


Visual Abstract

Abstract
Immune checkpoint blockade (ICB) has achieved groundbreaking results in clinical cancer therapy; however, only a subset of patients experience durable benefits. The aim of this study was to explore strategies for predicting tumor responses to optimize the intervention approach using ICB therapy. Methods: We used a bilateral mouse model for proteomics analysis to identify new imaging biomarkers for tumor responses to ICB therapy. A PET radiotracer was synthesized by radiolabeling the identified biomarker-targeting antibody with 124I. The radiotracer was then tested for PET prediction of tumor responses to ICB therapy. Results: We identified galectin-1 (Gal-1), a member of the carbohydrate-binding lectin family, as a potential negative biomarker for ICB efficacy. We established that Gal-1 inhibition promotes a sensitive immune phenotype within the tumor microenvironment (TME) for ICB therapy. To assess the pre-ICB treatment status of the TME, a Gal-1–targeted PET radiotracer, 124I-αGal-1, was developed. PET imaging with 124I-αGal-1 showed the pretreatment immunosuppressive status of the TME before the initiation of therapy, thus enabling the prediction of ICB resistance in advance. Moreover, the use of hydrogel scaffolds loaded with a Gal-1 inhibitor, thiodigalactoside, demonstrated that a single dose of thiodigalactoside–hydrogel significantly potentiated ICB and adoptive cell transfer immunotherapies by remodeling the immunosuppressive TME. Conclusion: Our study underscores the potential of Gal-1–targeted PET imaging as a valuable strategy for early-stage monitoring of tumor responses to ICB therapy. Additionally, Gal-1 inhibition effectively counteracts the immunosuppressive TME, resulting in enhanced immunotherapy efficacy.
Immunotherapy such as immune checkpoint blockade (ICB) using anti–programmed death receptor 1 (PD-1) and anticytotoxic T-lymphocyte–associated antigen 4 (CTLA-4) antibodies has produced promising clinical outcomes in melanoma, non–small cell lung cancer, and several other tumor types (1). Nevertheless, only a subgroup of patients experience positive outcomes with ICB therapy, with objective response rates spanning 5%–60% (2). It is imperative to identify biomarkers to effectively predict ICB efficacy and evaluate their expression levels using a reliable detection approach, which can facilitate patient stratification and immunotherapy optimization, aligning with the objective of achieving individualized precision treatment.
Although several biomarkers, including programmed cell death 1 ligand 1 (PD-L1), high microsatellite instability, tumor mutation burden, and mismatch repair system deficiency, associated with responses to ICB therapy have been reported, they cannot predict tumor responses effectively and accurately (3). This limitation arises from an insufficient understanding of the intricate and dynamic mechanisms affecting ICB efficacy (4). An essential factor to consider is the immune state of the tumor microenvironment (TME) before treatment. This condition can be broadly categorized as either inflammatory, enhancing subsequent cytotoxic CD8+ T-cell–mediated antitumor activity, or immunosuppressive, diminishing it. Therefore, identifying biomarkers capable of assessing the immune status of the TME before the initiation of therapy is crucial.
Several clinical criteria have been established to monitor responses and evaluate immunotherapy effectiveness. These include immune-related RECIST and immunotherapy RECIST (5,6). These criteria are based on anatomic information obtained from CT and MRI scans, which lack molecular insights into the immune response, resulting in a considerable delay between treatment initiation and response evaluation. The immunohistochemical assessment of PD-L1 expression in tumor biopsy specimens has been extensively used in predicting immunotherapy outcomes. This method helps to predict the treatment response to anti–PD-1 or anti–PD-L1 therapies, as patients with PD-L1–positive tumors typically exhibit favorable responses to therapy (7,8). However, several challenges hinder the adoption of this approach, including variations in the timing and biopsy site and the invasive and nonrepeatable nature of the examinations (9). Molecular imaging technology, especially PET, has emerged as a robust tool for predicting immunotherapy effectiveness through the longitudinal, quantitative, and noninvasive assessments of biomarkers (10). In clinical practice,18F-FDG is used to predict potential responses to immunotherapy and track treatment progress (11). However, distinguishing pseudoprogression from actual tumor progression with 18F-FDG PET is challenging, resulting in potential misinterpretations (12).
