


Abstract
PET neuroimaging of amyloid-β (Aβ) provides an in vivo biomarker for pathologic changes associated with Alzheimer disease (AD). Aβ-targeted agents have been approved by the Food and Drug Administration, with additional agents, most notably targeting tau, currently under clinical investigation and one approved in May 2020. These agents, along with nonscintigraphic biomarkers from blood and cerebrospinal fluid, have provided an opportunity to investigate the pathogenesis, prodromal changes, and time course of the disease in living individuals. The current understanding is that the neuropathologic changes of the AD continuum begin up to 25 y before the onset of clinical symptomatology. The opportunities afforded by in vivo biomarkers of AD, whether by serum, cerebrospinal fluid examination or PET, have transformed the design of AD therapeutic trials by shifting focus to the preclinical stages of disease. Future disease-modifying therapies, should they be forthcoming, will rely heavily on the use of approved biomarkers or biomarkers currently under investigation to confirm the presence of target pathology. Understanding the progressive neuropathologic changes that occur in AD—and how scintigraphic findings relate to these changes—will help the interpreting physician to fully appreciate the implications of the scintigraphic findings and provide a basis to interpret the examinations. The recently adopted National Institute on Aging–Alzheimer Association guidelines define postmortem AD neuropathologic changes as a composite score based on 3 elements. These elements are the extent of involvement (spread) by cerebral Aβ based on the progression model defined by the Thal Aβ phases, the extent of involvement (spread) by neurofibrillary tangles (composed of hyperphosphorylated tau proteins) based on the progression model defined by Braak, and the Consortium to Establish a Registry for Alzheimer’s Disease score, which describes the density of neuritic plaques based on certain key locations in the neocortex. This paper will review the 3 elements that define the National Institute on Aging–Alzheimer’s Association scoring system and discusses current evidence on how these elements relate to findings based on Aβ and tau PET scintigraphy.
- neurology
- PET/CT
- Alzheimer’s disease
- amyloid scintigraphic imaging biomarkers
- neuropathologic staging
- tau scintigraphic biomarkers
NOTEWORTHY
AD is characterized by the gradual deposition of Aβ protein plaques and NFTs (tau protein) in characteristic progressive stages.
The postmortem definition of AD is based on the stage of both Aβ protein plaques and NFTs, as well as the density of neuritic plaques.
In vivo PET scintigraphy is able to identify, with limitations, both Aβ and NFT pathology.
These new imaging modalities allow for in vivo confirmation of AD.
In 1906, Alois Alzheimer described the first documented case of what is now termed Alzheimer disease (AD) (1). At age 51, the patient Auguste Dieter began to experience memory loss, hallucinations, psychosocial impairment, and disorientation. From 1901 to 1906, she remained hospitalized with a rapidly progressive dementia syndrome, spending her final years in an apathetic bedridden state until her death in 1906. In 1907, Alzheimer published the clinical and neuropathologic findings based on her autopsy. Until the 1990s, AD was clinically differentiated from the more loosely defined senile dementia and was classified as a presenile dementia. Today, these diseases are known to represent different variants of the same pathologic state. Younger-onset cases are frequently caused by highly penetrant genetic mutations, whereas dementia with onset after the age of 65 y is typically governed by a combination of modifiable (primarily cardiovascular) and nonmodifiable (age, genetics, and sex) risk factors. It is now suspected that Auguste Dieter developed her condition due to a mutation in the presenilin 1 gene. The gene produces the protein presenilin, a component of an enzyme (γ-secretase) that plays a central role in the generation of amyloid-β (Aβ) peptides from the larger amyloid precursor protein molecule (2).
