in oncology and bears a closer relationship to tumor malignancy. Early attempts in the PET field used 11C-thymidine, initially labeled in the methyl group and later with the label in the 2-position (6–10). Although a correlation has been found between tracer uptake and malignancy grade, a reasonable assumption is that the uptake reflects primarily the entry of thymidine into the cells through a nucleoside transporter and not necessarily the DNA synthesis. The half-life of 11C seems too short to allow observation of DNA incorporation.
The thymidine analog bromodeoxyuridine (BrdU) has been used extensively in immunohistochemical methods for the assessment of proliferation (11). The use of 77Br-bromodeoxyuridine, 123I-iododeoxyuridine, and 131I-iododeoxyuridine as tracers has been described (12–18), but these tracers may be affected by factors such as fast elimination and rapid metabolism, leading to a short biologic half-life. Recent studies with an 18F-labeled thymidine analog have been encouraging (19).
76Br, a positron-emitting radionuclide with a half-life of 16 h, will allow an extended observation time (20), and studies in rats and pigs have suggested that 76Br-bromodeoxyuridine(76Br-BrdU) may allow a determination of proliferation potential in vivo using PET (21). A significant problem was found, however, when using this substance: a large fraction of the tissue radioactivity was constituted by 76Br-bromide. This 76Br-bromide is a metabolite of 76Br-BrdU and, soon after administration, dominates the radioactivity in plasma and in nonproliferating and slowly proliferating tissues. With forced diuresis, 76Br-bromide can to some extent be eliminated through the kidneys (22). Although such a method may facilitate the use of 76Br-BrdU as a PET tracer for DNA synthesis, the elimination of 76Br-bromide is far from complete and the use of diuresis makes the studies significantly more cumbersome for patients.
The metabolism of nucleosides is efficiently reduced by substituting a fluoride in the 2′-position of the deoxy-sugar (23). This substitution may lead to reduction of in vivo metabolism, formation of 76Br-bromide, and prolongation of the biologic half-life. We undertook our study so as to explore 5-(76Br)bromo-2′-fluoro-2′-deoxyuridine (BFU) as a potential in vivo tracer for DNA synthesis.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (age range, 12–15 wk; body weight range, 300–450 g) were used. The animals were housed under standard laboratory conditions (20°C and 50% humidity) during the entire experimental period, were allowed free access to food and water, and were kept unanesthetized. The Research Animal Ethics Committee of Uppsala University (permissions C 184/95 and C 241/98) approved the studies.
Labeling
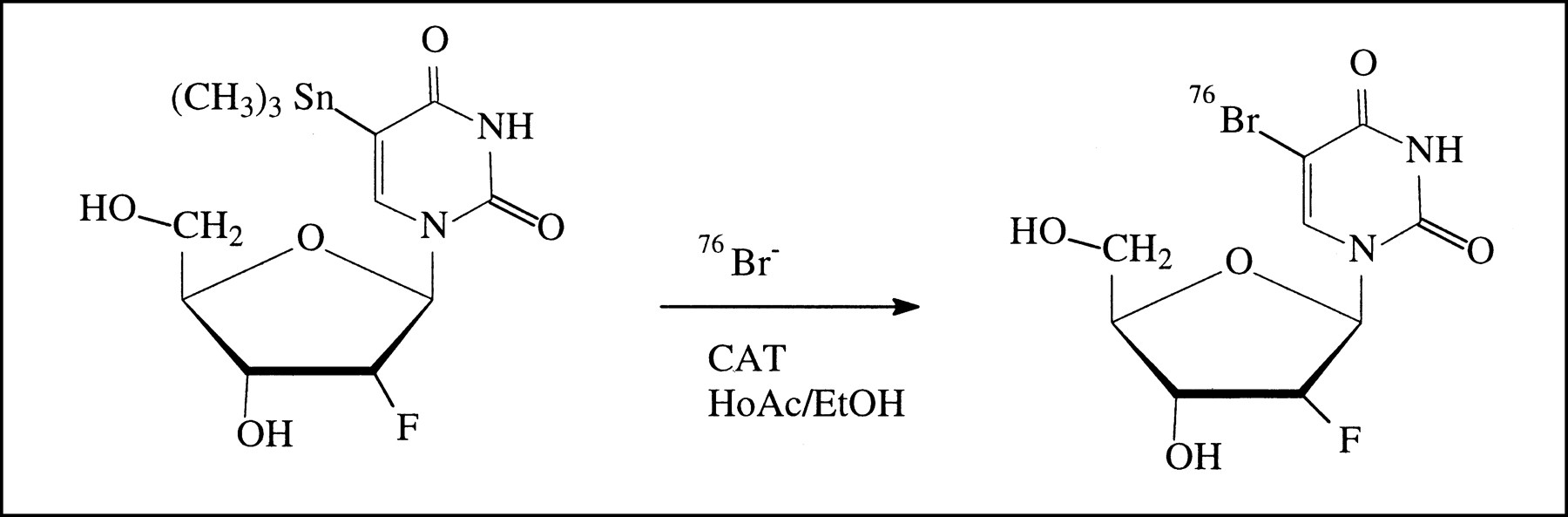
5-[76Br]bromo-2′-fluoro-2′-deoxyuridine was prepared from 5-trimethylstannyl-2′-fluoro-2′-deoxyuridine by an electrophilic substitution reaction using chloramine-T as an oxidizing agent (Fig. 1). The product was isolated in approximately 80% radiochemical yield and greater than 99% radiochemical purity within 45 min from the start of synthesis.