Despite significant advancements in PET imaging techniques targeting immune cells, including the T-cell phenotype, activation, and effector function (13–16), most radiotracers have been used after immunotherapy initiation to monitor treatment effectiveness by assessing antitumor immune activity. Accordingly, the use of noninvasive PET imaging for predicting therapeutic efficacy before immunotherapy holds significant potential. This approach helps in prestratifying patient groups and guiding clinical interventions to optimize treatment outcomes while reducing the risk of immune-related adverse events.
Therefore, the aim of this study was to explore strategies for predicting tumor responses and optimizing immunotherapeutic strategies. Through quantitative proteomic analysis, we discovered that tumors exhibiting low galectin-1 (Gal-1) expression respond positively to ICB therapy. Gal-1, which belongs to a class of endogenous lectins with an affinity for β-galactosides, plays critical roles in different cellular functions, including cell migration and angiogenesis (17). After confirming that Gal-1 inhibition enhances tumor responses to ICB therapy, we developed a radiotracer to perform Gal-1–targeted PET imaging. We demonstrated that Gal-1–targeted PET can be used to predict ICB efficacy before therapy. Additionally, we designed Gal-1 inhibitor–loaded hydrogel scaffolds that could reverse the immunosuppressive TME. Our findings suggest that Gal-1 inhibition may represent a valuable combination therapeutic strategy for enhancing immunotherapy efficacy.
MATERIALS AND METHODS
Detailed descriptions of the experimental procedures are provided in the supplemental materials (supplemental materials are available at http://jnm.snmjournals.org).
Animal Models
The protocols for all animal experiments were approved by the Institutional Animal Care and Use Committee at Peking University. Female BALB/c and C57BL/6 mice (5–6 wk old) were purchased from the Department of Laboratory Animal Science at Peking University. T-cell receptor–transgenic OT-I mice [C57BL/6-Tg(TcraTcrb)1100Mjb/J] were purchased from Shanghai Model Organisms Center Inc.
To establish the 4T1, Gal-1 knockdown (KD) 4T1 (4T1-KD), and CT26 tumor–bearing mouse models, we administered 106 tumor cells suspended in 100 μL of phosphate-buffered saline subcutaneously into the lower right flank (for single tumor-bearing models) or both flanks (for bilateral tumor-bearing models) of BALB/c mice. To generate a B16-OVA tumor–bearing mouse model, we subcutaneously administered 2 × 106 tumor cells into the upper right flank of C57BL/6 mice. Tumor growth was measured using a caliper, and tumor volume was calculated using the formula volume = length × width2/2.
Preparation of Gal-1–Targeted PET Radiotracer
Na124I was produced at the Department of Nuclear Medicine at Peking University Cancer Hospital using a medical cyclotron. For 124I radiolabeling, 100 μg of anti–Gal-1 antibody (αGal-1) (clone 201002; catalog number MAB12451; R&D Systems) and 74 MBq of Na124I dissolved in 600 μL of phosphate buffer (0.1 M, pH 7.4) were introduced into a reaction vial precoated with 1,3,4,6-tetrachloro-3α,6α-diphenylglycouril (40 μg; catalog number T0656; Sigma). The mixture was allowed to react for 10 min at room temperature before being purified using a PD-10 desalting column (GE Healthcare). The same protocol was adhered to in preparing a 124I-labeled isotype-matched control (124I-IgG). The radiochemical purities of 124I-αGal-1 and 124I-IgG were assessed using instant thin-layer chromatography with saline as the mobile phase.
Statistical Analysis
Quantitative data are presented as mean ± SD. Data obtained from 2 groups were compared using a 2-tailed unpaired Student t test. Tumor growth curves over time were compared using a 2-way ANOVA with a Tukey multiple-comparisons test. P values of less than 0.05 were considered statistically significant.
RESULTS
Tumors with Low Gal-1 Expression Before ICB Respond to ICB
Quantitative proteomic analysis was conducted using a bilateral tumor model (18,19) to identify predictive biomarkers of ICB efficacy before therapy intervention. This model was based on the biologic assessment of the resected tumor, allowing for inference of the therapeutic outcome on the basis of the remaining tumor treatment response. The tumor growth curve indicated that the bilateral tumor growth trend in each mouse was generally symmetric; however, each mouse exhibited varying responses to the anti–PD-1 plus anti–CTLA-4 treatment (Supplemental Fig. 1). One tumor was surgically excised before ICB treatment, whereas the other was observed to determine whether the mouse exhibited a responsive or nonresponsive reaction to the therapy. Mice were categorized on the basis of tumor size ratios from day 9 to day 0, with a ratio of less than 2 indicating responders and a ratio of greater than 2 indicating nonresponders (Fig. 1A). On the basis of this definition, 4 responders with significantly lower tumor volumes were distinguished from nonresponders (P < 0.001) (Fig. 1A).