In examining Auguste Dieter’s brain, Alzheimer described the two hallmark microscopic changes of the condition: extracellular plaques and characteristic intracellular bundles now known as neurofibrillary tangles (NFTs) (3). For the following 70 y, there was only minimal progress in the understanding of how Aβ plaques and NFTs relate to the pathophysiology of the disease. Medical students in the 1980s were instructed with little information other than that from Alois Alzheimer. The extracellular plaques were by then identified as containing a core of misfolded Aβ protein that are frequently but not always surrounded by abnormally configured neuronal processes (neurites). These neuritic plaques, in contrast to pure Aβ plaques, comprise a combination of Aβ and NFTs. The NTFs were in turn found to be formed by aggregation of a hyperphosphorylated form of tau, a neuronal structural protein in its dephosphorylated form. The rapid progression in the understanding of the pathology of AD and predementia states such as mild cognitive impairment (4) (broadly defined as objective impairment in at least one cognitive domain but preserved independence in functional abilities) over the ensuing 30 y now poses a challenge, as these medical graduates are today the senior physicians tasked with performing biomarker scintigraphy of AD. The present paper aims to address this potential gap in knowledge by reviewing what is known about the pathophysiology of AD and its relevance to the field of molecular imaging.
PATHOPHYSIOLOGY OF AD
Molecular biology has elucidated the biology of Aβ protein turnover: it begins with the production of amyloid precursor protein and its subsequent enzymatic cleavage into Aβ isomers. One of these isomers, Aβ42, is particularly prone to aggregation, and it is thought that overproduction or delayed clearance is what is responsible for the plaque generation. Variations in the genes governing the various stages of Aβ turnover have been associated with both early- and late-onset forms of the disease, giving credence to the notion that Aβ plays the critical initiating role in the disorder. Specifically, several rare autosomal dominant mutations (PSEN1, PSEN2, and APP genes) have been shown to invariably result in AD onset before the age of 65 y through dramatically increased Aβ42 production. In addition, individuals with an extra APP copy through chromosome 21 trisomy (Down syndrome) are at a significantly higher risk of developing dementia and invariably have sufficient levels of plaque and NFTs to warrant a pathologic diagnosis of AD by the age of 40 y (5). Genetic risk of late-onset AD is primarily mediated by the E4 and E2 alleles (increased and decreased risk, respectively) of the apolipoprotein gene that governs lipid transfer. In addition to increasing the risk for AD, APOE4 carriership also associates with earlier onset, therefore implicating its role in cases with onset before the age of 65 y (6).
The prominence of Aβ burden in AD led to the development of the so-called amyloid hypothesis: Aβ induces neuronal stress (by mechanisms that remain an area of intense research) with subsequent neuronal degeneration and formation of intracellular paired helical filaments (or so-called “neurofibrillary tangles”; NFTs) consisting of hyperphosphorylated tau. It follows that hyperphosphorylation of tau is a downstream effect triggered by Aβ cortical burden reaching a critical threshold (7). Across the full course of the disease, NFT plaques tend to colocalize more closely with regions of atrophy and hypometabolism than Aβ plaques, which has led to the current view that they underlie much of the neurodegeneration found in AD, although there is evidence that they can in turn promote Aβ’s own neurotoxic effects (8).
Extrapolation of longitudinal Aβ imaging, as well as other biomarker data (most notably cerebrospinal fluid), has led to the conclusion that AD follows a prolonged preclinical course of 15−25 y of Aβ deposition in the cerebral cortex without any evidence of cognitive impairment (9). As a result, Aβ-targeting therapeutic trials in AD have shifted their focus from the syndromic to the preclinical stages of the disease in an attempt to modify Aβ deposition before clinical symptoms are apparent (10), in a fashion analogous to the treatment model used to reduce the incidence of coronary artery disease through lipid modification by statins. Such a treatment does not aim to remove the present Aβ plaques but instead attempts to shift the biology toward greater removal or decreased production of the protein. An alternative strategy is to develop compounds interfering with the various stages of NFT formation: tau hyperphosphorylation, microtubule depolymerization, and aggregation (11). An effective tau-directed therapy aims to sever the links between Aβ accumulation and downstream neurotoxicity as mediated by NFTs. A further strategy that is under exploration is to interfere with neuroinflammation, which is thought to be a major conduit of the AD protein neurotoxic effects (12).