Scheme for preparation of 5-[76Br]bromo-2′-fluoro-2′-deoxyuridine.
[76Br]Br− was produced using the Scanditronix MC-17 cyclotron at the Uppsala University PET center using the 76Se(p,n)76Br nuclear reaction by proton irradiation of [76Se]Se-enriched (96.5% enrichment) Cu2Se. The [76Br]Br was separated from the Cu2Se pellet by a thermal diffusion procedure (24).
The precursor 5-trimethylstannyl-2′-fluoro-2′-deoxyuridine was synthesized from 5-iodo-2′-fluoro-2′-deoxyuridine according to a published procedure (25,26).
To a solution of 5-trimethylstannyl-2′-fluoro-2′-deoxyuridine (1–2 mg) in 1% acetic acid in ethanol (140 μL) was added the [76Br]Br− in ethanol (200 μL) followed by a chloramine-T solution in ethanol (2.2 mg/mL). The mixture was heated at 70°C for approximately 20 min, diluted with saline, and purified by preparative high-performance liquid chromatography (HPLC) on an Ultrasphere C-18 column (10 × 250 mm; Beckman Coulter, Inc., Fullerton, CA). Sterile saline with 5% ethanol and a gradient to 10% ethanol during 5 min was used as the mobile phase, and the flow rate was 7 mL/min. The fraction eluting with a retention time of approximately 9 min was collected and filtered to sterility. Radiochemical purity and identity were assessed using analytic HPLC with the addition of an authentic reference material. An Ultrasphere C-18 column (4.6 × 250 mm) with water:acetonitrile (95:5 to 92:8 gradient during 8 min) as the mobile phase and a flow rate of 2 mL/min was used. The retention time was approximately 8 min.
Chemicals
Hydroxyurea was purchased from Sigma Chemical Company (St. Louis, MO), and cimetidine (Acinil; A/S GEA, Fredriksburg, Denmark) was obtained from the Uppsala University hospital pharmacy. DNAzol reagent was obtained from Life Technologies Co. (Grand Island, NY).
Radioactivity Distribution
Forty rats were used. The rats were randomly assigned to 2 control groups and 2 treatment groups. Each group consisted of 10 rats. All rats were given a bolus injection of 76Br-BFU at a dose of 5 MBq through the tail vein. In the treatment group, the animals were given a dose of 200 mg hydroxyurea intravenously 30 min before injection of 76Br-BFU. At 6 and 16 h after the administration of radioactivity, the rats were killed after CO2 inhalation. One control group and 1 treated group were included at each time. The heart, lung, liver, kidney, spleen, and intestine were removed, and samples of blood were taken at the same time. The radioactivity of the samples was measured in a calibrated well counter, and their weight was recorded. The radioactivity concentration of the organs was presented as a standardized uptake value (SUV) [SUV = (radioactivity of the organ/weight of the organ)/(total given radioactivity/rat body weight)]. As a further standardization, the organ radioactivity concentration was given in relation to the whole-blood radioactivity concentration (ratio = SUV of the organ/SUV of the blood). The groups were compared statistically using the ANOVA program in StatView (SAS Institute, Cary, NC). Fisher's protected least significant difference test checked for significance at the 5% level.