Identification and validation of Gal-1 as predictive biomarker for monitoring ICB therapy. (A) Time line of proteomics analysis and ICB therapy in 4T1 bilateral tumor model and criteria for classifying mice as responders and nonresponders. Individual growth curves depict remaining tumors in 4T1 mice, categorized into responder (n = 4) and nonresponder (n = 9) groups on basis of established criteria. Quantitative comparison of relative tumor volumes on day 9 (V9/V0) of remaining tumors in responder and nonresponder groups is shown. (B) Heatmap illustrating log2 fold changes (nonresponders vs. responders) in 27 differentially expressed membrane and secretory proteins related to immune functions. These proteins were identified through proteomics analysis of resected tumors in corresponding groups (n = 3/group; scale bars: white, 0; red, >1.6 log2 fold change; blue, <1.6 log2 fold change). (C) Western blotting bands and quantification of Gal-1 levels in resected tumors from both groups (n = 4/group). All numeric data are presented as mean ± SD. *P < 0.05; ***P < 0.001. α = anti-; GAPDH = glyceraldehyde 3-phosphate dehydrogenase; i.p. = intraperitoneally.
To characterize the pretreatment TME, tumor proteins extracted from each group were analyzed using mass spectrometry–based proteomics. With the application of the criterion of a fold change of greater than 1.6, an unsupervised hierarchical clustering analysis of the top 60 differentially expressed proteins revealed a clear demarcation between responders and nonresponders (Supplemental Fig. 2A). Pathways related to the immune response, including interferon signal transduction and antigen presentation, were markedly enriched in the responder group (Supplemental Fig. 2B).
To identify readily detectable biomarkers receptive to PET imaging using radiotracers, we focused on the differentially expressed membrane or secretory proteins associated with antitumor immune activity. Gal-1 emerged as a potential target for further investigation, primarily because of its differential expression pattern between the 2 groups (Fig. 1B). Ex vivo quantitative reverse transcription–polymerase chain reaction (P < 0.05) (Supplemental Fig. 3A), Western blotting (P < 0.05) (Fig. 1C), and enzyme-linked immunosorbent assay (P < 0.01) (Supplemental Fig. 3B) all confirmed that Gal-1 expression was significantly lower in the responder group than in the nonresponder group. The interferon-γ fraction was also significantly higher in the responder group than in the nonresponder group (Supplemental Fig. 3C). Kaplan–Meier survival analysis revealed a significant correlation between high Gal-1 expression and shorter overall survival in patients with several tumor types (Supplemental Figs. 4A–4C). These findings suggest that low Gal-1 expression in pretreatment tumors is associated with a response to ICB therapy, highlighting its potential as a predictive biomarker of the efficacy of immunotherapy.
Gal-1 Inhibition Improves ICB Efficacy by Fostering Immune-Favorable TME
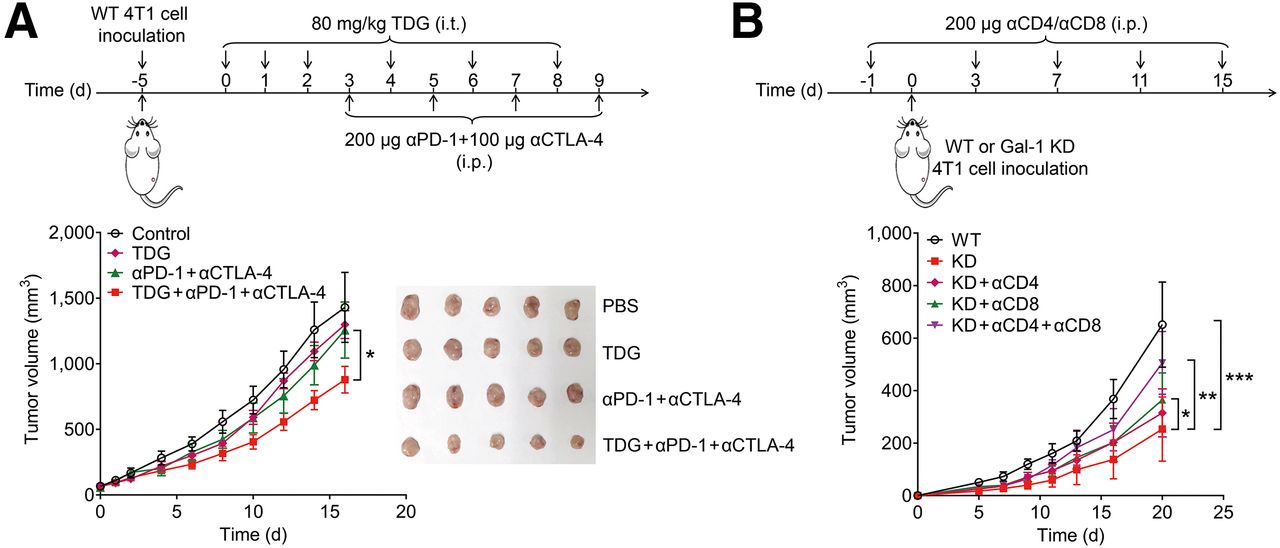
To investigate whether Gal-1 inhibition promotes a tumor response to ICB therapy, a Gal-1 inhibitor, thiodigalactoside (TDG), was combined with anti–PD-1 and anti–CTLA-4 antibody administration in 4T1 tumor–bearing mice (Fig. 2A). Tumor growth curves showed that combining TDG and ICB treatment was more effective than ICB treatment alone (P < 0.05) (Fig. 2A). The synergistic effect of TDG on ICB treatment was further corroborated in CT26 tumor–bearing mice (Supplemental Fig. 5).