NEUROPATHOLOGIC ASSESSMENT OF AD
Until recently, the only method to definitively diagnose AD as a cause of dementia was through postmortem examination. The standard for this was redefined in 2012 by the National Institute on Aging and the Alzheimer’s Association (13,14). It relies on a tiered evaluation of brain regions for 3 pathologic hallmarks of AD: diffuse Aβ plaque burden (based on Thal phases), NFT burden (based on Braak stages), and neuritic plaque location and density (based on the Consortium to Establish a Registry for Alzheimer’s Disease [CERAD] score). Immunohistochemistry is the preferred method for the first 2 elements, and the neuritic plaque evaluation technique described in the modified CERAD protocol recommends thioflavin S or silver stain (15). The distinction between diffuse and neuritic plaques is imperfect, as they tend to contain varying levels of Aβ protein, which affects their reactivity. Each of the 3 components is scored 0–3 (ABC score: A for Aβ, B for Braak, and C for CERAD, based on and translated from the Thal, Braak, and CERAD scoring systems, respectively), and a final combined score relating to AD pathology is generated, which corresponds to the level of probability (none, low, intermediate, or high) and is used to assess the likelihood that AD neuropathologic changes can be considered causative of the cognitive impairment or dementia in a particular patient. The novel scintigraphy methods for assessing AD pathology offer an exciting opportunity to bring these diagnostic procedures to the living patient, with a subsequent impact on decision making for disease-modifying therapy should this become available in the future. The biological definition of AD has been proposed by use of in vivo biomarkers for Aβ, tau and neurodegeneration, the so-called AT(N) framework (16).
AMYLOID BETA STAGING
The long preclinical stage of AD is characterized by the gradual accumulation of Aβ plaques across the cerebral cortex. This accumulation is thought to be a result of imbalance between the production and removal of extracellular Aβ protein. Aβ accumulates in the form of Aβ-only diffuse plaques (amorphous, irregularly shaped Aβ collections lacking surrounding neurites), as well as the combined Aβ–NFT pathology of neuritic plaques.
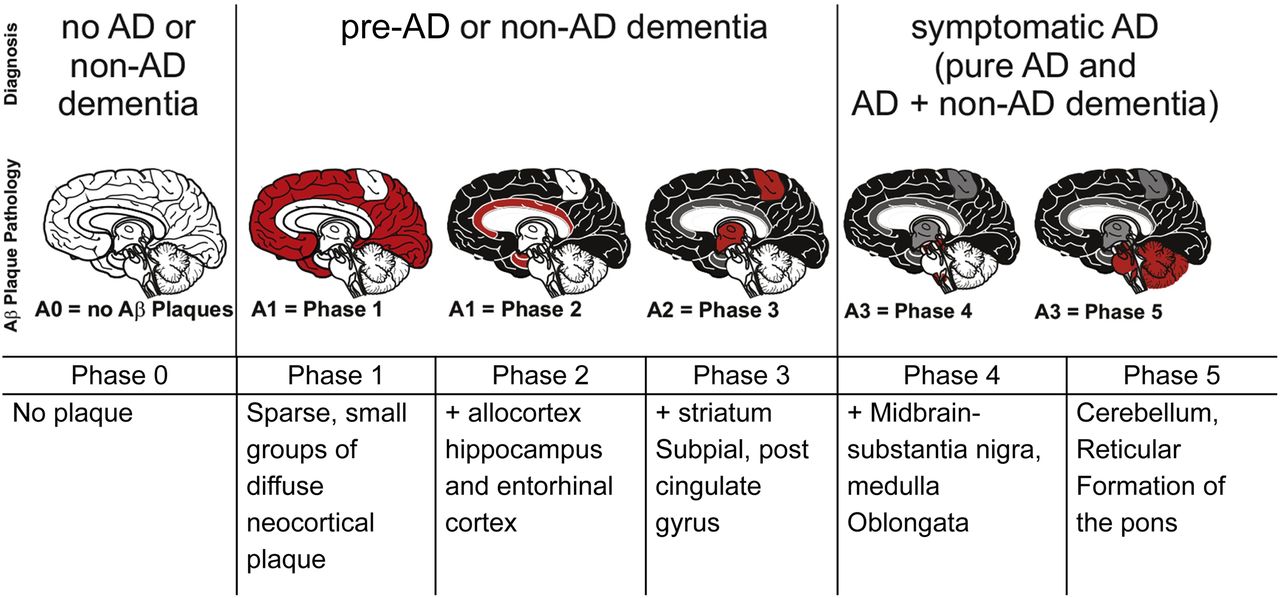
In 2002, Dietmar Thal and coauthors published autopsy results for 51 carefully selected patients ranging from cognitively normal to severely demented (17). The authors identified progressive cerebral changes, which were classified into 5 phases of cerebral β-amyloidosis. These stages have subsequently been coined Thal phases. The phases are based on a single parameter, the presence or absence of Aβ deposits in specific regions of the brain, without regard to the quantity of Aβ present in a location or the type of Aβ plaque (i.e., diffuse or neuritic) (Fig. 1).