DNA Separation
The samples were processed for DNA separation after the radioactivity of the organs had been measured. Two hundred milligrams tissue were taken from the spleen and small intestine, and to each sample were added 2.2 mL gnomic DNA isolation reagent. Homogenization using a Polytron homogenizer (Kinematica AG, Lucern, Switzerland) followed. After the homogenate had been centrifuged at 2500 rpm for 6 min, 1 mL supernatant was taken, and the radioactivity of the supernatant was measured and used to define the concentration of radioactivity in tissue. One-half milliliter 99.5% ethanol was added to the supernatant, and the sample was mixed by inversion and stored at room temperature for 3 min. When the band containing the DNA fraction was visible, the sample was centrifuged at 15,000 rpm and 4°C for 10 min to precipitate DNA. The pellet was washed once more with 0.5 mL 99.5% ethanol, the supernatant was removed, and renewed centrifugation for 5 min followed. The radioactivity of the pellet, representing the DNA fraction, was measured, and the DNA-incorporated radioactivity was calculated as the percentage of radioactivity in tissue ([radioactivity of pellet/radioactivity in tissue] × 100%).
Urinary Excretion of 76Br-BFU
In a few experiments, 2 groups with 6 rats in each were included. All animals were given 76Br-BFU at a dose of 5 MBq through the tail vein, and in the treatment group the animals were pretreated with 200 mg hydroxyurea, given intravenously 30 min before injection of 76Br-BFU. After injection of radioactivity, each animal was kept separately in a cage floored with tissue paper, in which the rat's urine was collected. The tissue paper was changed every hour until 6 h, and the radioactivity collected in the tissue paper was measured. The accumulated radioactivity in the urine was calculated as the percentage of the total radioactivity injected ([Σ radioactivity from tissue paper/total given radioactivity] × 100%).
Treatment with Cimetidine
In these experiments, the rats were divided into 4 groups, with each group including more than 8 animals. All rats in the 4 groups were given 5 MBq 76Br-BFU in the tail vein. In the 3 treated groups, the animals were given different doses of cimetidine (2, 6, or 20 mg/kg) at the same time as the radioactivity injection. Twenty-four hours later, the animals were killed after CO2 inhalation, and organ radioactivity and DNA fraction were determined as above. The radioactivity concentration of the organs was presented as SUV, and the DNA percentage was calculated.
HPLC Analysis of Urine
In untreated rats and in rats treated with cimetidine, the urine was collected until 2 h after administration of the tracer. The radioactivity content in the urine samples was analyzed using HPLC. An Ultrasphere C-18 column (4.6 × 250 mm) with water:acetonitrile (95:5 gradient to 92:8 during 8 min) as the mobile phase and a flow rate of 2 mL/min was used.
Whole-Body Autoradiography
Selected rats from the control group (16- and 24-h) and the group treated with 6 mg/kg cimetidine were studied by whole-body autoradiography. After the animals were killed, they were frozen in a mold and sectioned using a freezing microtome with a slice thickness of 50 μm. The slices were exposed on phosphor imaging plates for 60 h. Scanning and imaging were performed using ImageQuant software (Molecular Dynamics, Sunnyvale, CA).
RESULTS
Radioactivity Distribution
When the tracer was given alone, the highest organ uptake in the 6-h control group was in the spleen (SUV = 1.0, SD = 0.28, n = 10). The next highest organ uptake was in the small intestine (SUV = 0.53, SD = 0.11, n = 10). In the 16-h control group, the highest organ uptakes were in the small intestine (SUV = 0.46, SD = 0.14, n = 10) and spleen (SUV = 0.4, SD = 0.19, n = 10). All other organs had an SUV lower than 0.09 in the 2 groups. In the rats pretreated with hydroxyurea, the SUVs of all organs were lower than 0.12 and a pronounced inhibition was found in the small intestine and spleen (P < 0.0001). When the organ radioactivity was represented as a fraction of blood radioactivity, a significant difference (P = 0.0001) was shown in the small intestine between the 6-h control group and the 16-h control group (Fig. 2).
