


Abstract
Since the invention of 18F-FDG as a neurochemical tracer in the 1970s, 18F-FDG PET has been used extensively for dementia research and clinical applications. FDG, a glucose analog, is transported into the brain via glucose transporters and metabolized in a concerted process involving astrocytes and neurons. Although the exact cellular mechanisms of glucose consumption are still under investigation, 18F-FDG PET can sensitively detect altered neuronal activity due to neurodegeneration. Various neurodegenerative disorders affect different areas of the brain, which can be depicted as altered 18F-FDG uptake by PET. The spatial patterns and severity of such changes can be reproducibly visualized by statistical mapping technology, which has become widely available in the clinic. The differentiation of 3 major neurodegenerative disorders by 18F-FDG PET, Alzheimer disease (AD), frontotemporal dementia (FTD), and dementia with Lewy bodies (DLB), has become standard practice. As the nosology of FTD evolves, frontotemporal lobar degeneration, the umbrella term for pathology affecting the frontal and temporal lobes, has been subclassified clinically into behavioral variant FTD; primary progressive aphasia with 3 subtypes, semantic, nonfluent, and logopenic variants; and movement disorders including progressive supranuclear palsy and corticobasal degeneration. Each of these subtypes is associated with differential 18F-FDG PET findings. The discovery of new pathologic markers and clinicopathologic correlations via larger autopsy series have led to newly recognized or redefined disease categories, such as limbic-predominant age-related TDP-43 encephalopathy, hippocampus sclerosis, primary age-related tauopathy, and argyrophilic grain disease, which have become a focus of investigations by molecular imaging. These findings need to be integrated into the modern interpretation of 18F-FDG PET. Recent pathologic investigations also have revealed a high prevalence, particularly in the elderly, of mixed dementia with overlapping and coexisting pathologies. The interpretation of 18F-FDG PET is evolving from a traditional dichotomous diagnosis of AD versus FTD (or DLB) to a determination of the most predominant underlying pathology that would best explain the patient’s symptoms, for the purpose of care guidance. 18F-FDG PET is a relatively low cost and widely available imaging modality that can help assess various neurodegenerative disorders in a single test and remains the workhorse in clinical dementia evaluation.
Since the development of FDG (2-deoxy-2-[F-18]fluoro-d-glucose [18F-FDG]) as a radiopharmaceutical to measure brain metabolism with PET imaging in the 1970s, both research and clinical applications in neurodegeneration continue to grow. Although the number of research publications concerning 18F-FDG PET and dementia has been surpassed recently by proteomic-specific imaging such as amyloid and tau PET, 18F-FDG PET remains a major workhorse for the clinical evaluation of cognitive disorders.
18F-FDG AS A NEUROCHEMICAL TRACER
Glucose is the principal source of energy for the mammalian brain. In adult humans, the brain represents approximately 2% of the total body mass, yet it uses approximately 20%–25% of the glucose consumed daily (1). Regional changes in neuronal activity due to neurodegeneration can be sensitively reflected by regional brain glucose consumption. A structural analog of glucose, 2-deoxy-d-glucose, was labeled with 14C, 11C, and 18F and used as a tool to investigate energy metabolism in the brain in both preclinical and clinical studies in the 1970s (2,3). Applications of the tracer were quickly extended to the investigations of myocardial and cancer glucose metabolism, and the tracer is now widely available and used in clinical nuclear medicine practices worldwide.
Glucose is transported from plasma to the brain via glucose transporters (GLUTs), primarily GLUT1 expressed in the blood–brain barrier/astrocytes and GLUT3 expressed in neurons (4). Glucose is then phosphorylated by hexokinase to glucose-6-phosphate in cells and metabolized in the glycolytic pathway to produce adenosine triphosphate (2). However, unlike glucose-6-phosphate, deoxyglucose-6-phosphate cannot be used as a substrate for further glycolysis and subsequently accumulates in cells because glucose-6-phosphatase (the reverse enzyme) activity in the brain is low (5).
CELLULAR MECHANISMS OF 18F-FDG UPTAKE IN THE BRAIN
18F-FDG uptake in the brain is considered to represent general neuronal activity. The cellular mechanisms of brain glucose metabolism and 18F-FDG uptake have been further elucidated over the past several decades. Although energy consumption in neurons occurs for various signaling and neurotransmitter processes, synaptic currents and action potentials seem to comprise the greater part of the consumption (6). This observation coincides with deoxyglucose uptake in the axonal terminals when neurons are stimulated electrophysiologically (7). Excitatory synapses, in particular glutamatergic, predominate among cortical synapses and thus consume a large portion of the 18F-FDG uptake in the human brain (6). Recent studies indicate that glucose is transported from the capillaries to the astrocytes and metabolized into lactate, which subsequently serves as an energy source for neurons (“astrocyte-neuron lactate shuttle”) (8). For simplicity, the terms hypometabolism and decreased 18F-FDG uptake on 18F-FDG PET are used interchangeably hereafter.
REMOTE EFFECTS ON 18F-FDG UPTAKE (DIASCHISIS)
18F-FDG uptake not only represents local neuronal/synaptic activity, but also can reflect remote effects via deactivation of projection neurons without local neuronal injury. One classic example of such phenomena is crossed cerebellar diaschisis (CCD) (Fig. 1A) initially discovered in stroke patients (9). Decreased 18F-FDG uptake/perfusion in the contralateral cerebellar hemisphere is caused by deactivated ponto–cerebellar neurons secondary to the primary injury in corticopontine neurons from the supratentorial lesion. Such remote effects (diaschisis) can also be seen in neurodegeneration such as Alzheimer disease (AD) (10), frontotemporal dementia (FTD) (11), corticobasal syndrome, and semantic and logopenic variants of primary progressive aphasia (PPA) (12). In AD, decreased 18F-FDG uptake due to diaschisis can be seen in the cerebellar hemisphere contralateral to the predominant cortical involvement as well as in the ipsilateral thalamus and basal ganglia (13). Similarly, decreased 18F-FDG uptake in the caudate nucleus is often seen in FTD. Although the imaging modalities of 18F-FDG PET and MRI are combined under the term Neurodegeneration (N) as a category in the recent National Institute on Aging and Alzheimer's Association (NIA-AA) Research Framework (14), careful translation of 18F-FDG PET findings to a pathophysiologic interpretation is needed for a better understanding of such disease processes. When interpreting 18F-FDG PET scans, it is crucial to understand the underlying anatomy of white matter tracts and fiber projections so that the primary and remote pathologies can be inferred correctly.

18F-FDG and cerebral blood flow (CBF) PET. Crossed cerebellar diaschisis (CCD) (A) seen in a right (Rt.) middle cerebral artery (MCA) stroke. Decreased glucose metabolism (18F-FDG) and perfusion (CBF) in contralateral left (Lt.) cerebellar hemisphere (white arrow) where no ischemic injury is present. Decreased 18F-FDG uptake and CBF are secondary to deactivation of cortico–ponto–cerebellar tract (“remote effect”). Such remote effects are known to occur in neurodegenerative disorders. 18F-FDG uptake not only reflects local pathology, but also could reflect remote pathology. Knowledge of cortical pathways is crucial for scan interpretation. (B) Coupling between glucose metabolism (18F-FDG) and CBF in AD measured by PET and 3D-SSP analysis. Statistical t maps (top 2 rows) represent regional hypometabolism and hypoperfusion seen in a group of AD patients. Both 18F-FDG and CBF PET show similar regional changes, though CBF PET appears slightly less sensitive. Ratio maps between CBF and 18F-FDG (bottom row) indicate that metabolic-flow coupling is relatively preserved in areas both affected and not affected by AD. This observation supports use of various flow-related measurements such as perfusion SPECT, early flow images obtained as a part of amyloid PET or tau PET, or MRI-based perfusion imaging for dementia evaluation.