Gal-1 inhibition augments ICB efficacy. (A) Treatment time line and tumor growth of TDG combined with ICB therapy in 4T1 tumor–bearing mice. Representative photographs of tumors harvested at study endpoint for 4T1 tumor–bearing mice subjected to various treatments are shown (n = 5/group). (B) Treatment time line and tumor growth curves of mice inoculated with WT and 4T1-KD tumors after specified treatments (n = 7/group). Data are presented as mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. α = anti-; i.p. = intraperitoneally; i.t. = intratumorally; PBS = phosphate-buffered saline.
We further investigated whether Gal-1 inhibition enhances the efficacy of ICB therapy by affecting the immune status of the TME. 4T1 cells were stably transfected with 2 Gal-1–specific small-hairpin RNAs. Both Gal-1 KD 4T1 cell lines showed decreased Gal-1 expression, with Gal-1 KD-2 4T1 cells showing a more pronounced reduction (∼40%) in protein expression (Supplemental Fig. 6A). Therefore, Gal-1 KD-2 4T1 (4T1-KD) cells were chosen and used for further experiments. Enzyme-linked immunosorbent assay results revealed that Gal-1 secretion from 4T1-KD cells was significantly impaired (P < 0.0001) (Supplemental Fig. 6B).
4T1-KD and wild-type (WT) 4T1 cells were then administered to WT BALB/c mice. Tumor growth was significantly slower in mice inoculated with 4T1-KD cells than in those inoculated with WT 4T1 cells (P < 0.001) (Fig. 2B). Depletion of CD8+ T cells partially reversed 4T1-KD tumor growth (P < 0.05) (Fig. 2B), and the tumor suppression induced by Gal-1 KD was abrogated after CD4+ and CD8+ T-cell depletion (P < 0.01) (Fig. 2B). Moreover, 4T1-KD tumor slices exhibited an elevated CD3+ T-cell count (Supplemental Fig. 7A) and decreased Gal-1 expression (Supplemental Fig. 7B). Additionally, as detected using Ki-67 immunofluorescence staining, the tumor cell proliferation index decreased significantly (Supplemental Fig. 7A). Therefore, we proposed that inhibiting tumor-induced Gal-1 might trigger T-cell–dependent antitumor immunity, rendering tumors more sensitive to ICB therapy.
Compared with that in WT 4T1 tumors, the percentage of regulatory T cells (CD45+ CD4+ FoxP3+) in 4T1-KD tumors decreased (Supplemental Fig. 8A), whereas that of CD8+ T cells increased (Supplemental Fig. 8B). We stained CD8+ interferon-γ+ cells to analyze the CD8+ T-cell immunophenotype further and observed an increased level of activated T cells in 4T1-KD tumors (Supplemental Fig. 8C). As secreted interferon-γ can upregulate PD-L1 expression (20), we examined PD-L1 expression in tumor cells (CD45− PD-L1+) and found that it was upregulated in 4T1-KD tumors (Supplemental Fig. 8D). In addition, we examined the dynamics of myeloid cells, which play crucial roles in promoting the immunosuppressive TME (21). The percentages of granulocytic–myeloid-derived suppressor cells (Supplemental Fig. 8E) and M2 macrophages (Supplemental Fig. 8F) were also significantly lower in 4T1-KD tumors than in WT 4T1 tumors. Together, Gal-1 inhibition reprogrammed the TME into a phenotype sensitive to ICB therapy. This transformation was characterized by an enhanced cytotoxic CD8+ T-cell immune response and attenuated immunosuppressive activity.