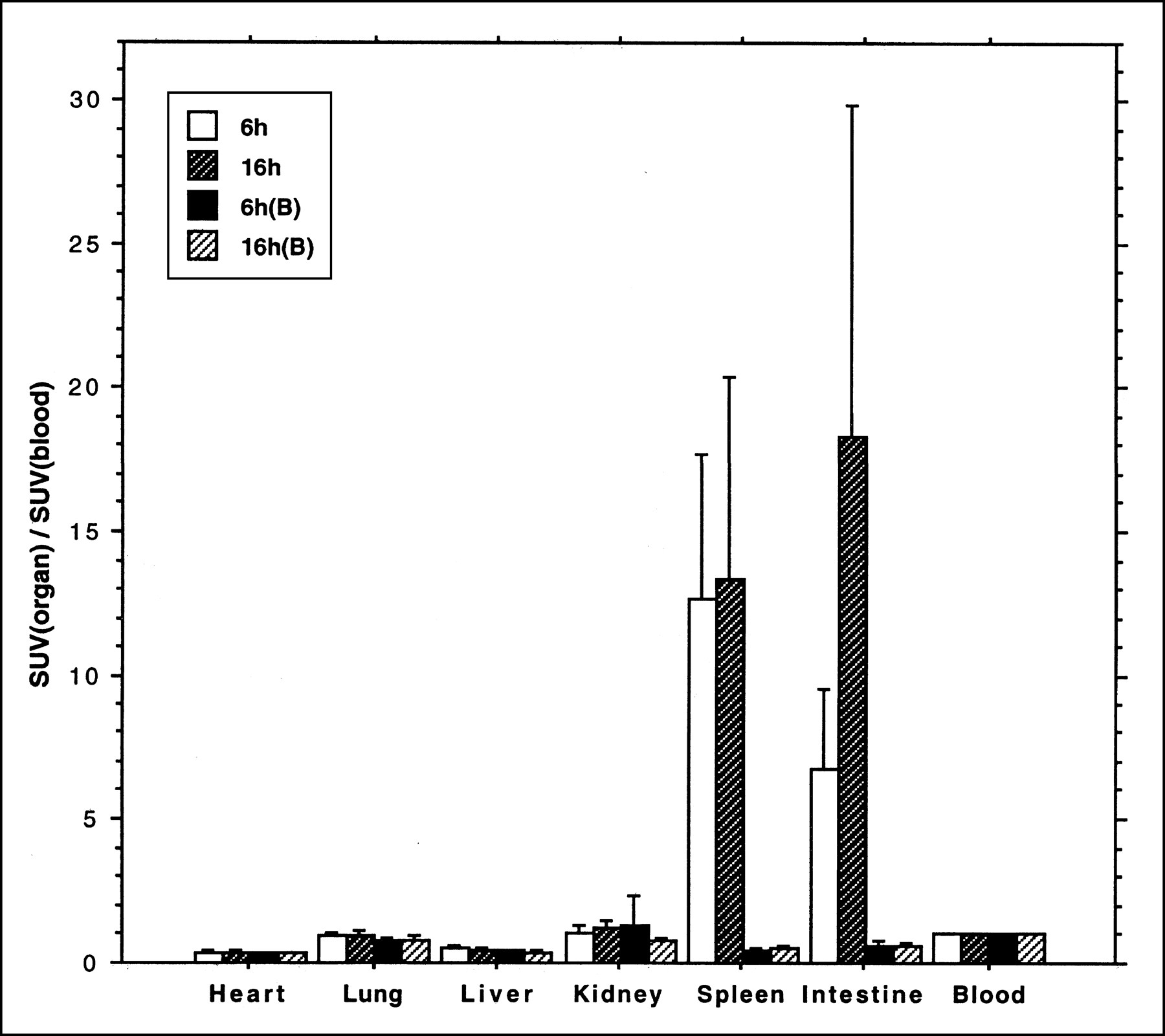
Uptake of 76Br-BFU, expressed as ratio to blood radioactivity concentration (mean ± SD), in rat organs. Animals were killed after 6 or 16 h. Separate animals (B) were given hydroxyurea 30 min before radioactivity.
DNA Separation
Analysis of the radioactivity incorporated into the DNA fraction in the 6-h control group showed a value of 95% (SD = 5%, n = 8) in the spleen and 89% (SD = 9%, n = 8) in the small intestine. In the hydroxyurea-treated group, the percentage was lower than 10%. The reduction in the percentage of DNA incorporation was statistically significant (P < 0.0001; Fig. 3). Almost the same result was found for the 2 time points (6- and 16-h).

Fraction of 76Br-BFU radioactivity, expressed as percentage of total tissue radioactivity (mean ± SD), recovered in DNA fraction in rat organs. Separate animals (B) were given hydroxyurea 30 min before radioactivity.
Urinary Excretion of 76Br-BFU
At 6 h after administration of 76Br-BFU, the accumulated radioactivity in the urine was 71% of the total given radioactivity in the control group (n = 6). In the group pretreated with hydroxyurea, the fraction was 67% (n = 6). No significant difference was seen between the 2 groups.
