


Significant advances continue in the development of cancer-specific molecular imaging agents and modalities for diagnosing, staging, and treating various cancer types. For these advances to translate successfully to widespread clinical use, both regulatory approval by the U.S. Food and Drug Administration (FDA) and completion of the reimbursement approval process with the Centers for Medicare & Medicaid Services (CMS) must be achieved. To this end, methods for expediting these processes are being addressed by both agencies. This report summarizes a 1-d meeting hosted by the National Cancer Institute (NCI) on May 15, 2017, with officials from the Clinical Trials Branch of the Cancer Imaging Program, FDA, and CMS; members of the Society of Nuclear Medicine and Molecular Imaging (SNMMI), the World Molecular Imaging Society, the American College of Radiology, the Radiologic Society of North America, the International Society for Strategic Studies in Radiology, and the Medical Imaging and Technology Alliance; molecular imaging scientists from some major hospitals and institutions; and industry representatives, who joined to learn about the pathways of approval, coverage, and payment decisions.
This 2017 meeting grew out of one held on May 4, 2016, that focused primarily on FDA approval requirements and the expectations for approval of novel drugs and devices, packaged separately or in combination, within the context of optical surgical navigation (1,2) for cancer detection.
The goals of the 2017 meeting were formulated by prior active collaboration among the meeting participants along with colleagues from the European Society of Radiology/European Congress of Radiology and the European Society for Molecular Imaging. The meeting’s focus was threefold. The first focus was to broaden the conversation to other agents or modalities and to address both the reimbursement and the regulatory components. The second was to present a current view of FDA regulatory innovations in cancer imaging development, a past and present view of the PET and molecular imaging agent approval processes, and a current view of CMS reimbursement protocols. The third focus was to discuss scientific issues involved in orchestrating agency-required phase III clinical trials as presented through 3 candidate agents as case studies: a PET probe targeting the prostate-specific membrane antigen (PSMA) for imaging prostate cancer (presented by Thomas Hope, University of California, San Francisco), an optical antibody probe for image-guided head and neck cancer surgery targeting epidermal growth factor receptor (presented by Eben Rosenthal, Stanford University), and ultrasonography with targeted microbubbles against a neovasculature target, kinase insert domain receptor, for ovarian cancer (presented by Sanjiv S. Gambhir, Stanford University). The conceptual design for these 3 case studies was distributed in advance to participants. The 3 agents themselves were currently in phase II development, but the presenters were asked to think broadly and, through a slide presentation, discuss what would be the ideal larger or definitive phase II trial, as well as the pivotal phase III trial. The presenters were to address what the primary objective of the trial was, what the investigator was trying to do, how the trial would be implemented, how much of a difference would be made versus the standard of care, how much evidence would be required to support either approval or reimbursement, and whether the risk of standard-of-care alterations would be justified by a benefit to clinical care regarding diagnosis and staging, potential prognostic and predictive markers, response assessment, and cancer surveillance.
Additionally, the meeting served to provide more clarity to researchers on trial design by delineating more uniformly required steps investigators should take to successfully translate their innovations. Reported here are the meeting’s discussion items and recommendations for continued honing of the approval and reimbursement processes investigators must navigate.
The meeting was videocast by the National Institutes of Health and added to the National Institutes of Health website videocast archive (https://videocast.nih.gov/summary.asp?Live=23377&bhcp=1).
FDA CURRENT VIEW: REGULATORY INNOVATIONS IN CANCER IMAGING DEVELOPMENT
Phillip B. Davis, clinical reviewer in the Division of Medical Imaging Products, Center for Drug Evaluation and Research, FDA, presented as an FDA representative.
Through collaboration and engagement, the FDA aims to improve the processes used for collecting and analyzing evidence needed for approval of new imaging products. This is evidenced by the FDA’s willingness now to parallel-track with CMS in the “Program for Parallel Review of Medical Devices” (3). Its vision is that by having all stakeholders share in the responsibility of product development, greater collaboration will be achieved, thus speeding innovation, a core component of the FDA’s mission. Interest in this mission is found throughout government as discussed in a 2016 New England Journal of Medicine publication (4). These authors discussed how broad collaboration and participation between the FDA, NCI, and CMS represents the wave of the future. It is also necessary to ensure that the best possible evidence is available to make important decisions about the nation’s health care. Key principles of this new paradigm include engaging multiple stakeholders and systems and considering evidence from multiple study sites such as using uniform FDA-reviewed protocols that are shared among sites and investigators via professional societies.