Gal-1–Targeted PET Imaging Can Be Used to Predict Efficacy of ICB Therapy
Considering the significance of Gal-1 in generating an immunosuppressive TME, we hypothesized that a radiotracer targeting Gal-1 could noninvasively detect Gal-1 expression in vivo to allow early prediction of the efficacy of ICB therapy before the commencement of treatment. To test this hypothesis, we developed a Gal-1–specific radiotracer, 124I-αGal-1, by radiolabeling αGal-1 with 124I (Supplemental Fig. 9A). 124I-αGal-1 exhibited favorable in vitro stability (Supplemental Fig. 9B) and Gal-1–binding specificity (Supplemental Fig. 9C). Next, we conducted small-animal PET imaging and biodistribution studies to assess the in vivo tumor-targeting specificity of 124I-αGal-1. 124I-αGal-1 uptake in WT 4T1 tumors was significantly higher than that in 4T1-KD tumors and 124I-IgG uptake in WT tumors at 72 h after injection (Figs. 3A and 3B). To further validate the Gal-1–targeting specificity of 124I-αGal-1, we treated 4T1 tumor–bearing mice with nanoparticle albumin–bound paclitaxel, which can upregulate Gal-1 expression (Supplemental Fig. 10) (22). Biodistribution studies showed significantly higher 124I-αGal-1 uptake in nanoparticle albumin–bound paclitaxel paclitaxel–treated tumors than in control tumors at 24 and 72 h after injection (Fig. 3B; Supplemental Fig. 11).

In vivo Gal-1 specificity of 124I-αGal-1. (A) PET/CT images and quantified tumor uptake at 4, 8, 24, 48, and 72 h after 124I-αGal-1 and 124I-IgG administration in 4T1 or 4T1-KD tumor–bearing mice (n = 3/group). White arrows indicate tumors. (B) Biodistribution of 124I-αGal-1 and 124I-IgG in WT 4T1 (with or without nanoparticle albumin–bound paclitaxel [Nab-paclitaxel] treatment) or 4T1-KD tumor–bearing mice at 72 h after injection (n = 4/group). All numeric data are presented as mean ± SD. *P < 0.05; **P < 0.01. α = anti-; %ID/g = percentage injected dose per gram.
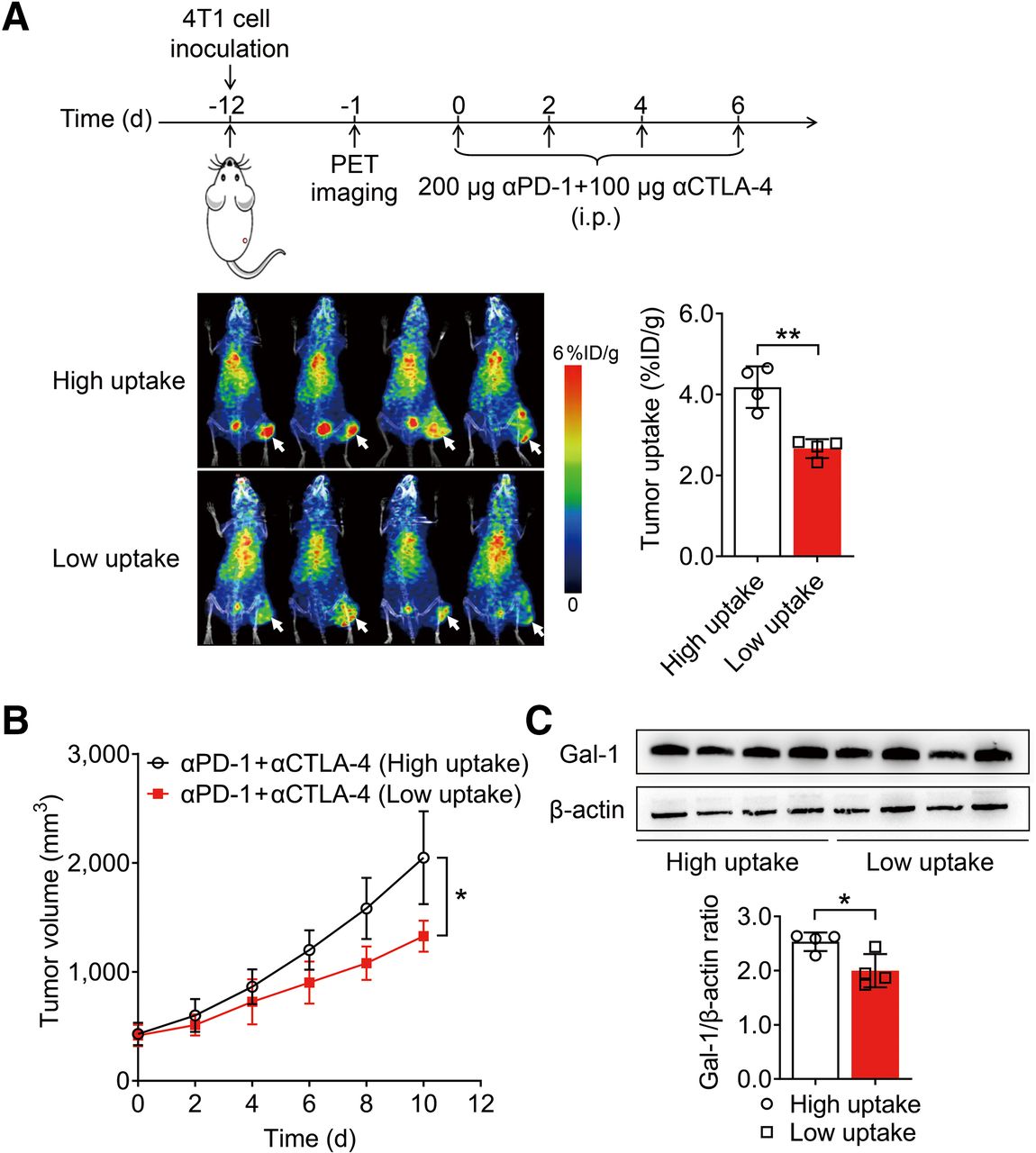