Treatment with Cimetidine
In these experiments, the relative distribution of the tracer was similar to that shown for the control group in the 16-h experiment. The small intestine, closely followed by the spleen, had the highest concentration. However, the absolute magnitude of the SUVs was considerably higher than in untreated animals for all 3 treatment groups (Fig. 4). Analysis showed that, for the spleen and small intestine, more than 90% of radioactivity was incorporated into the DNA fraction in the treated and untreated groups.
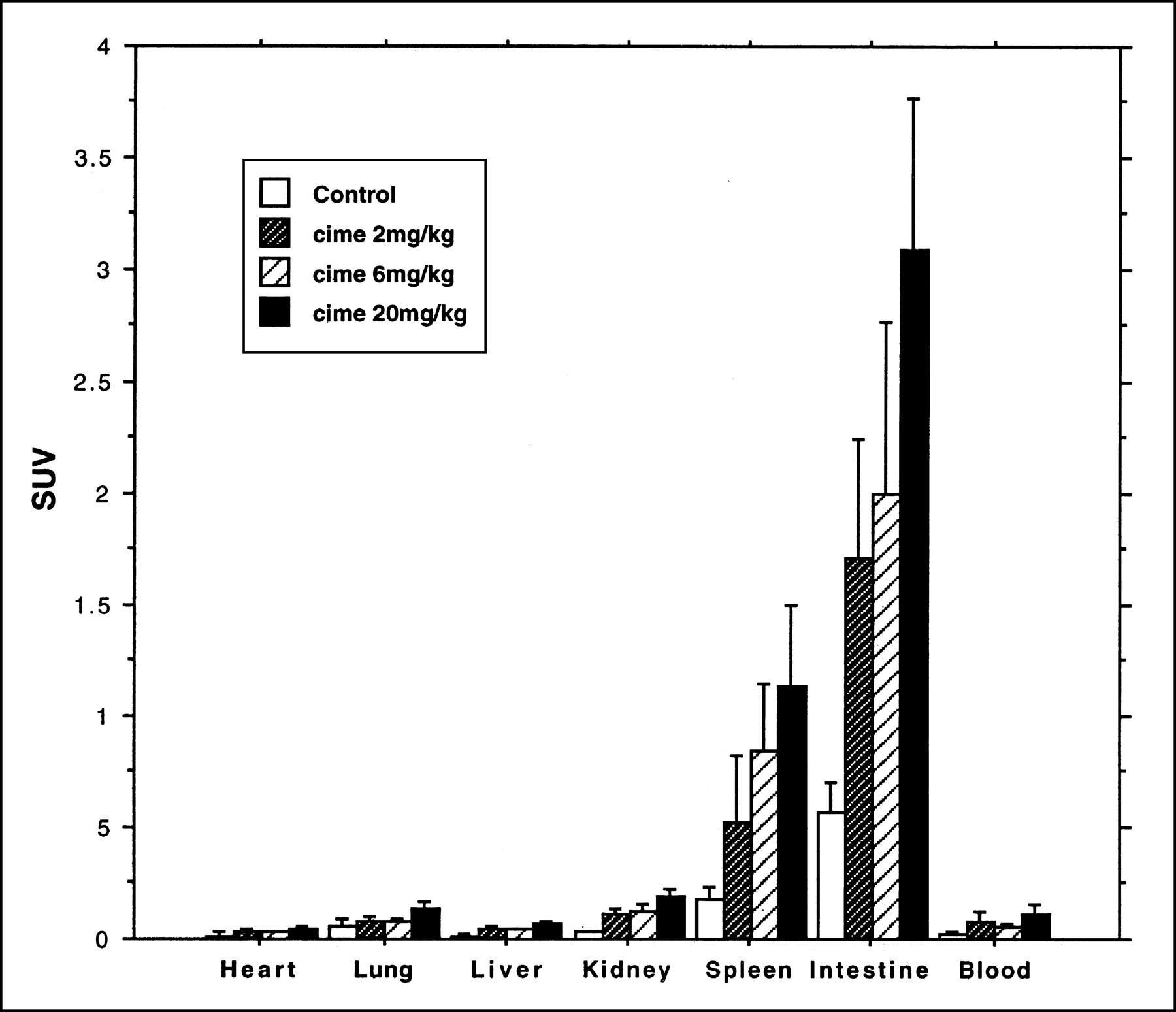
Uptake of 76Br-BFU, expressed as SUV (mean ± SD), in rat organs. Animals were killed after 24 h. Separate animals were given cimetidine together with 76Br-BFU.