Representation of Thal phases of Aβ plaque accumulation and their correspondence to clinical status (22).
In phase 1, sparse Aβ deposits are identified in the frontal, parietal, temporal, or occipital cortex (i.e., neocortex), appearing as focal or small groups. No other areas of Aβ plaque deposition are noted. The presence of neocortical Aβ, but not hippocampus, entorhinal, or midbrain Aβ, irrespective of plaque density, is considered phase 1.
There is a progressive increase in Aβ deposition in cortical and subcortical regions during phases 2–5, beginning with structures of the medial temporal lobe. Phase 2 includes the entorhinal cortex, the cornu ammonis 1 portion of the hippocampus, and, to a lesser extent, the cingulate gyrus, the amygdala, and extension into the subpial layer in the regions of phase 1.
Progression to phase 3 is characterized by the presence of Aβ plaques in deeper subcortical nuclei, including the caudate, putamen, claustrum, thalamus, and hypothalamus, as well as white matter and greater bandlike deposition in the regions of phases 1 and 2. Phase 4 involves midbrain structures (including the substantia nigra and red nucleus) and the inferior olivary nucleus in the medulla. Finally, phase 5 disease includes additional brain stem nuclei as well as the cerebellum.
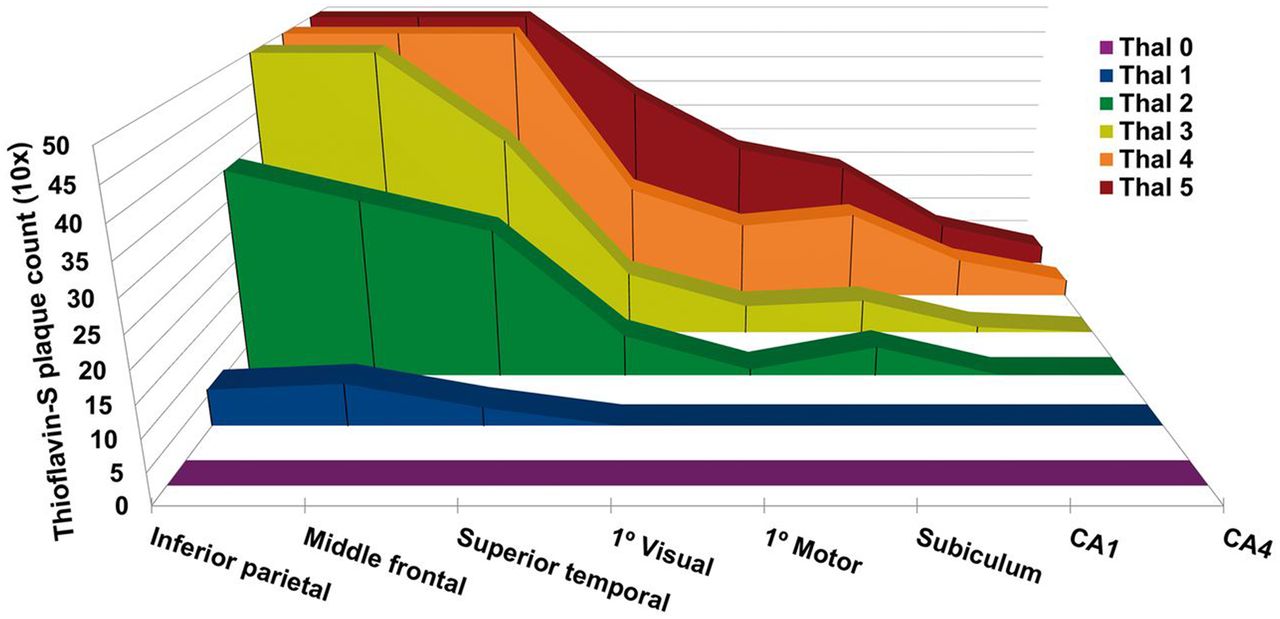