IMAGE INTERPRETATION USING STATISTICAL MAPPING TECHNIQUES
One significant advancement for 18F-FDG PET interpretation in neurodegeneration is the widespread use of statical mapping for image interpretation and analysis. Various neurodegenerative dementias are known to affect specific regions of the brain. PET scans can demonstrate altered 18F-FDG uptake corresponding to the regions affected directly or indirectly. For the differential diagnosis of dementing disorders, interpreters must recognize the spatial distribution of the changes in 18F-FDG uptake, and the magnitude of such regional changes. This interpretational task may not be straightforward due to several factors, including: the anatomic information on PET is tracer-specific and relatively limited as compared with that on structural imaging such as MRI; the image presentation (e.g., image orientation, color scale) can be variable in clinical settings; the metabolic changes associated with neurodegeneration can be subtle and diffuse; and it is difficult to consistently assess the extent and severity of metabolic alterations by visual inspection of the reconstructed images. To overcome these challenges, a quantitative statistical mapping approach for scan interpretation was developed (15).
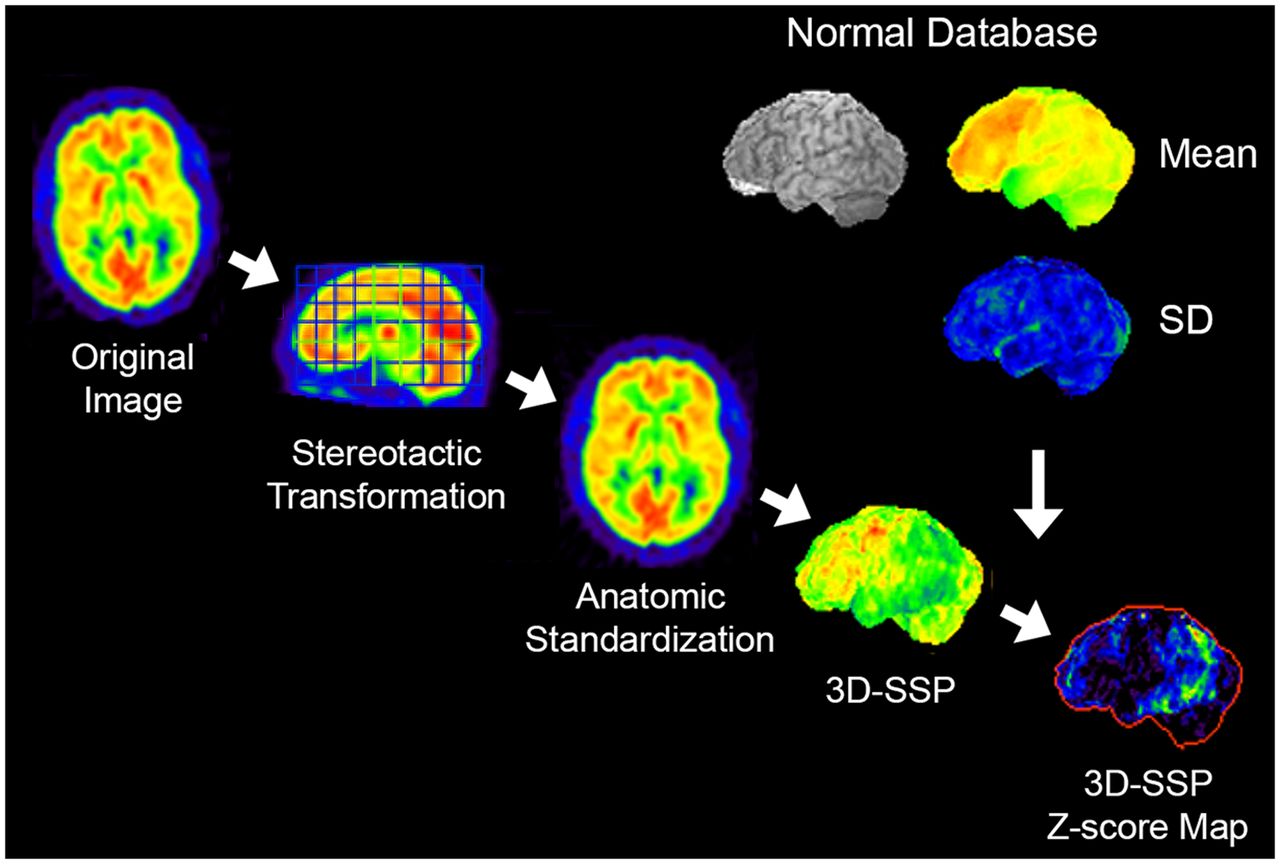
In statistical mapping, the original reconstructed images are realigned in the stereotactic coordinate system (16), and individual anatomic differences across subjects are minimized by anatomic standardization (17,18). The anatomically standardized image set from an individual subject is compared with a normal database composed of similarly processed image sets from multiple age-similar cognitively normal subjects, and the differences between the individual scan compared with the normal database are visualized as a z score map (15). To further reduce the residual anatomic differences across subjects and to minimize effects of cortical atrophy on this comparison, a 3-dimensional stereotactic surface projection (3D-SSP) algorithm can be applied as a data extraction/reduction method (Fig. 2) (15). z score maps demonstrate the pattern of altered 18F-FDG uptake in a standardized format, which can be used for scan interpretation in conjunction with the original reconstructed images. This approach not only improves overall diagnostic accuracy (Fig. 3), but also offers quantitative information on the significance level of detected abnormalities, helps to standardize interpretation, allows cross-institutional comparisons, and helps support consistent scan interpretation by physicians with different levels of experience (19). Statistical mapping relies on the consistency of image quality that can be improved by standardized imaging protocols and image reconstruction.
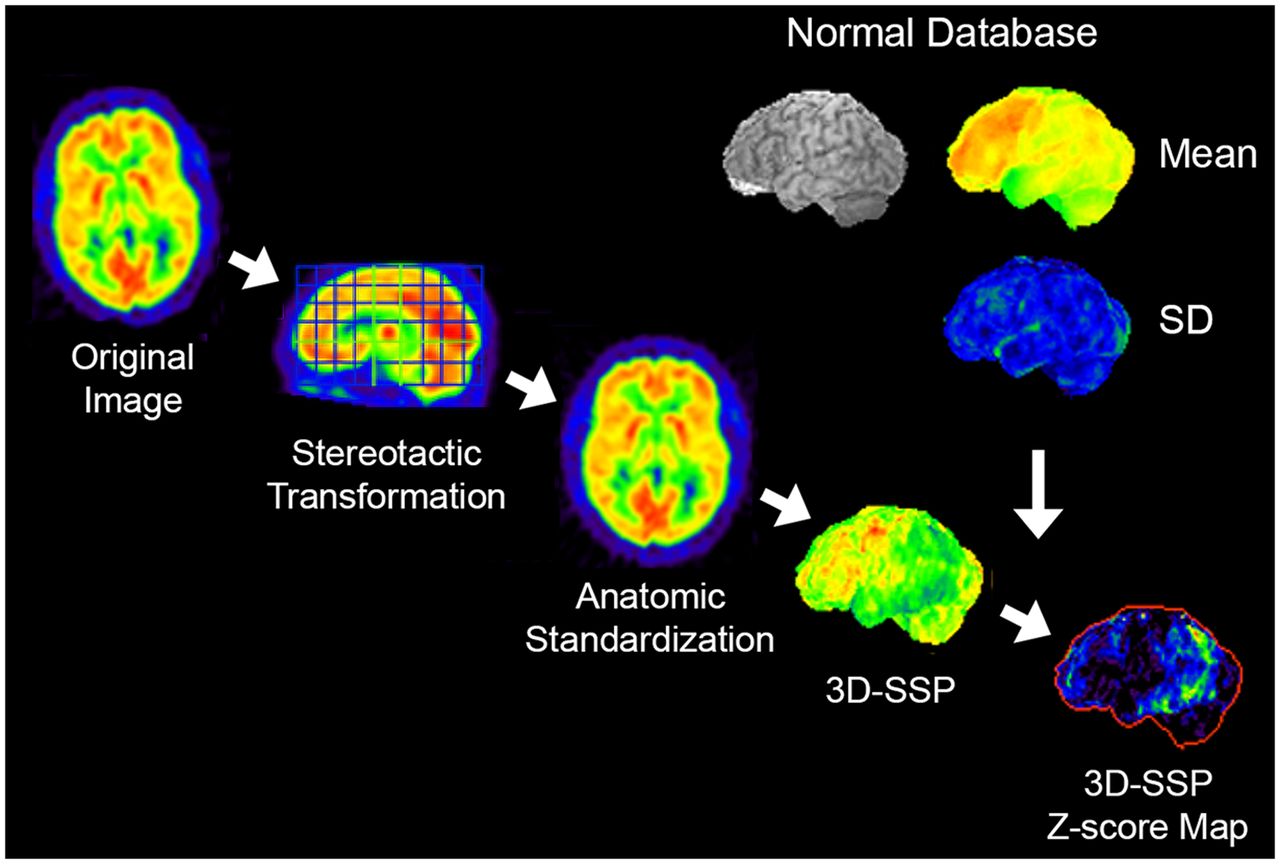
3D-SSP. Original transaxial images are anatomically standardized in the stereotactic coordinate system, and gray matter activity is extracted on a pixel-by-pixel basis by the 3D-SSP algorithm. Extracted data are then compared with a normal database (mean and SD), which is composed of similarly processed PET scans from multiple normal subjects. Differences between individual data and normal database are expressed as z score maps.