HPLC Analysis of Urine
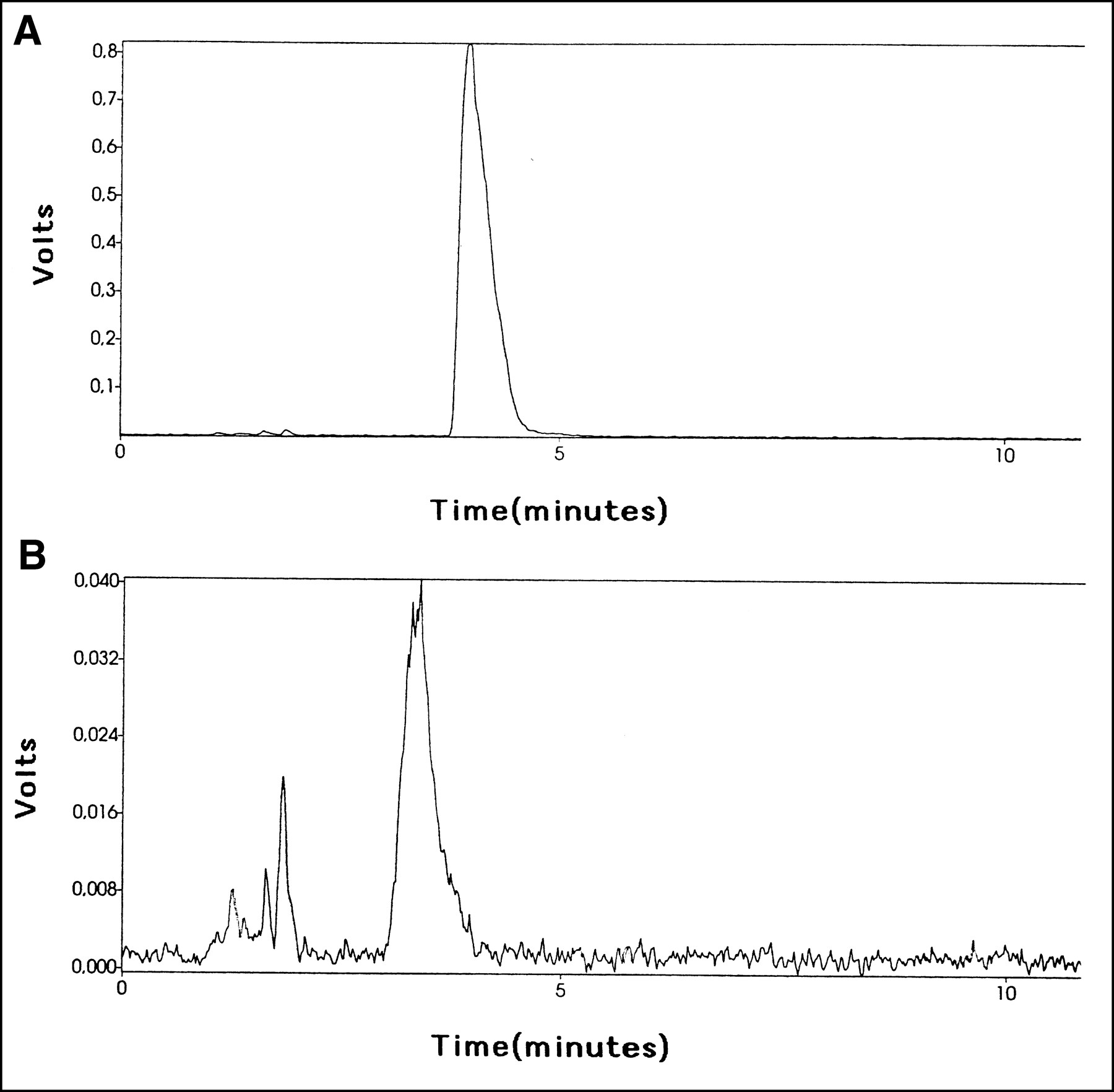
The chromatogram from urine samples showed a dominant peak of 76Br-BFU in the control group (>95%) and approximately 60% of intact 76Br-BFU in the group treated with cimetidine, with the rest of the radioactivity consisting of metabolites more polar than 76Br-BFU (Fig. 5). The retention time was shorter (approximately 4–6 min) when urine was included in the samples, compared with the analysis after synthesis. The radioactivity concentration was markedly lower after cimetidine treatment.