Subsequently, we explored whether PET imaging of 124I-αGal-1 could monitor the therapeutic efficacy of ICB before therapy. Mice were markedly distinguished into high- and low-uptake groups on the basis of the tumor uptake value for 124I-αGal-1, with a median cutoff of 3.4 percentage injected dose per gram (P < 0.01) (Fig. 4A). These mice were then treated with anti–PD-1 and anti–CTLA-4 antibodies, and tumor growth was significantly slower in the low-uptake group than in the high-uptake group (P < 0.05) (Fig. 4B), indicating that tumors with low Gal-1 expression respond well to ICB therapy. Ex vivo Western blotting confirmed that Gal-1 expression levels were significantly higher in the group with high 124I-αGal-1 uptake than in the group with low 124I-αGal-1 uptake (P < 0.05) (Fig. 4C). Flow cytometric analysis of tumor tissues revealed that the levels of CD4+ T cells in the groups with high and low 124I-αGal-1 uptake were comparable (Supplemental Fig. 12A). Conversely, the levels of tumor-infiltrating CD8+ T cells (P < 0.05) (Supplemental Fig. 12B) and CD8+ interferon-γ+ cells (P < 0.001) (Supplemental Fig. 12C) were significantly higher in the group with low 124I-αGal-1 uptake than in the group with high 124I-αGal-1 uptake. These findings are consistent with the results characterizing the sensitivity of the TME to ICB therapy in Gal-1 KD tumors (Supplemental Fig. 8). Further, they reveal that 124I-αGal-1 PET imaging may be used to distinguish the level of Gal-1 in the tumors, potentially facilitating early prediction of tumor responses to ICB treatment.

PET imaging of 124I-αGal-1 predicts ICB therapy. (A) PET/CT images and quantified tumor uptake of 124I-αGal-1 72 h after injection in 4T1 tumor–bearing mice, classified into high- and low-tumor-uptake groups (cutoff, 3.4 percentage injected dose per gram [%ID/g]) (n = 4/group). White arrows indicate tumors. (B) Tumor growth curves for 4T1 tumor–bearing mice in high- and low-tumor-uptake groups of 124I-αGal-1 after ICB therapy (n = 5/group). (C) Western blot analysis and quantitative assessment of Gal-1 levels in high- and low-tumor-uptake groups of 124I-αGal-1 (n = 4/group). All numeric data are presented as mean ± SD. *P < 0.05; **P < 0.01. α = anti-; i.p. = intraperitoneally.