18F-FDG PET scan of patient with progressive mild cognitive decline. Original transaxial images (top 6 rows, black and white) demonstrate very mild metabolic reductions in parietal association cortex and posterior cingulate cortex/precuneus. However, such mild changes cannot be appreciated consistently. 3D-SSP z score maps (bottom row, from left to right: right lateral, left lateral, superior, inferior, anterior, posterior, right medial, and left medial views) from same patient demonstrate apparent metabolic reductions in posterior cingulate cortex/precuneus as well as parietal association cortex bilaterally seen on lateral, superior, and posterior views. Statistical mapping, if used appropriately, improves diagnostic accuracy and consistency.
Statistical mapping with 18F-FDG PET was also used to discover metabolic signatures unique to different types of dementing disorders. Hypometabolism in the posterior cingulate cortex was found in an early stage of AD (20,21) and in APOE4 homozygote subjects (22). Hypometabolism in the primary visual cortex and the occipital lobe was found in Parkinson disease with dementia (23) as well as autopsy-proven DLB (24). These findings are now hallmarks in the clinical interpretation of 18F-FDG PET scans.
When applying imaging biomarkers to better understand the pathologic changes occurring in AD (25), differences in the detection threshold of each imaging modality or biomarker need to be carefully incorporated into the interpretation. The detection threshold is also significantly affected by the method used for the analysis of the imaging data. According to a metaanalysis based on the imaging biomarker framework of “Metrics” and “Submarkers” (26), the diagnostic positive likelihood ratio of 18F-FDG PET analyzed by 3D-SSP was equivalent to that of an amyloid PET distribution volume ratio (DVR) analysis. However, when 18F-FDG PET was interpreted visually, the diagnostic positive likelihood ratio decreased precipitously. Different statistical mapping methods can also yield different outcomes (27). If biomarker findings are used to infer the pathogenesis of the disease and the time course for progression, such differences in the detection threshold simply due to the analytic method can cause a significant bias in research.
CLINICAL IMAGING ALTERNATIVES TO 18F-FDG PET FOR DEMENTIA EVALUATION
Regional energy metabolism and cerebral blood flow are tightly coupled in physiologic or nonacute pathologic conditions (28), and such coupling may be generally preserved in aging (29) and neurodegeneration (Fig. 1B). These observations serve as the physiologic basis to use cerebral blood flow as measured by perfusion SPECT (or PET) as an alternative to 18F-FDG PET for dementia evaluation. However, perfusion SPECT suffers from technical factors, such as a lower spatial resolution and sensitivity, inaccuracy in the attenuation correction, and difficulties in standardizing image quality and quantification. Recent efforts to translate perfusion SPECT/PET techniques to MRI-based perfusion measurements, such as arterial spine labeling MRI, are ongoing (30,31). Perfusion-equivalent information can also be obtained from early-phase dynamic PET imaging of amyloid (32,33) or tau (34) PET. The diagnostic accuracy of these alternative techniques needs to be established prospectively among dementia patients seen in clinical settings.
DIFFERENTIAL DIAGNOSIS OF AD, FTD, AND DLB: STANDARD OF CARE
The most common use of 18F-FDG PET in the context of a dementia evaluation is to differentiate AD, FTD, and DLB (Table 1). The clinical course, complications, and clinical management of these conditions differ. An accurate and specific diagnosis guides both clinicians and family care partners, permitting early interventions and proactive planning. Cholinesterase inhibitors and memantine are effective in AD, but not in FTD (35). Amyloid targeting immunotherapy has the potential to be disease modifying only in AD. 18F-FDG PET offers improved diagnosis of DLB, sometimes not initially considered by the clinician. Adverse events are more frequently associated with neuroleptics in DLB and Parkinson disease with dementia (36). Therefore, the clinical differential diagnosis of major neurodegenerative disease categories, AD, FTD, and DLB, becomes critical in terms of appropriate medication and patient management. In the United States, the clinical use of 18F-FDG PET for the differential diagnosis of AD versus FTD can be covered by the Centers for Medicare & Medicaid Services.
Differential Diagnosis of Neurodegenerative Dementing Disorders by 18F-FDG PET
18F-FDG PET imaging features associated with AD, FTD, and DLB have been characterized by numerous investigators over the last 4 decades. Different types of neurodegenerative disorders tend to affect specific brain regions (selective vulnerability) while relatively sparing other regions. The spatial patterns of decreased versus relatively preserved 18F-FDG uptake in the brain give differential clues to the specific neurodegenerative substrate (Fig. 4).
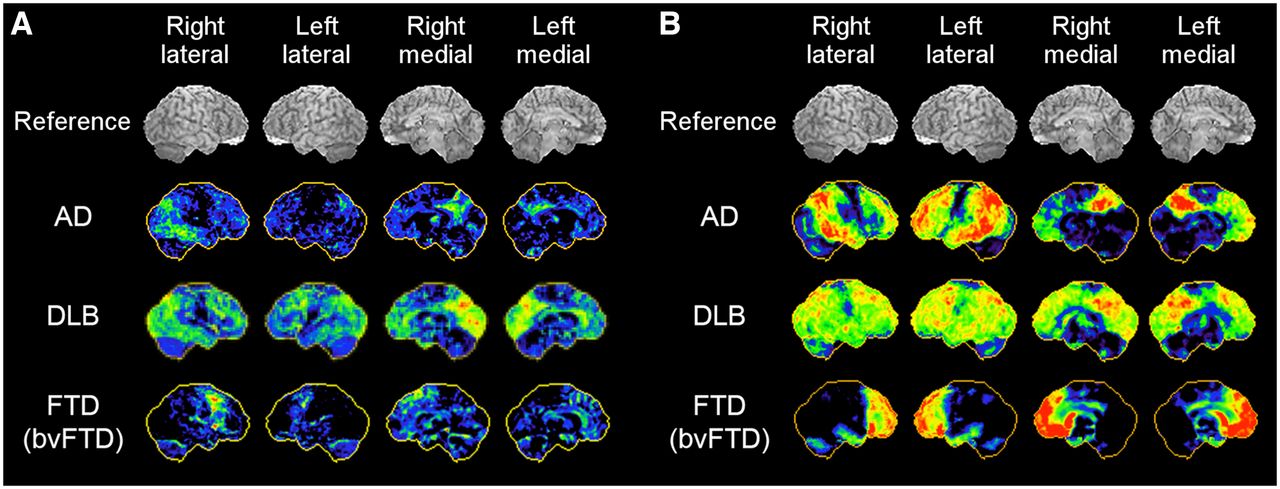
18F-FDG PET 3D-SSP maps from representative cases of AD, DLB, versus bvFTD. Red indicates more severe hypometabolism. Mild cases (A) and severe cases (B) are shown.