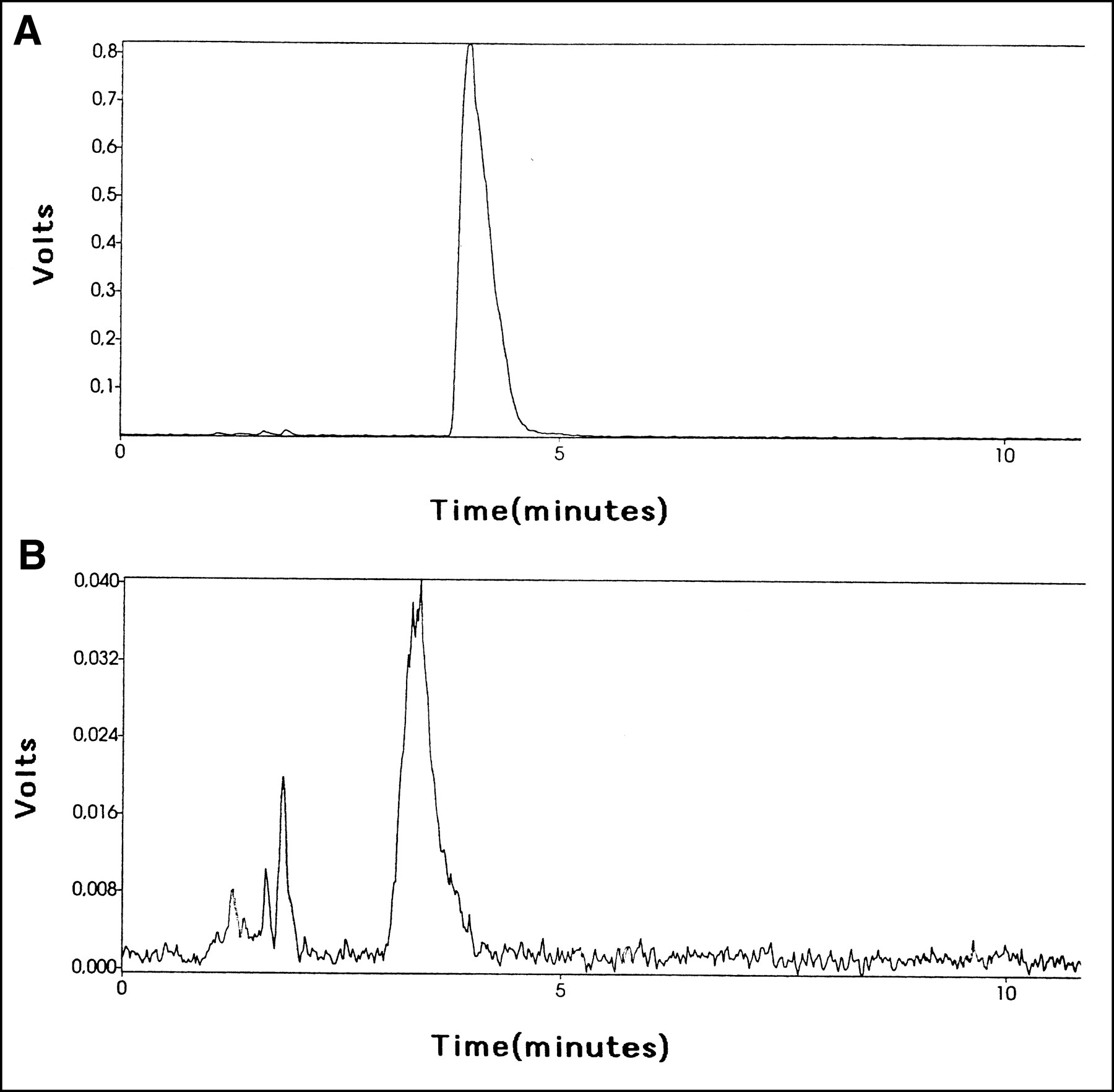
HPLC analysis of urine after administration of 76Br-BFU alone (A) or with simultaneous administration of cimetidine (B).
Whole-Body Autoradiography
When only 76Br-BFU was injected into the rat, the images clearly showed the small intestine and bone (Fig. 6). Radioactivity uptake was also observed in the spleen. Delineation of the small intestine was clearer in the 24-h control group than in the 16-h control group. The best explicit image of the small intestine was in the group treated with 6 mg/kg cimetidine (Fig. 6C). High radioactivity uptake was also found in skin and bone in the same picture.