Gal-1 Inhibition with Single Dose of TDG-Loaded Hydrogel Potentiates ICB and Adoptive Cell Immunotherapies
We devised an effective rescue strategy for treating tumors with high Gal-1 expression. Biomaterials, such as hydrogels, can serve as carriers for high-concentration drug loading and retain sustainable drug release. Therefore, the Gal-1 inhibitor TDG was incorporated into a chitosan–chitosan quaternary ammonium salt solution, and then β-glycerophosphate disodium was added dropwise to form TDG-hydrogel. The gel solution remained transparent as a liquid at 4°C but underwent transformation into a nontransparent semisolid hydrogel after 15 min of incubation at 37°C (Supplemental Fig. 13A). TDG was slowly released over a 96-h period, with approximately 60% of the TDG being released cumulatively (Supplemental Fig. 13B).
In vivo therapy studies showed that neither single-dose TDG therapy, ICB therapy, nor a single dose of TDG combined with ICB exhibited any antitumor effects. In contrast, administering a single dose of TDG-hydrogel significantly enhanced the tumor response to ICB therapy (P < 0.0001) (Fig. 5), surpassing the outcomes achieved with multiple TDG administrations in combination with ICB treatment (Fig. 2A). Immunofluorescence staining confirmed decreased Gal-1 expression and tumor cell proliferation index after TDG-hydrogel treatment (Supplemental Fig. 14). A multiplex immunofluorescence assay revealed a significant increase in CD8+ T cells (P < 0.001) and a decrease in CD206+ (P < 0.01) and Ly6G+ (P < 0.001) cells (Supplemental Fig. 15). Notably, single-dose TDG (Fig. 5) had a much lower potentiating effect on ICB therapy than a continuous low-dose administration of TDG (Fig. 2A), suggesting that continuous low-dose TDG may more effectively remodel the immunosuppressive TME.

Single-dose TDG-hydrogel administration potentiates ICB therapeutic efficacy. Synthetic representation of process for TDG-hydrogel, time line for combining TDG-hydrogel with ICB therapy in 4T1 tumor–bearing mice, and tumor growth curves of 4T1 tumor–bearing mice after indicated treatments (n = 5–7/group). Data are presented as mean ± SD. ****P < 0.0001. α = anti-; i.p. = intraperitoneally.
We further investigated whether TDG-hydrogel could enhance the efficacy of adoptive cell transfer (ACT) therapy. TDG-hydrogel combined with ACT of CD8+ T cells generated from transgenic OT-I mice significantly inhibited tumor growth compared with TDG-hydrogel or ACT therapy alone (P < 0.05) (Fig. 6A). ACT effectiveness depends on the proliferation, activation, and effector function of CD8+ T cells, and granzyme B secretion is a biomarker of CD8+ T-cell effector function (23). We thus performed granzyme B–targeted PET imaging using our previously described granzyme B–specific radiotracer, 68Ga-grazytracer (16), to further assess the ability of TDG-hydrogel to enhance the efficacy of ACT therapy. The results showed that the tumor uptake of 68Ga-grazytracer was higher in the TDG-hydrogel group or the ACT therapy group than in the control group. 68Ga-grazytracer showed the highest tumor uptake in the group treated with TDG-hydrogel plus ACT therapy (Fig. 6B). This result represents the most substantial tumor-killing effect of CD8+ cytotoxic T cells induced after TDG-hydrogel in combination with ACT treatment. The sustained release of TDG through a single dose of TDG-hydrogel efficiently remodeled the immunosuppressive TME, potentiating ICB and ACT therapy (Fig. 6C).

Single-dose TDG-hydrogel administration augments adoptive CD8+ T-cell transfer therapy. (A) Illustration of time line for TDG-hydrogel, ACT therapy, and PET imaging in B16-OVA tumor–bearing mice and tumor growth curves of B16-OVA tumor–bearing mice after specified treatments (n = 5 or 6/group). (B) Granzyme B PET/CT images and quantified tumor uptake using radiotracer 68Ga-grazytracer in B16-OVA tumor–bearing mice after subjection to indicated treatments (n = 5 or 6/group). White arrows indicate tumors. (C) Schematic representation of TDG-hydrogel sensitizing TME to augment immunotherapeutic efficacy. All numeric data are presented as mean ± SD. *P < 0.05; ***P < 0.001. α = anti-; DC = dendritic cell; %ID/g = percentage injected dose per gram; i.v. = intravenously; MDSC = myeloid-derived suppressor cell; Treg = regulatory T cells.