In AD, the parietotemporal association cortices as well as the posterior cingulate cortex and precuneus are commonly involved. However, the metabolic activity in the primary sensorimotor and primary visual cortices as well as the basal ganglia, thalamus, pons, and cerebellum is relatively preserved (15). The areas of relatively preserved activity, such as the pons, can be used as a reference region for pixel normalization in quantitative image analysis (37). As the disease progresses, the frontal association cortex becomes involved. However, in a fraction of AD patients with prominent behavioral symptoms (behavioral variant of AD [bvAD]), the frontal lobe involvement can be distinguished at an early stage of the disease, and it is difficult to differentiate from the behavioral variant of FTD (bvFTD) on 18F-FDG PET imaging (38).
FTD or classic Pick’s disease generally affects the frontal association cortex and the anterior temporal lobe. Additional decreased uptake can be seen progressively in the caudate nucleus and thalamus (39). The frontal involvement in bvFTD is often sharply demarcated, and was initially described as “lobar atrophy” on CT or MRI (40). Asymmetric involvement of the hemispheres appears to be quite common (41).
DLB shows 18F-FDG PET findings similar to those of AD, but additional hypometabolism is seen in the primary visual cortex in the medial occipital lobe where activity is relatively preserved in AD (24). Such metabolic reductions in the occipital lobe make the differential diagnosis between DLB and posterior cortical atrophy somewhat challenging (42). Metabolic activity in the posterior cingulate cortex seems less affected in DLB than AD (“cingulate island sign”) (43). Both occipital hypometabolism and cingulate island sign on 18F-FDG PET are considered as supportive features in the consensus diagnostic criteria for DLB (36).
PREDICTIVE VALUE OF 18F-FDG PET IN EVALUATION OF COGNITIVE DECLINE
One significant value of 18F-FDG PET has been attributed to the short-term prediction of impending dementia in subjects with mild cognitive impairment (MCI). Particularly, metabolic reductions in the posterior cingulate cortex have been demonstrated to have high predictive value (21,44). Interestingly, the added predictive value of 18F-FDG PET over amyloid PET imaging has also been demonstrated, as it was shown that amyloid-positive MCI patients without hypometabolic abnormalities maintained clinical stability over years (45). It has been discussed frequently that 18F-FDG PET also represents an ideal tool for disease staging and follow-up due to its tight association with patient symptoms and clinical severity (46). 18F-FDG PET has found entrance into various expert recommendations within this context (47,48).
CLINICAL SUBTYPES OF AD
Suspected AD patients may show atypical clinical features and 18F-FDG PET findings. Recent investigations using molecular imaging biomarkers have contributed to the consideration of AD subtypes, which are not as infrequent as previously assumed. AD subtypes are accompanied by characteristic patterns of hypometabolism on 18F-FDG PET, closely reflecting symptomatic features. Investigations demonstrated similar spatial variations of neuropathology in AD (49). These observations also indicate that, in later stages of the disease, different subtypes may converge to a common pattern of disease topography.
AD subtypes with visual symptoms (posterior cortical atrophy), a frontal executive or behavioral variant (bvAD, discussed earlier in the article), and a language-dominant variant (logopenic variant PPA, discussed later in the article) have been described. Clinically, posterior cortical atrophy is initially characterized by dominant visual-constructive deficits (50). On 18F-FDG PET, a distinct bilateral occipitoparietal hypometabolism has been described (42). In the fraction of AD patients with the frontal/executive subtype (bvAD), behavioral symptoms dominate the initial clinical appearance, and, also in 18F-FDG PET, the frontal lobe involvement can be prominent from an early stage of the disease. In a fraction of patients clinically diagnosed with a logopenic form of aphasia (lvPPA) and often assigned clinically to FTD (see below), amyloid pathology was confirmed as one of the underlying neuropathologies (51).
CLINICAL SUBTYPES OF FRONTOTEMPORAL LOBAR DEGENERATION (FTLD)/FTD
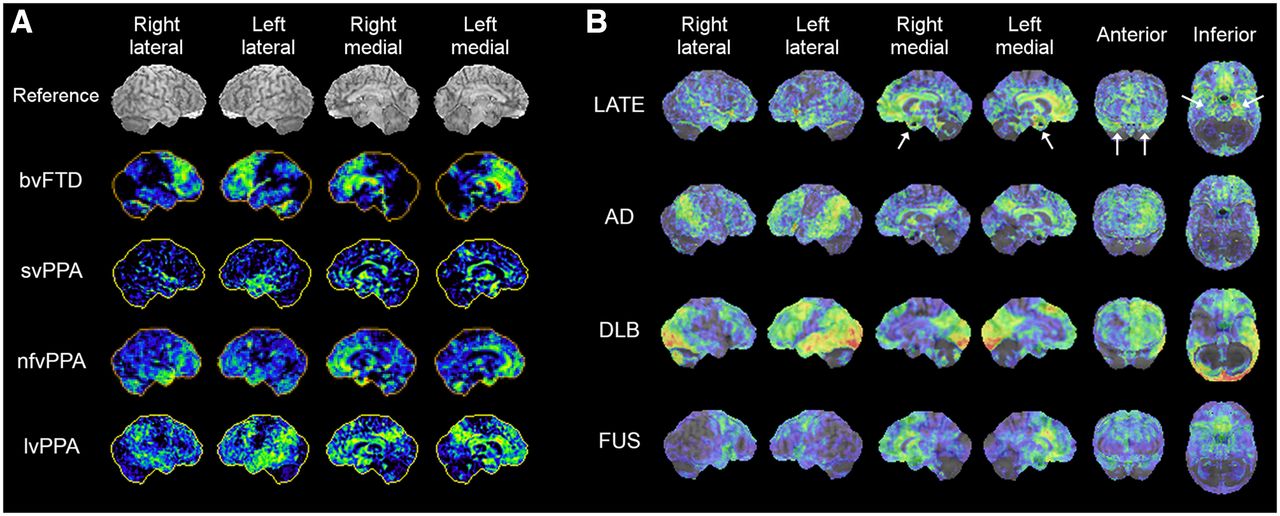
Over the last few decades, there has been an evolving recognition of different clinical presentations and neuropathologies in frontotemporal dementia, as well as a better understanding of the clinicopathologic and imaging correlations. FTLD, an umbrella term used for several different neurodegenerative disorders, is characterized by neurodegeneration predominantly involving the frontal and temporal lobes (52). FTLD and FTD are sometimes used interchangeably. They present heterogeneous clinical features and underlying pathologies not in one-to-one correspondence, creating a complex clinicopathologic relationship. Generally, FTLD/FTD encompasses clinical presentations of bvFTD, often referred to as simply FTD or Pick’s disease; PPA; and atypical parkinsonism/movement disorders such as progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) (Table 1). PPA is further subclassified based on clinical features of the language disturbance into semantic variant PPA (svPPA), also called semantic dementia (SD); nonfluent variant PPA (nfvPPA); and lvPPA. The underlying pathologic changes of the PPA subtypes are heterogeneous. Classification of PPA subtypes also provides information valuable to patient management. Although there are no disease pathology–modifying treatments yet available for PPA, an accurate diagnosis of PPA subtypes facilitates the appropriate patient management including speech therapy and planning of care, as each subtype presents with a different time course of disease progression and clinical complications (53). There has been increasing evidence of differential findings on 18F-FDG PET associated with FTLD/FTD subtypes (Fig. 5A) relevant to clinical interpretation (54–56). Consequently, a recent expert consensus recommends 18F-FDG PET as a first-line PET examination for workup of patients with suspected non–AD-type dementia (48).