Ex vivo autoradiography in rats injected with 76Br-BFU. (A) Animal given only tracer and killed after 16 h. (B) Animal given only tracer and killed after 24 h. (C) Animal given 6 mg/kg cimetidine simultaneously with tracer and killed after 24 h.
DISCUSSION
Previous work from our laboratory suggested that the thymidine analog 76Br-BrdU could be used as a tracer for DNA synthesis. However, the rapid metabolism and a residual high content of 76Br-bromide pose significant problems (21,22). Forced diuresis using a loop diuretic may improve the latter aspect but introduces additional discomfort to the patient. Therefore, this tracer may have limited use in clinical practice.
The study presented in this article has revealed that 76Br-BFU, with a fluorine atom in the 2′ position of 2′-deoxyribose in the 76Br-BrdU molecule, was much more stable than 76Br-BrdU. Only limited amounts of 76Br-bromide were produced. In the organ distribution experiments, the highest concentration of radioactivity was in organs with active DNA synthesis, such as the spleen and intestines. Pretreatment of animals with the DNA synthesis inhibitor hydroxyurea (27) reduced uptake in these organs markedly—to a level similar to that in organs with limited DNA synthesis, such as the heart, lungs, and liver. The experiments with DNA separation also showed that a dominant portion of tissue radioactivity was incorporated into DNA in the organs with active DNA synthesis. All our results suggest that 76Br-BFU is superior to 76Br-BrdU as a proliferation marker.
We did observe, however, that the SUV of 76Br-BFU was lower than that of 76Br-BrdU. The reason was the elimination of 70% of total given radioactivity as intact compound by the kidneys through urinary secretion within 6 h after 76Br-BFU injection. A dominant portion of this urinary excretion occurred within the first 3 h.
Some authors have suggested that cimetidine, an inhibitor of the organic cation secretory system, may inhibit the secretion of some nucleosides (28–30). According to the pharmacokinetics of cimetidine, its effect after a single dose may remain for 2–3 h. This time should be sufficient for incorporation of radioactivity into DNA. After our rats received cimetidine, the spleen and small intestine showed a pronounced increase of SUV while retaining a very high proportion of the radioactivity incorporated into DNA. Simultaneously, the amount of radioactivity recovered in the urine decreased markedly. HPLC analysis of the urine also showed that, without cimetidine treatment, the urinary radioactivity was predominantly composed of intact 76Br-BFU and, with cimetidine treatment, the intact 76Br-BFU was markedly reduced, leaving some polar metabolites in the urine.
We observed a pronounced difference in ex vivo autoradiography (Fig. 6) with respect to imaging of the proliferating organs. Although the mucosa of the small intestine was visualized after giving only a tracer dose of 76Br-BFU, the uptake was low and the autoradiograph was unclear. In cimetidine-administered animals, on the other hand, strong uptake rendered excellent visualization of the organs with active DNA synthesis, such as the intestines, bones, skin, and spleen.
CONCLUSION
The studies show that the radioactivity of 76Br-BFU after in vivo administration to rats is predominantly incorporated into DNA, and high contrast is observed between organs of high proliferation and organs of low proliferation. An otherwise relatively rapid and pronounced urinary elimination of the tracer is largely blocked if cimetidine is given in conjunction with the tracer. These facts suggest that 76Br-BFU may have great potential as a PET tracer for the assessment of proliferation in vivo.
Acknowledgments
This study was supported in part by grants from the Swedish Cancer Society. The valuable discussions and support of Dr. Ronald Blasberg, Sloan-Kettering Memorial Institute, New York, NY, are greatly acknowledged.
Footnotes
Received Nov. 5, 1999; revision accepted Mar. 28, 2000.
For correspondence or reprints contact: Mats Bergström, PhD, PET Center, Uppsala University Hospital, 751 85 Uppsala, Sweden.
REFERENCES
In this issue




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Imaging of Cell Proliferation: Status and Prospects
- Evaluation of 76Br-FBAU as a PET Reporter Probe for HSV1-tk Gene Expression Imaging Using Mouse Models of Human Glioma
- Biodistribution and Radiation Dosimetry Estimates of 1-(2'-Deoxy-2'-18F-Fluoro-1-{beta}-D-Arabinofuranosyl)-5-Bromouracil: PET Imaging Studies in Dogs
- Rat Studies Comparing 11C-FMAU, 18F-FLT, and 76Br-BFU as Proliferation Markers
- Rationale of 5-125I-Iodo-4'-Thio-2'-Deoxyuridine as a Potential Iodinated Proliferation Marker