Subsequent investigations have further elaborated on the Thal staging system, with attention to the Aβ plaque density present in each phase. Murray et al. evaluated regional distribution of Aβ plaque density in neocortical areas and the hippocampus in 3,618 autopsies. In addition to the autopsy results, 35 cases had recent antemortem 11C-labeled Pittsburgh compound B (PiB) PET amyloid Aβ scans available for correlation (18). The study demonstrated that between Thal phases 1 and 2 there is a pronounced increase in Aβ plaque counts in the association neocortex, reaching a plateau by phase 3 in most areas of the neocortex evaluated (Fig. 2). Correlation of the Thal phases with 11C-PiB clearly confirms increased Aβ PET signal as individuals progress along the Thal phases. This increase is demonstrated in Figure 3, which compares representative 11C-PiB images over a range of Thal phases. PiB is quantified using SUV ratios, with cerebellum as the reference range. Patients with a clinical and pathologic diagnoses of Alzheimer dementia invariably had Aβ deposition patterns consistent with phases 3−5.

Progression of amyloid plaque density in Thal stages 1–5 (18).
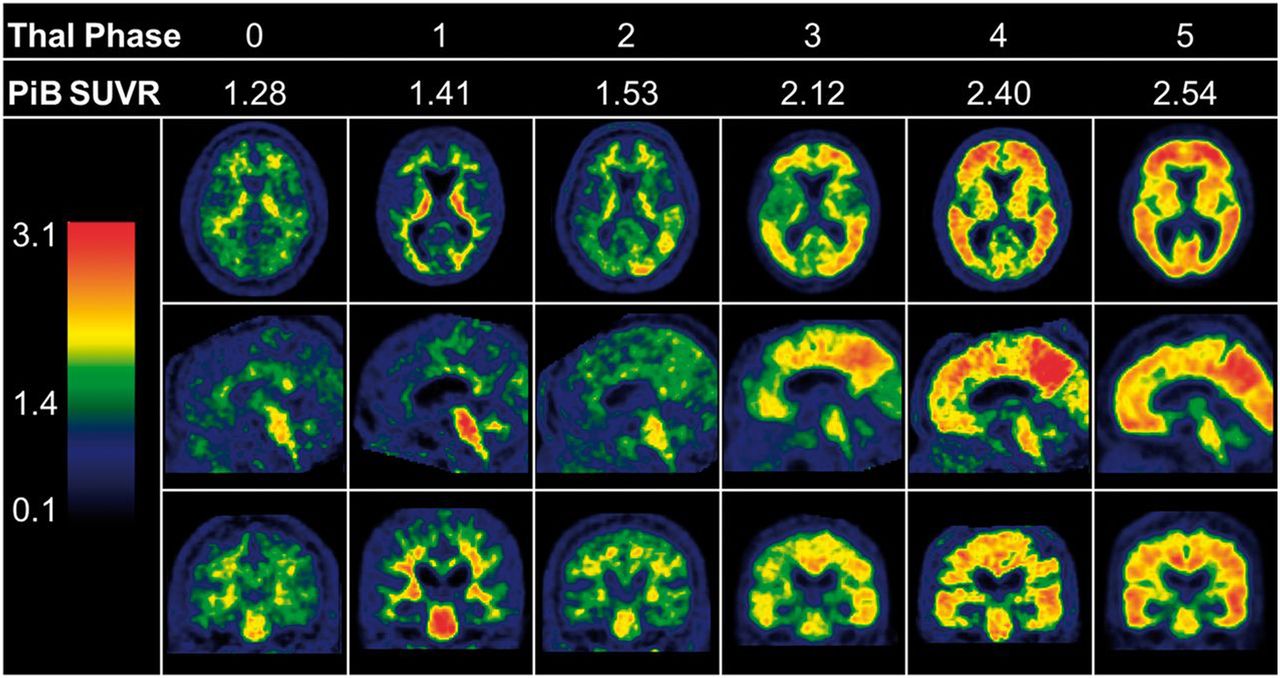
Correlation of Thal stages with amyloid plaque signal as measured by 11C-PiB SUV ratio (SUVR) (18).