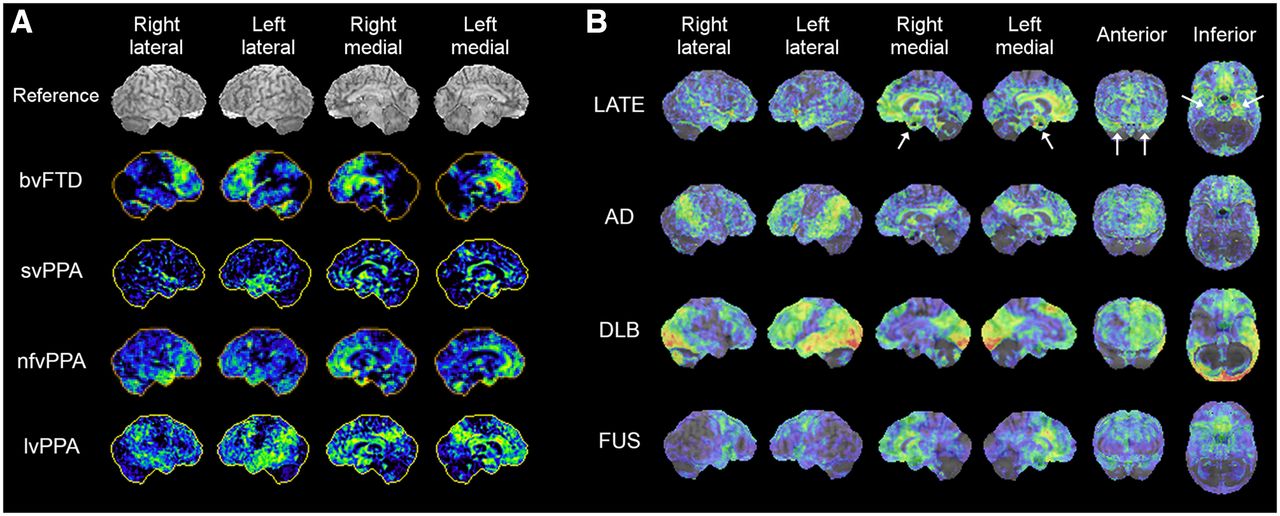
18F-FDG PET 3D-SSP z score maps of representative cases of FTLD/FTD variants, bvFTD, svPPA, nfvPPA, lvPPA, and PSP (A) and LATE, AD, DLB, and FUS (B, z score maps superimposed on reference MR image). The case of nfvPPA presented here shows bilateral temporofrontal involvement with right side slightly more prominent than left. LATE demonstrates prominent involvement of medial temporal lobe and hippocampus (white arrows) and medial and orbital frontal cortices. 18F-FDG uptake in medial temporal lobe is relatively mild in AD and DLB. FUS demonstrates pattern involving frontal and anterior temporal lobes, resembling FTLD/FLD spectrum.
PPA: THREE SUBTYPES
Patients with svPPA or SD present with a loss of semantic memory in both verbal and nonverbal domains. 18F-FDG PET findings are fairly characteristic, involving the anterior temporal lobe bilaterally, but often the left temporal lobe is more severely hypometabolic than the right (Fig. 5A, svPPA). Hypometabolism in the anterior temporal lobe is distinct from the pattern of temporal lobe involvement seen in AD, in which the mid to posterior lateral temporal cortex is often affected. A similar pattern of decreased 18F-FDG uptake can be seen in the right anterior temporal lobe (right-sided SD) often presenting with a different language/behavioral profile than that of left-sided SD (57), and the differential diagnosis from bvFTD can become challenging (58).
Patients with nfvPPA or progressive nonfluent aphasia experience difficulty in speaking, apraxia of speech, agrammatism, and impaired comprehension of complex sentences, as well as difficulty in swallowing and other motor symptoms sometimes apparent in other types of FTD. Decreased 18F-FDG uptake is often seen in the left lateral posterior frontal and superior medial frontal cortices, as well as the insula (Fig. 5A, nfvPPA) (55,59), which is distinct from the anterior temporal lobe hypometabolism seen in svPPA/SD.
Patients with lvPPA or logopenic progressive aphasia often manifest impaired naming and sentence repetition and an inability to retain complex verbal information, as the disease progresses. They also tend to exhibit more cognitive and behavioral symptoms as compared with other types of PPA (59), likely reflecting a frequent underlying AD pathology (59), but other pathologies have also been identified in lvPPA (60). Decreased 18F-FDG uptake is typically seen in the posterior temporal cortex and inferior parietal lobule in lvPPA, with the left hemisphere often more severely affected than the right (Fig. 5A, lvPPA). These findings are somewhat similar to those of AD (55,56). reflecting the underlying pathology.
ATYPICAL PARKINSONIAN MOVEMENT DISORDERS (PSP AND CBD)
Atypical parkinsonism caused by neurodegeneration including PSP and CBD can manifest with variable motor and cognitive disorders. PSP and CBD affect the frontal and temporal lobes and are often considered FTD variants. Both conditions are marked by tauopathy. PSP and CBD show diminished radiotracer uptake in the striatum on dopamine transporter SPECT imaging or presynaptic dopaminergic PET imaging, unlike AD and most FTD (61). Although dopamine transporter SPECT findings cannot differentiate PSP and CBD, patterns of decreased 18F-FDG uptake can provide a clue for the differential diagnosis among these Parkinson-plus syndromes.
The antemortem diagnosis of PSP versus other cognitive disorders can be challenging. 18F-FDG PET shows decreased cortical uptake in the medial frontal and anterior cingulate cortices as well as caudate nuclei and thalami (62). In addition, distinct focally decreased uptake is seen in the midbrain (63) before atrophy of the midbrain tegmentum detected by MRI, which is known as the Hummingbird sign (64). CBD is characterized by rigidity, apraxia, uncontrollable limb movement (“alien limb syndrome”) and cognitive impairment. Recently, the term corticobasal syndrome has been used for the clinical classification of this entity because of a growing recognition that different neuropathologies including AD pathology can underlie this symptom-complex (65). 18F-FDG PET shows decreased uptake in the frontoparietal regions without sparing of the sensorimotor cortex, basal ganglia, and thalamus in CBD, and is often noticeably asymmetric, which is consistent with the pattern of clinical symptoms (66).
RECENTLY RECOGNIZED NEURODEGENERATIVE DEMENTING DISORDERS
Recent efforts in the development and identification of new pathologic markers, investigations based on large series of clinicopathologic correlations, and consensus efforts have resulted in the recognition of new categories for neurodegenerative disorders. The prevalence of some of these disorders is greater than previously suspected. Molecular imaging plays a major role in the antemortem characterization of such disorders, and the differential findings from 18F-FDG PET need to be incorporated into the interpretation (Table 1).
LIMBIC-PREDOMINANT AGE-RELATED TDP-43 ENCEPHALOPATHY (LATE) AND HIPPOCAMPUS SCLEROSIS (HS)
The recent identification of the transactive response DNA binding protein of 43 kDa (TDP-43) proteinopathy has led to a new category of neurodegenerative dementia occurring late in life, namely LATE. The anatomic manifestation of LATE can be a profound atrophy in the hippocampus, likely making up a significant proportion of cases previously classified as HS (67). Frequent occurrence of HS in dementia patients was later recognized in some large autopsy series (68,69). Phosphorylated TDP-43 was initially identified in FTLD and amyotrophic lateral sclerosis (70) and subsequently identified in patients with AD and HS (71), particularly among elderly patients above the age of 80 y. These observations have led to the recognition of TDP-43 proteinopathy, which is often associated with HS and cognitive impairment, as a distinct disease entity (72) despite significant overlap with other neurodegenerative disorders such as AD and FTLD. A consensus working group report describing LATE has been published recently (73).
LATE mainly affects people older than 80 y and manifests with cognitive impairment, which mimics amnestic dementia similar to AD. LATE neuropathologic changes exist in more than 20% (ranging from 5% to 50%) of subjects in an autopsy series, and the overall public health impact of LATE is considered to be the same order of magnitude as AD (73). LATE is often associated with hippocampal atrophy as seen by MRI. In elderly patients with Alzheimer dementia, hippocampal volume was more strongly associated with TDP-43/HS than AD (74).
