


Abstract
A 76-y-old man with hypoosmolar hyponatremia of unknown origin was referred to the nuclear medicine department for 18F-FDG PET/CT to exclude a malignant cause. Increased 18F-FDG uptake in both adrenal glands was observed and investigated.
PART 1
Hyponatremia, defined as a serum sodium concentration below 135 mmol/L, is usually caused by a disturbance in the urinary diluting mechanism. The algorithm for diagnostic assessment of patients with hyponatremia is well documented. Nevertheless, the final diagnosis can be challenging because multiple factors can contribute to hyponatremia.
Here, we present a case of hyponatremia in which nuclear imaging shed light on the differential diagnosis.
Case Report
A 76-y-old man presented to the emergency department after referral by his family physician due to general discomfort, cognitive impairment, and hypoosmolar hyponatremia (sodium, 112 mmol/L; osmolality, 226 mmol/kg H2O). He was euvolemic (blood pressure, 114/65 mm Hg; heart rate, 72 beats per minute; weight, 55 kg; height, 150 cm) and had no severe neurologic manifestations.
His past medical history revealed chronic hyponatremia. Two weeks earlier, he had been admitted to the hospital for a subdural hematoma due to a fall under unclear circumstances. In addition to a 40-pack-year history of smoking, his medical records included arterial hypertension and peripheral arterial disease. At presentation, he was taking amlodipine (5 mg/d), metoprolol (50 mg/d), simvastatin (20 mg/d), calcium, and vitamin D. Acetylsalicylic acid (80 mg/d) and ramipril (5 mg/d) were stopped during his previous admission.
Additional laboratory findings showed normal renal function (serum creatinine, 0.59 mg/dL) and a normal serum potassium concentration. Urine sodium (102 mmol/L) with high osmolality (335 mmol/kg H2O) was observed. Exclusion of severe hypothyroidism and cortisol deficiency was based on a normal level of thyroid-stimulating hormone and morning cortisol (respectively, 1.15 mIU/L and 10.2 μg/dL). The tentative diagnosis was syndrome of inappropriate antidiuretic hormone secretion.
Cranial CT was repeated and showed stable findings concerning the subdural hematoma. Subsequently, 18F-FDG PET/CT was performed to exclude a malignant cause associated with syndrome of inappropriate antidiuretic hormone secretion.
To follow up on the increased glucose metabolism of the adrenal glands, additional hormonal screening was performed (Table 1).
Hormonal Adrenal Testing
Discussion
The 18F-FDG PET/CT revealed hypermetabolic adrenal glands (Fig. 1). On CT, the adrenal glands had an enlarged aspect but were within the upper limit of the normal size range and showed high-contrast uptake. A myelolipoma (well-defined mass with lower- and higher-attenuation areas and no hypermetabolism) was visualized within the left adrenal gland (Fig. 1E). These lesions are most prevalent in patients with endocrine disorders (1). In the lower lobe of the right lung, a hypermetabolic spiculated lesion was detected (Fig. 2). Bilateral apical pleural thickening and slightly hypermetabolic hilar and mediastinal lymph nodes were also observed.
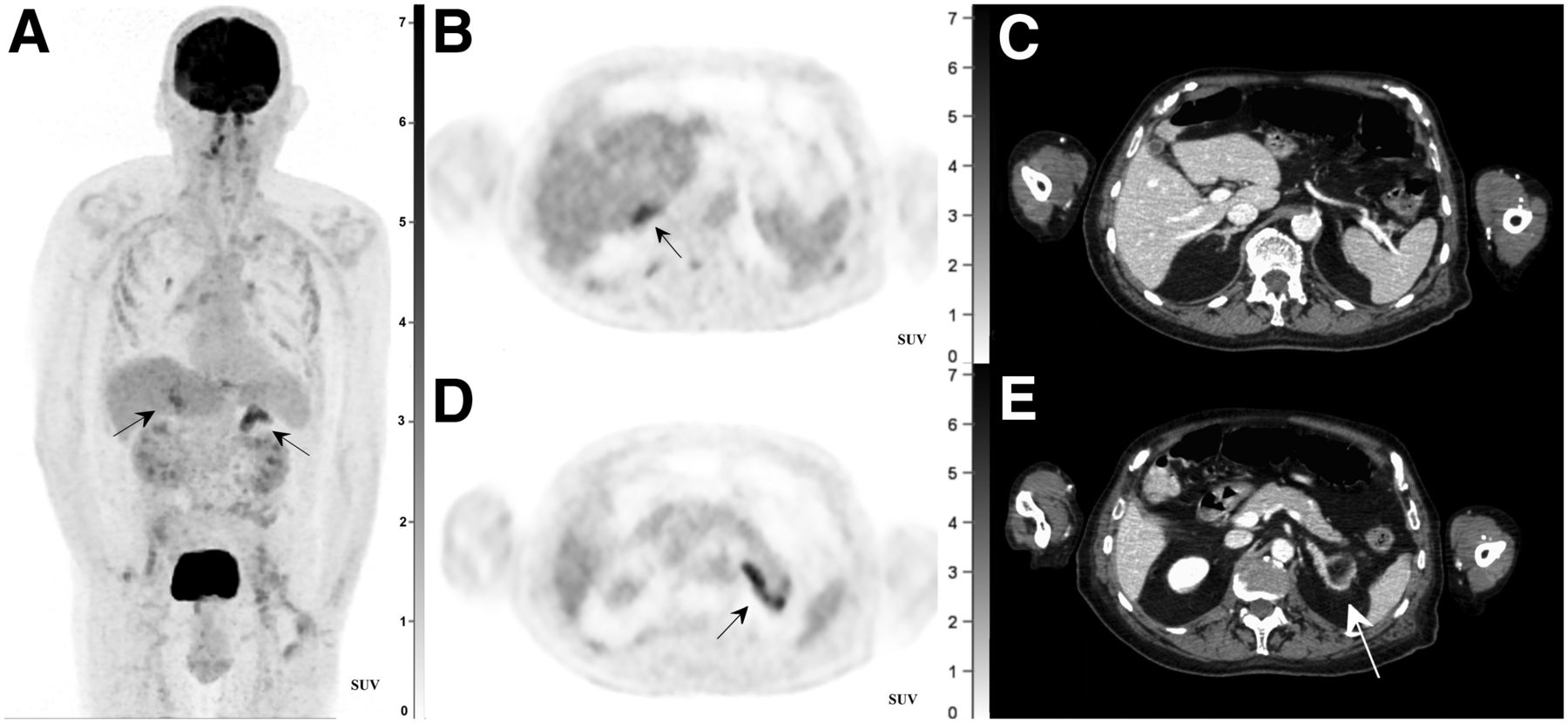
(A) High 18F-FDG uptake in both adrenal glands on maximum-intensity-projection PET image. Further findings are physiologic uptake in brain, liver, spleen, kidneys, and urinary bladder, as well as light uptake in both shoulder joints (degenerative arthropathy), intercostal musculature, and bilaterally around neck of total hip prostheses (inflammatory reaction). (B and D) Axial 18F-FDG PET images showing hypermetabolic right (B) and left (D) adrenal glands (arrows). (C and E) Axial CT images showing high-contrast uptake in right (C) and left (E) adrenal glands. Myelolipoma (arrow) is observed within left adrenal gland.