Aβ Imaging of Thal Phases
All currently available Aβ PET tracers bind to the fibrillar Aβ structure and thus have specific affinity for both neuritic and diffuse plaques, nonspecific binding to cerebral white matter, and binding to instances of non-AD pathology, such as amyloid-laden blood vessels in cerebral amyloid angiopathy (19,20). The high affinity to Aβ plaques is highlighted by the strong inverse correlation between 11C-PiB signal and cerebrospinal fluid levels of Aβ42, thought to underlie much of the plaque formation (21). Aβ imaging is capable of distinguishing between lack of Aβ signal (Thal phases 0–2) and the presence of high neocortical Aβ (Thal phases 4–5) based on visual analysis of the signal generated in the neocortex. Scans for individuals in Thal phase 3 tend to be reported as intermediate in signal strength. Thal et al. reported a binary positive read of 33% in these cases (22). Three Aβ tracers have so far been approved by the Food and Drug Administration (23): 18F-florbetapir (Amyvid; Eli Lilly/Avid Radiopharmaceuticals), 18F-florbetaben (Neuraceq; Piramal Imaging), and 18F-flutemetamol (Vizamyl; GE Healthcare).
A more fine-grained distinction between Thal phases 0–5 using PET Aβ imaging is not possible because of 4 related factors. The first is the presence of tracer binding nonspecific to AD, such as to amyloid in blood vessels in cerebral amyloid angiopathy or off-target binding to white matter. The second is an insufficient Aβ density to generate sufficient signal strength. The third is that areas of distinction among stages 3, 4, and 5 are small subcortical areas affected by both partial-volume effects and interference by white matter nonspecific binding. The fourth is that Aβ PET tracers bind to both diffuse Aβ and neuritic plaques, complicating the distinction between Thal and CERAD scores. For example, according to Thal et al., phase 4 is defined by presence of Aβ plaques in the medulla oblongata and red nucleus whereby “there are often only one to three plaques in the entire anatomic structure” (17). In any case, the clinical utility of differentiating accurately between the advanced Thal stages in vivo is questionable.
The evaluation of Aβ pathology brings to light the inherent conflict between pathologic findings and scintigraphic signal in general, which applies to all imaging examinations. Imaging is limited by a signal threshold below which there is no appreciable visual signal. This threshold is determined not only by the amount of signal present in the substrate and the level of interfering nonspecific activity but also by the spatial resolution and count sensitivity of the imaging system. Aβ binding radiotracers all have a relatively high degree of nonspecific binding to white matter, leading to difficulties identifying subtle signal changes that may occur between Thal phases. A final barrier to equating scintigraphic and pathologic examinations is that unlike PET Aβ signal, Thal staging is not affected by plaque density in the areas of interest.