The number of reports of 18F-FDG PET findings in autopsy-confirmed LATE is increasing, and findings are also extrapolated from observations in HS and amyloid-negative, tau-negative dementia patients (suspected non-Alzheimer disease pathophysiology (14)). These investigations demonstrated significant hypometabolism in the medial temporal lobe including hippocampus, whereas in AD hippocampal 18F-FDG uptake is less affected (75); hypometabolism in the medial temporal lobe and the superior medial frontal and orbital frontal cortices (76); and decreased uptake in the medial and lateral temporal lobes as well as the prefrontal cortex (Fig. 5B) (77). The ratio of inferior temporal metabolism over medial temporal metabolism was significantly higher in HS patients (75). However, how specific these findings are to LATE is not currently known. As described in the next section, other neurodegenerative disorders affecting the medial temporal lobe also demonstrate decreased uptake, which can mimic LATE findings on 18F-FDG PET imaging.
When an amnestic patient has 18F-FDG PET findings not typical of AD, FTD, or DLB, LATE could be a differential diagnostic consideration particularly in elderly patients. Hippocampal atrophy detected by MRI suggests not only AD, but also LATE. Currently, there is no specific imaging biomarker or TDP-43 ligand, and TDP-43/LATE-neuropathologic changes pathology could be indirectly speculated by exclusion (negative amyloid PET and negative tau PET). Alternatively, 18F-FDG PET may provide supportive evidence of LATE by demonstrating medial temporal lobe abnormalities. Further investigations on the clinicopathologic and imaging correlations of LATE are clearly warranted.
ARGYROPHILIC GRAIN DISEASE (AGD), PRIMARY AGE-RELATED TAUOPATHY (PART), AND FUSED IN SARCOMA (FUS)
Recent advances in tau PET are shedding new light on neurodegenerative disorders that have been described by neuropathologists in the past 2 decades. AGD is characterized by pathologic features of small argyrophilic inclusions in the hippocampus, which are largely composed of 4-repeat tau isoforms (78). Patients manifest a slowly progressive MCI, often accompanied by behavioral symptoms suggesting FTD. The prevalence was reported as high as 30% in an autopsy series of patients with dementia and also can be found in normal control individuals (79), and AGD often coexists with other neurodegenerative disorders such as AD and FTLD (80).
PART is characterized by neurofibrillary tangles without coexisting Alzheimer neuritic amyloid plaques and appears to be common in elderly patients (81). Previously described as “tangle-only dementia,” the neurofibrillary tangles seen in PART are composed of 3-repeat and 4-repeat isoforms similar to AD, and the changes are typically seen in the temporal lobe (80). It has been debated if PART is a distinct disease entity or within the spectrum of AD. PART patients demonstrate slower decline in memory, language, and visuospatial functions than patients with AD (82).
The presence of tau deposition in PART can be depicted by tau PET (83). A recent investigation using both amyloid and tau PET revealed a significant fraction of patients with negative amyloid and positive tau PET findings (A−T+N+), which might be consistent with a high prevalence of PART (84). The diagnostic features of 18F-FDG PET are still under investigation but thus far demonstrated medial temporal hypometabolism with extension into the frontolimbic regions in amyloid-negative and very slowly progressing amnestic MCI patients with suspected PART, AGD, or LATE pathologies.
Abnormal deposition of FUS protein is found in amyotrophic lateral sclerosis as well as in FTLD and recognized relatively recently as a form of neurodegenerative dementia (70). Clinicopathologic characterization is still under investigation, and some report indicated caudate atrophy as one of the features of FUS (85). 18F-FDG PET findings of FUS have not been characterized systematically, but the pattern of hypometabolism appears to conform to the FTLD spectrum (Fig. 5B).
MIXED DEMENTIA AND COEXISTING PATHOLOGIES
There has been renewed enthusiasm to better characterize overlapping and coexisting neurodegenerative disorders because of the discovery of new pathologic markers, molecular imaging, and investigations of clinicopathologic correlations. It has been long appreciated that 2 or more pathologies can often coexist in dementia patients. For example, AD and vascular dementia (VaD), 2 common causes of dementia, often coexist (86) and this is referred to as “mixed dementia,” for which the prevalence may be quite high. With the development of new pathologic markers, mixed dementia with coexisting multiple neurodegenerative disorders, in addition to vascular changes, is becoming recognized more frequently, particularly in elderly patients. A recent autopsy investigation showed 94% with one or more pathologies, 78% with 2 or more, 58% with 3 or more, and 35% with 4 or more pathologies, among elderly patients with cognitive impairment (87). The presence of major pathologic markers such as amyloid, tau, α-synuclein, and TDP-43 overlaps and coexists in such dementia patients (Fig. 6). Various mixed dementia with multiple comorbid neurodegenerative disorders as defined by pathologic markers have been reported (Table 2; Fig. 7).
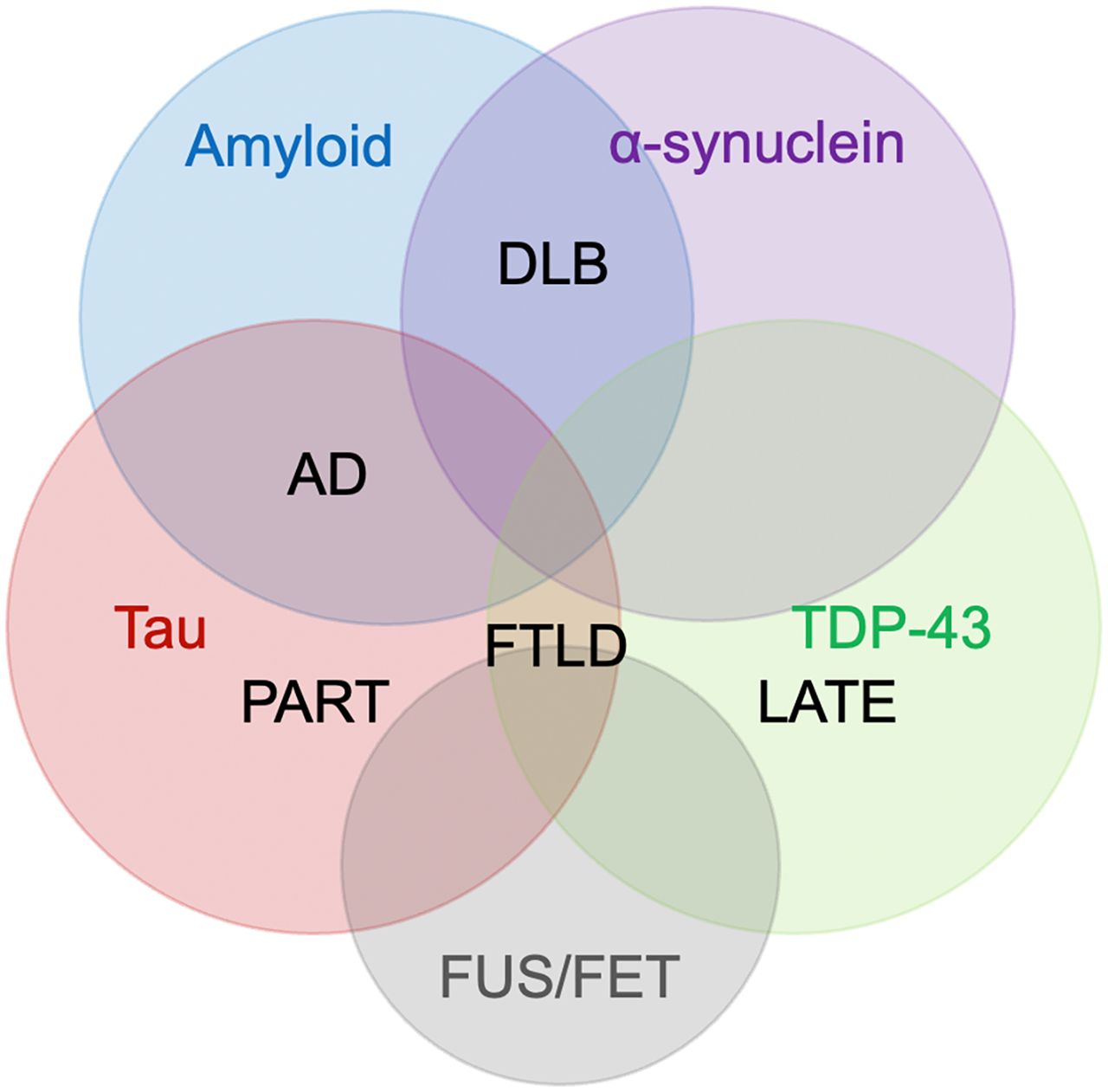
Cooccurrence of 4 major proteinopathies: amyloid, tau, α-synuclein, and TDP-43, and overlapping neurodegenerative disorders: AD, DLB, FTLD, PART, and LATE. Emerging proteinopathies such as fused in sarcoma (FUS), which belongs to the FET family of proteins (FUS, EWS, TAF15), have been identified in FTLD, comorbid with other proteinopathies, and awaiting further characterization. Pathologic diagnosis of neurodegenerative disorders involves new markers, and further investigations of clinicopathologic correlations including imaging will allow more precise antemortem diagnosis in the future.