DISCUSSION
Early prediction of immunotherapy efficacy is crucial to precisely guide patient selection and optimize the therapeutic paradigm (24). In the present study, we revealed that low Gal-1 expression levels in the ICB pretreatment tumors correlated with a favorable response to ICB and therapeutically altering the immunosuppressive TME through a single-dose administration of TDG-loaded hydrogel rendered the TME phenotype more sensitive to potentiate ICB efficacy. These findings suggest that using tumor baseline Gal-1 PET imaging may serve as a noninvasive imaging biomarker for predicting ICB therapy and that Gal-1 is a promising target for combination therapy with immunotherapy.
Gal-1 has garnered increasing attention because of its upregulated expression in various tumor tissues (25). Studies have consistently shown that Gal-1 expression correlates with tumorigenesis, progression, metastasis, and recurrence (26). Moreover, Gal-1 may influence the immune milieu within the TME by regulating antitumor immune responses, as confirmed by our observation of enhanced effector T-cell activity and attenuated myeloid cell activity in Gal-1 KD tumors. Evidence suggests that Gal-1 can predict the response to anti–PD-1 therapy in clear cell renal carcinoma (27), consistent with our findings in the 4T1 triple-negative breast cancer model.
Considering the role of Gal-1 in fostering an immunosuppressive TME, Gal-1–targeted imaging may hold promise for early monitoring of the therapeutic efficacy of immunotherapy. Previous studies showed the potential of Gal-1 imaging for tumor detection (28) and predicting radiotherapy resistance (29). In the present study, a Gal-1–specific antibody was radiolabeled with 124I, and proof-of-concept PET imaging using 124I-αGal-1 demonstrated its role in monitoring immunotherapy efficacy. Notably, 124I-labeled intact antibodies are not ideal radiotracers for clinical PET imaging because of the possible deiodination and slow clearance of antibodies in vivo. Radiometals such as 89Zr-labeled antibodies may have better in vivo stability and higher tumor uptake (30). In addition, 68Ga- or 18F-labeled small-molecule–based radiotracers targeting Gal-1 may also hold promise for potential clinical PET imaging because of the favorable in vivo pharmacokinetics. Recently, we developed a galectin-targeted radiotracer, 68Ga-galectracer, that could detect Gal-1 and Gal-3 expression in patients (29). Further optimization of Gal-1 selectivity and affinity as well as the pharmacokinetics of PET radiotracers targeting Gal-1 may eventually generate radiotracers with great potential in predicting the efficacy of immunotherapy and guide immunotherapeutic combinations in the clinic.
CONCLUSION
We demonstrated the feasibility of Gal-1–targeted PET to noninvasively monitor an immunosuppressive TME. This approach could enable the early prediction of tumor responses to immunotherapy and facilitate the selection and design of new combination regimens for efficient tumor immunotherapy.
DISCLOSURE
This work was supported by the National Key R&D Program of China (2023YFC3404600 to Zhaofei Liu), the National Natural Science Foundation of China (81920108020 and 82325028 to Zhaofei Liu), the Beijing Natural Science Foundation (Z220011 and Z220014 to Zhaofei Liu), and the Beijing Nova Program Interdisciplinary Cooperation Project (20220484182 to Hua Zhu and Zhaofei Liu). No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can new imaging biomarkers be explored for noninvasively predicting early tumor responses to ICB therapy?
PERTINENT FINDINGS: Gal-1 expression reflects an immunosuppressive TME, and Gal-1 inhibition improves immunotherapeutic outcomes. Gal-1 PET imaging using 124I-αGal-1 offers a sensitive approach to determining tumor responses to ICB therapy.
IMPLICATIONS FOR PATIENT CARE: Gal-1 PET opens avenues for the early prediction of ICB efficacy before treatment initiation and facilitates the precision design of combinational regimens.
ACKNOWLEDGMENT
We are grateful to the cyclotron team at Peking University Cancer Hospital and Institute for 124I production.
Footnotes
Published online Mar. 21, 2024.
- © 2024 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication October 17, 2023.
- Revision received February 20, 2024.
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.