As a result of the above considerations, the outcome of Aβ imaging is currently restricted to labeling of scans as positive or negative. However, as discussed earlier, this exposes the issue of scans that are reported as being of intermediate positivity. Although such results may introduce an unhelpful ambiguity in the clinical setting, it nonetheless reflects the evidence that subthreshold Aβ burden associates with cognitive change (24) and that the rate of Aβ accumulation may point to a particularly high-risk group for dementia (25).
The clinical application of Aβ PET scanning remains an area of investigation. The current consensus points to value in confirming AD diagnosis (30% of cases undergo a change of diagnosis after Aβ PET) and the increase in diagnostic certainty by 60% (26). This consensus has led to the adoption of amyloid positivity (i.e., suprathreshold amyloid burden as evidenced by cerebrospinal fluid or PET) as a standard inclusion criterion for trials of disease-modifying agents in AD (11). In addition, a set of clinical scenarios has been highlighted as appropriate for Aβ scintigraphy by a consensus statement (27). Although PET Aβ is useful in distinguishing AD from non-AD causes of cognitive impairment, among cognitively healthy adults amyloid positivity ranges from 10% at age 50 to 44% at age 90 (28). This difference in prevalence across age groups may mean that Aβ PET offers greater diagnostic utility when clarifying cases of cognitive impairment in younger people (29,30). In terms of prognostic power, the best evidence is for subjects with mild cognitive impairment, who have significantly higher progression rates to AD if scintigraphic findings confirm the presence of suprathreshold cortical Aβ plaque burden (31). More broadly, long-term follow-up data of amyloid-positive individuals have shown a tendency for greater cognitive decline relative to those with no significant Aβ burden (32,33). It has been proposed that, in future, information on Aβ burden will need to be complemented by measures of tau pathology (cerebrospinal fluid or PET) as well as neurodegeneration (MRI, 18F-FDG PET) to define the extent to which a neurodegenerative process is due to AD and to provide an indication of its prognosis (16).
Neuritic Plaque Density (CERAD Staging)
Neuritic plaques are Aβ plaques surrounded by dystrophic neurons rich in NFTs. These plaques are strongly associated with postmortem confirmation of AD through the CERAD scoring system (15). Specific biomarker targets for neuritic plaque have not been identified, but Aβ tracers have been shown to have affinity for both diffuse Aβ and neuritic plaques (19,34), because the substrate for all known tracers is the β-pleated sheet conformation of fibrillary Aβ (19). Currently available Aβ PET tracer signal therefore reflects both the diffuse and the neuritic plaque burden.
NEUROFIBRILLARY TANGLE STAGING
The landmark investigation of Heiko and Eva Braak on the staging of AD NFTs was first published in 1991 (35).This paper described the progressive and predictable sequence of buildup of NFTs with AD disease progression. There are 6 Braak stages of NFT propagation that are characterized by both the location and the density of NFTs. Importantly, NFTs evolve separately from amyloid plaques, and the topographic distribution of these two AD findings therefore differs considerably (Fig. 4).

Representation of Braak stages 1–6. CA1 = cornu ammonis 1. (Reprinted with permission of (56).)
The earliest deposition of NFTs is noted in the anterior medial temporal lobe involving the transentorhinal cortex (stage I) and the adjacent entorhinal cortex (stage II). Stages I and II are termed the transentorhinal stages. These early stages are considered part of the normal cerebral aging process, and findings are present in the nondemented aging population (36–38). For example, one large autopsy study found that NFTs are present in the entorhinal cortex and hippocampus regardless of clinical status at time of death (cognitively healthy, mild cognitive impairment, or dementia). In contrast, severe pathologic changes in the inferior temporal lobe were present only in those with dementia (37). Studies on younger individuals (i.e., under the age of 30) have demonstrated the presence of NFTs in the transentorhinal regions from as early as age 6 (39). This is evidence either that AD is a lifelong, slowly progressive condition or that the NFT accumulation in these regions is part of nonpathologic aging (9).
Stages III and IV are termed the limbic stages and refer to progression of NFTs to the limbic structures (amygdala and hippocampus) and subsequently the thalamus. Clinically, the spread of NFTs to stages III and IV is associated with early signs of cognitive impairment.