Examples of Mixed/Comorbid Neurodegenerative Dementing Disorders
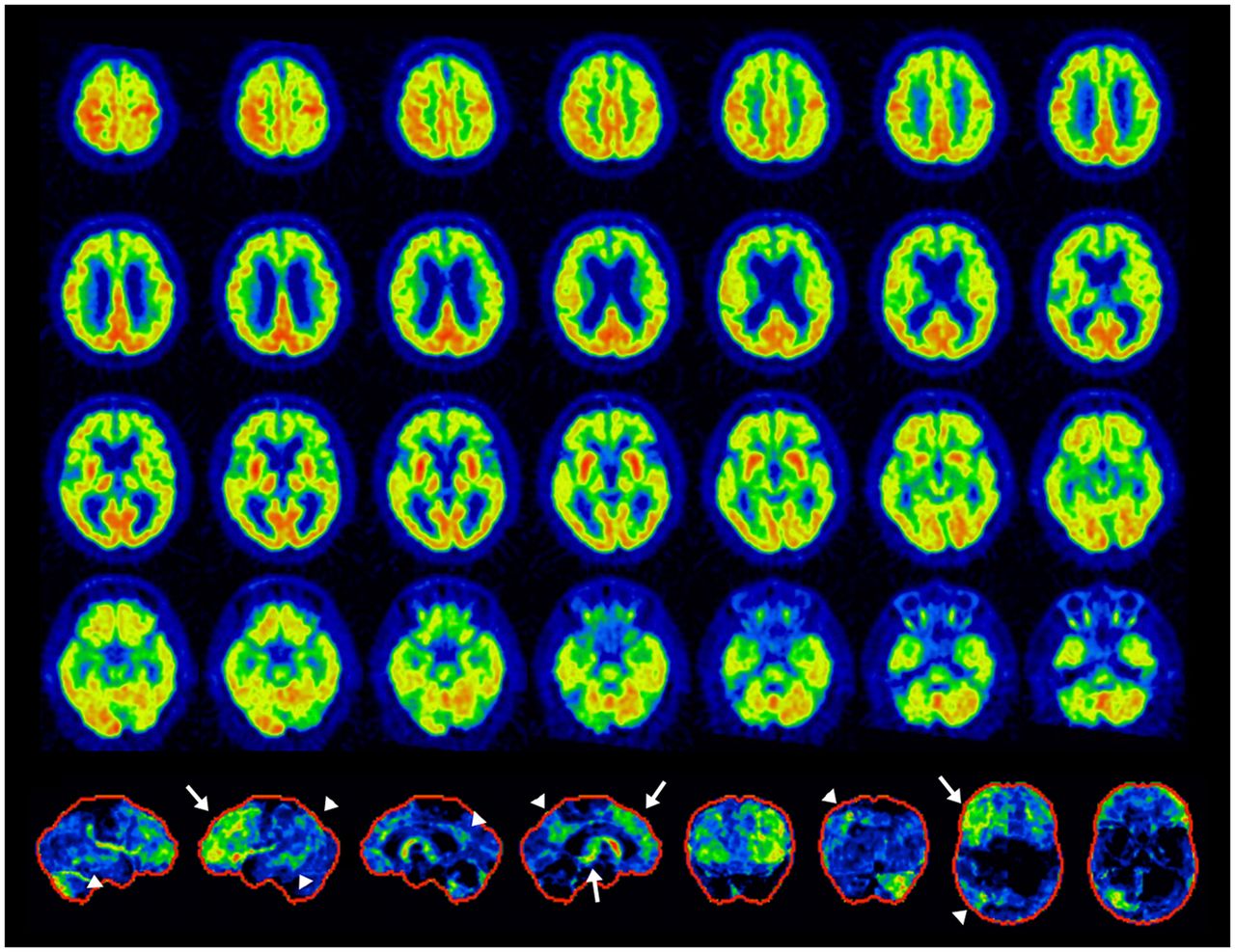
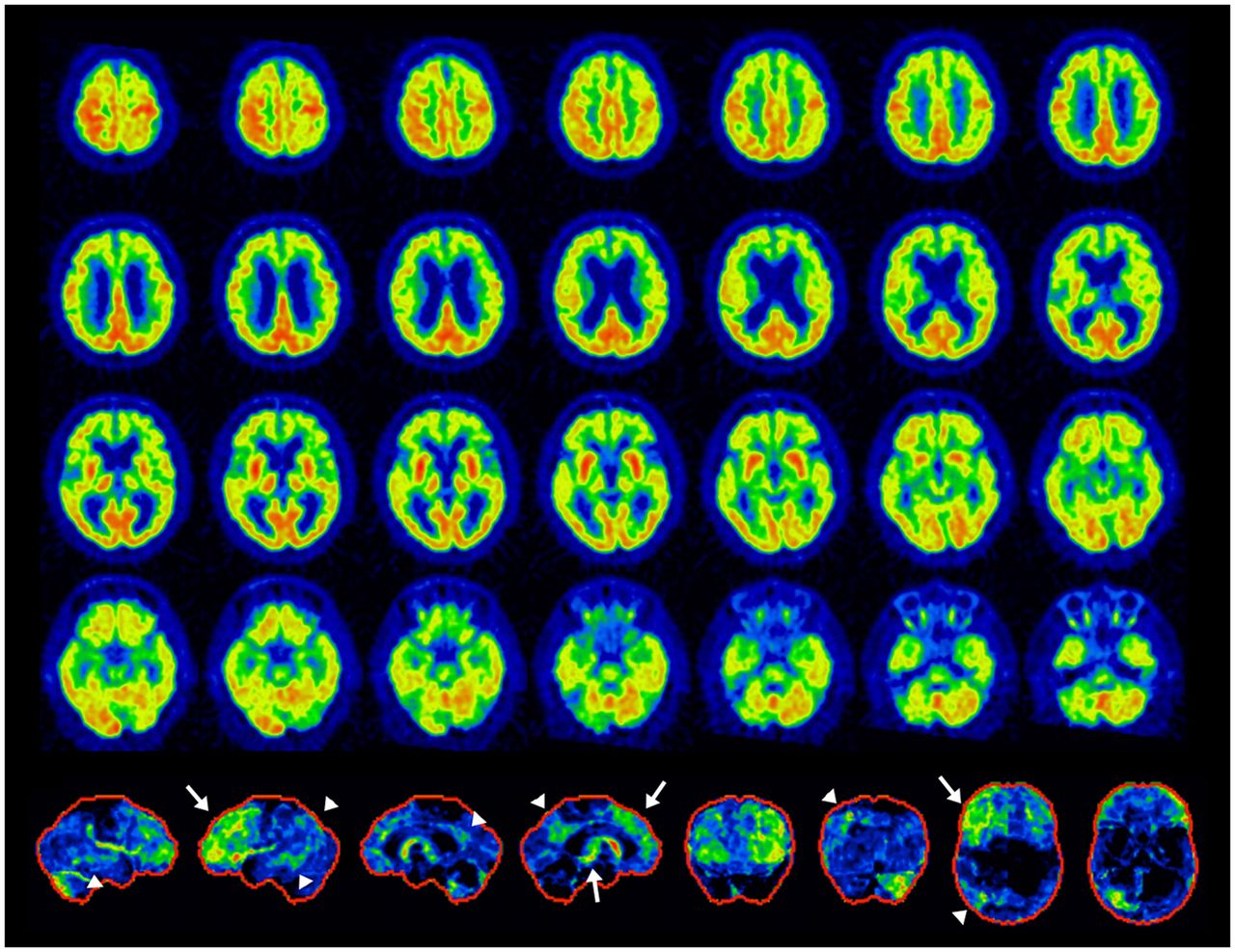
Example of mixed dementia, PSP+AD. Antemortem 18F-FDG PET: transaxial images (top 4 rows) and 3D-SSP z score maps (bottom row, from the left to right: right lateral, left lateral, right medial, and left medial, anterior, posterior, superior, inferior views). Findings are consistent with PSP (white arrows) and AD (arrowheads). Left-dominant pathologies associated with crossed-cerebellar diaschisis in right cerebellar hemisphere. It is important to note that these findings do not necessarily preclude other mixed pathologies. However, the frontal findings best explained the patient’s clinical symptoms.