(A) Axial 18F-FDG PET image showing high uptake in lesion (large arrow) in apex of right lower lobe of lung, as well as moderate uptake in intercostal muscles (small arrows) and hilar and mediastinal lymph nodes. (B) CT image revealing spiculated lesion with diameter of 11 mm (arrow).
Laboratory findings were concordant with adrenal hyperandrogenism, hypogonadotropic hypogonadism, and low normal cortisol levels in the context of a hospitalised patient despite highly increased adrenocorticotropic hormone (Table 1). The differential diagnosis was a syndrome of inappropriate antidiuretic hormone secretion secondary to a subdural hematoma or a primary lung cancer with adrenal metastases (2), a bilateral adrenal malignancy (primary or secondary) (3), or a partial primary adrenocortical insufficiency due to congenital adrenal hyperplasia (CAH).
PART 2
Final Diagnosis
Bilateral symmetric hypermetabolism together with contrast enhancement and nonnodular enlargement of the adrenal glands on the CT images was more suggestive of adrenal hyperplasia or primary adrenal lymphoma. Combined with the clinical history of the patient, we therefore did not consider adenomas, endothelial cysts, or pheochromocytomas in our differential diagnosis. The latter entities usually present as nodular enlargements and often have an asymmetric or unilateral appearance (3).
Nuclear imaging combined with the clinical presentation and laboratory findings of the patient led to the final diagnosis of CAH. This case of CAH presented with an intermediate phenotype between the salt-wasting form and the nonclassic form, with increased adrenocorticotropic hormone–driven adrenal androgen production and partial insufficiency of glucocorticoid and mineralocorticoid production.
Genetic testing confirmed the diagnoses of CAH due to 2 heterozygote-inactivating mutations in the CYP21A2 gene (pathogenic heterozygous c.290-13C>G [IVS2-13C>G] mutation and pathogenic heterozygous c.5151T>A [p.IIe172Asn] mutation) encoding for 21-hydroxylase. After substitution with hydrocortisone and fludrocortisone, the sodium levels normalized. In parallel, the adrenal hyperandrogenism decreased and testosterone and luteinizing hormone levels increased.
A follow-up CT scan revealed a decrease in volume of the pulmonary lesions, excluding malignancy and pointing toward an inflammatory or infectious cause.
Conclusion
CAH is a group of autosomal recessive disorders characterized by adrenal hyperandrogenism and variably impaired cortisol and aldosterone synthesis resulting from the deficiency of 1 of the 5 enzymes required for adrenocortical steroid hormone synthesis. The most common type is 21-hydroxylase deficiency (4). The classic form is rare but clinically overt. The nonclassic form represents one of the most common autosomal recessive disorders; it can, however, remain clinically occult for many years, as patients will retain some enzyme activity (4).
An 18F-FDG PET/CT study leading to the diagnosis of CAH at an older age is rare, with only 1 case described in the literature (5). Our case demonstrates that bilaterally increased glucose metabolism in the adrenal glands on 18F-FDG PET/CT with associated hyponatremia should prompt biochemical screening for excess adrenal androgens and should raise suspicion of undiagnosed CAH, even in older patients.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
↵* Contributed equally to this work.
Published online July 22, 2021.
- © 2021 by the Society of Nuclear Medicine and Molecular Imaging.
- Received for publication April 3, 2021.
- Accepted for publication June 14, 2021.
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.