Stages V (neocortical association cortices) and VI (primary motor, visual, and sensory areas) are the end stages of AD, with large numbers of NFTs throughout the neocortex, corresponding to the neuropathic diagnosis of AD. Cerebral atrophy is present, with the greatest destruction in the neocortical association areas (the prefrontal, parietal, and temporal lobes) and relative sparing of the primary visual, motor, and sensory areas.
TAU IMAGING OF BRAAK STAGES
The superior performance of neuropathologic Braak staging of AD relative to Aβ neuropathology with regard to clinical diagnosis and level of cognitive impairment is well established (38,40–42). In addition, atrophy on MRI colocalizes with NFTs on subsequent autopsy (41,43), including instances in which clinical subtypes of AD are concerned (44). PET imaging of tau binding is an active area of research, with numerous candidates currently being studied (45). Tau ligand design is complicated by the various pathologic isoforms of tau and the morphology of the fibrillar aggregates present (46), whereby compounds sensitive to tau AD pathology show surprisingly variable affinity to depositions in primary tauopathies (47,48).
The most widely studied agent, T807, now renamed 18F-AV-1451 (flortaucipir; Tauvid), licensed from Siemens to Eli Lilly, was Food and Drug Administration–approved in May 2020. The agent has been approved “to estimate the density and distribution of aggregated NFTs in adult patients with cognitive impairment who are being evaluated for AD.” Recently, postmortem data confirming tau binding of this agent have been published (47–50). Multiple second-generation tau agents under investigation show substantially less nonspecific binding and higher affinities to primary tauopathies and may be useful in identifying earlier Braak stages (45). These agents include MK-6240 (Merck & Co), PM-PBB3 (Aprinoia Therapeutics), PI-2620 (Life Sciences) (51), and RO-948 (Roche) (31).
Early studies in patients demonstrated that tau PET signal is an accurate predictor of AD diagnosis, cognitive impairment, and rate of atrophy and that its predictive ability is not improved by MRI data (52). Regional tau PET ligand uptake was shown to vary with clinical phenotype, aggregating to a larger extent in the cortical areas hypothesized to be affected on the basis of the profile of cognitive impairment (53). Intriguingly, investigators have demonstrated that tau PET can provide information on the topographic spread of NFTs along the Braak regions, thus suggesting a role for the tracer in staging the progress of AD pathology in vivo (54). From a clinical perspective, a recent study showed a high degree of concordance between visually rated tau PET scans and the subsequent extent and topography of NFTs (55).
SUMMARY
The postmortem diagnosis of AD requires the presence of Aβ plaques, tau deposition, and neuritic plaques. A revised postmortem classification system (ABC, for amyloid, Braak, and CERAD) based on these 3 components has emerged. Depending on the level of each component, the pathologic findings are scored as indicating no likelihood of AD or indicating a low, intermediate, or high likelihood.
Scintigraphic biomarkers can elucidate early-stage (Thal stages 0 and 1) or late-stage (Thal stages 3 and 4) presence of Aβ in the form of diffuse and neuritic plaques. They therefore reflect burden contributing to both Thal and CERAD staging components. Second-generation tau imaging agents with lower nonspecific binding may allow discrimination of the Braak stages, likely using a simplified 4-score system. These new in vivo biomarkers of AD pathology allow for a biological detection of the neuropathic changes of AD ante mortem. The advances in in vivo demonstration of AD pathology has led to a conceptual reformulation of AD (16) that draws a distinction between the terms AD pathologic change and AD pathology. The former indicates presence of cortical Aβ but not tau while the latter indicates presence of both sufficient Aβ and tau signal with or without evidence for neurodegeneration. This new development opens the way to targeted interventions based on biomarker readouts aimed at halting the disease process in its preclinical stages. Scintigraphic confirmation of preclinical AD may therefore become a major work stream for nuclear medicine specialists working in centers that specialize in AD disease modification clinical trials.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Aug. 6, 2020.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 1, 2020.
- Accepted for publication June 30, 2020.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.