When 18F-FDG PET does not demonstrate typical patterns of known neurodegenerative disorders or when multimodal imaging results are incongruent, mixed dementia with multiple copathologies should be a clinical consideration (88,89). Even in cases with characteristic patterns of 18F-FDG PET, it is important to recognize that the topography of neuronal dysfunction does not necessarily indicate specific or exclusive neuropathologies or proteomic abnormalities. However, 18F-FDG PET may be able to confirm the clinically congruent predominant cause of dementia, even in the presence of copathologies (90).
THE EVOLVING ROLE OF 18F-FDG PET IN DEMENTIA EVALUATION
18F-FDG PET has been used as a research tool as well as a clinical diagnostic tool in dementia evaluation. It can depict early metabolic changes associated with neurodegenerative disorders before the structural changes seen on CT or MRI. Early diagnosis of AD, FTD, and DLB has been, in part, complemented by the detection of specific pathologies on imaging biomarkers such as amyloid PET, tau PET, and dopamine transporter SPECT. However, access to amyloid and tau PET biomarkers is limited clinically, in part due to limited reimbursement. Currently, 18F-FDG PET is used primarily for the differential diagnosis of dementia and prediction of further cognitive decline among patients with MCI. It is relatively inexpensive, is widely available, and can differentiate multiple neurodegenerative disorders in a single test when images are interpreted accurately by trained clinicians using validated statistical mapping technology.
The dichotomous distinction of AD versus FTD by 18F-FDG PET is no longer sufficient or possible, given our increasing knowledge of disease subtypes and a recognition of the overlapping cooccurrence of multiple pathologies, particularly in elderly patients. It becomes less relevant to fit 18F-FDG PET findings into a single category of neurodegeneration. 18F-FDG PET findings supporting the cause of a patient’s main clinical features and identifying possible copathologies are important for clinical management. Such an approach is often required when interpreting cases with complex or atypical clinical presentations, which tend to be referred for advanced imaging by dementia specialists.
Currently, extensive efforts are under way to demonstrate the clinical effectiveness of amyloid-targeting immunotherapy to treat AD. When such treatments become widely available, the evaluation of copathologies in patients with amyloid-positive PET may become critical since antiamyloid monotherapy in such situations is likely to have limited efficacy. The use of imaging and nonimaging biomarkers, if available, is one way to address this issue. However, 18F-FDG PET may be a less expensive and more direct way to provide insight into the cause of typical and atypical clinical presentations and identify concurrent neurodegenerative disorders, thus helping guide the use of disease specific therapeutics.
SUMMARY
18F-FDG PET has been used for dementia research and clinical applications for more than 4 decades. Although the detailed cellular mechanisms of glucose metabolism are still under investigation, 18F-FDG PET can sensitively display the distinct patterns of neuronal and synaptic dysfunction associated with neurodegeneration, allowing early diagnosis and prediction of further cognitive decline. Differential patterns of altered 18F-FDG uptake can provide important differential diagnostic clues to clinicians, particularly when the interpretation of images is aided by statistical mapping technologies, which have become widely available in the clinic. The recognition by 18F-FDG PET of the 3 major neurodegenerative disorders, AD, FTD, and DLB, has become a standard of care. In addition, the subtypes of FTD/FTLD have been more widely recognized clinically, as well as pathologically, and are distinguishable by 18F-FDG PET and other molecular imaging techniques. New disease categories, such as LATE, PART, AGD, and FUS, have also been identified, and need to be incorporated into the differential diagnoses. Mixed dementia, not only AD and VaD but also multiple neurodegenerative disorders as defined by pathologic markers, is prevalent, particularly in elderly patients. By integrating recent insights into the modern interpretation of 18F-FDG PET, the potential of this diagnostic tool can be extended beyond established applications as a precise imaging biomarker of functional disease endophenotype and can contribute to care management. When 18F-FDG PET scans do not demonstrate typical features of known neurodegenerative disorders or typical features with atypical secondary findings, considerations for mixed dementia with copathologies or a newly recognized form of dementia such as LATE are needed in the clinical interpretation of 18F-FDG PET.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
- © 2022 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.
- 92.
- 93.
- 94.
- 95.
- 96.
- 97.
- 98.
- 99.
- 100.
- 101.
- 102.
- Received for publication February 1, 2022.
- Revision received April 22, 2022.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- 18F-FDG AS A NEUROCHEMICAL TRACER
- CELLULAR MECHANISMS OF 18F-FDG UPTAKE IN THE BRAIN
- REMOTE EFFECTS ON 18F-FDG UPTAKE (DIASCHISIS)
- IMAGE INTERPRETATION USING STATISTICAL MAPPING TECHNIQUES
- CLINICAL IMAGING ALTERNATIVES TO 18F-FDG PET FOR DEMENTIA EVALUATION
- DIFFERENTIAL DIAGNOSIS OF AD, FTD, AND DLB: STANDARD OF CARE
- PREDICTIVE VALUE OF 18F-FDG PET IN EVALUATION OF COGNITIVE DECLINE
- CLINICAL SUBTYPES OF AD
- CLINICAL SUBTYPES OF FRONTOTEMPORAL LOBAR DEGENERATION (FTLD)/FTD
- PPA: THREE SUBTYPES
- ATYPICAL PARKINSONIAN MOVEMENT DISORDERS (PSP AND CBD)
- RECENTLY RECOGNIZED NEURODEGENERATIVE DEMENTING DISORDERS
- LIMBIC-PREDOMINANT AGE-RELATED TDP-43 ENCEPHALOPATHY (LATE) AND HIPPOCAMPUS SCLEROSIS (HS)
- ARGYROPHILIC GRAIN DISEASE (AGD), PRIMARY AGE-RELATED TAUOPATHY (PART), AND FUSED IN SARCOMA (FUS)
- MIXED DEMENTIA AND COEXISTING PATHOLOGIES
- THE EVOLVING ROLE OF 18F-FDG PET IN DEMENTIA EVALUATION
- SUMMARY
- DISCLOSURE
- REFERENCES
- Figures & Data
- Info & Metrics
Related Articles
Cited By...
- Macroscopic cerebral energy efficiency corresponds to neuron reorganization in awake and anesthetized mice
- BOLD Amplitude Correlates of Preclinical Alzheimers Disease
- SNMMI Procedure Standard/EANM Practice Guideline for Brain [18F]FDG PET Imaging, Version 2.0
- Evaluation of Neurodegenerative Disorders with Amyloid-{beta}, Tau, and Dopaminergic PET Imaging: Interpretation Pitfalls
- Glucose intolerance induces anxiety-like behaviors independent of obesity and insulin resistance in a novel model of nutritional metabolic stress
- Molecular Imaging of Neurodegeneration: The Way to New Horizons