Current and Ongoing Initiatives
Current and ongoing initiatives include the Medical Imaging Drug Advisory Committee, which recently discussed an optical imaging agent under review that the FDA believes addresses an unmet medical need. The Medical Imaging Drug Advisory Committee brings together the FDA, industry, expert clinicians, and the public to discuss regulatory questions, clinical trial design and endpoints, and recommendations on specific products under FDA review.
In 2016, the NCI held its first workshop on medical imaging products (1). It focused on optical imaging drugs used in surgical oncology, discussing safety first and associated early-phase studies. This led to a White Paper publication outlining specific meeting recommendations, and the submission of additional optical imaging products to the FDA division.
The FDA is also engaged in ongoing discussions with the CMS to pursue collaborative reviews that lead to efficient regulatory decision making in which multiple goals can be achieved. The FDA has collaborated with academic sponsors of 68Ga-PSMA-11 studies to develop uniform protocols and share these with multiple investigators through the SNMMI. Currently, FDA collaboration with the CMS is at an early stage, an effort that was made public in 2016 (3). One goal is to move away from sequential data review and toward concurrent reviews by the separate agencies. Current policy goals include developing confirmatory studies designed to meet both approval and coverage objectives, performing parallel review activities, and coordinating on postmarketing studies that will allow data collection to be used by both agencies. Importantly, this information sharing does not create a new process requirement for industry. It also does not establish new evidence standards for either agency, nor does it influence either agency’s decision making. The anticipated benefits are enhanced efficiency and the shortening of review timelines.
Recent Drug Approvals Highlighting Endpoints and Studies Acceptable for Successful Marketing Authorization
NETSPOT
NETSPOT (Advanced Accelerator Applications) (first kit for the preparation of 68Ga-DOTATATE injection, a radioactive diagnostic agent for PET imaging) was a priority review product with orphan drug status and a successful example of how academic centers generated data under expanded access and then handed it to manufacturing sponsors for pursuit of marketing authorization. This radioactive probe helps to locate tumors in adult and pediatric patients with the rare somatostatin receptor–positive neuroendocrine tumors (5). Use of advanced imaging techniques to detect neuroendocrine tumors at an early stage in patients is critical. NETSPOT provides another diagnostic tool to help clinicians determine the location and extent of tumor, important for planning appropriate therapy.
Axumin
Axumin (Blue Earth Diagnostics) (18F-fluciclovine) is a radioactive diagnostic agent that targets the amino acid transporters LAT-1 and ASCT2. Axumin is indicated for PET imaging in men with suspected prostate cancer recurrence after prior treatment based on elevated blood prostate-specific antigen levels. Conventional imaging tests are often not able to determine the location of recurrent prostate cancer when the prostate-specific antigen is at very low levels. Axumin is shown to provide another accurate imaging approach for these patients (6). This tracer was approved in 2016 under priority review for prostate cancer imaging. The efficacy endpoint used for approval included comparison to another approved drug or reference standard, and academic investigators initially performed the studies and then handed the data rights to the sponsor. The sponsor then conducted its own analyses of these data. Here, again, no patient outcome data were submitted.
These oncologic products can be covered for Medicare beneficiaries on FDA approval by the local Medicare Administrative Contractors (MACs), without the need for a National Coverage Determination (NCD) by the CMS. However, for nononcologic products that will be subject to an NCD for Medicare coverage, the opportunity for a study to be designed for both regulatory and reimbursement approval is important. Sometimes products, including Axumin and NETSPOT (both priority review), are brought to market under less than ideal collaborative conditions in order to meet critical unmet patient needs. Medicare reimbursement for Axumin and NETSPOT is occurring under local, and not national, coverage policy.
The FDA website provides further guidance on clinical trial design for medical imaging products, as well as the 3 main guidance documents used by sponsors daily (7).
PET AND MOLECULAR IMAGING AGENT APPROVAL PROCESSES: PAST AND PRESENT
Terri Wilson, senior director of Patient Access and Healthcare Policy at Blue Earth Diagnostics and vice-chair of the PET Group of the Medical Imaging and Technology Alliance, presented as a representative of the Medical Imaging and Technology Alliance.
General Coverage Overview
Medicare coverage for new products or services can occur through 1 of 2 pathways: local coverage through the MACs or through an NCD (Fig. 1). Regardless of the pathway, a coverage request cannot be initiated until the product receives FDA approval. NCDs must be followed by all MACs and supersede local coverage decisions. In medical imaging, the only products or services that are required to go through the NCD are PET radiopharmaceuticals (except those for oncologic uses) and screening tests, such as low-dose CT for lung cancer screening.
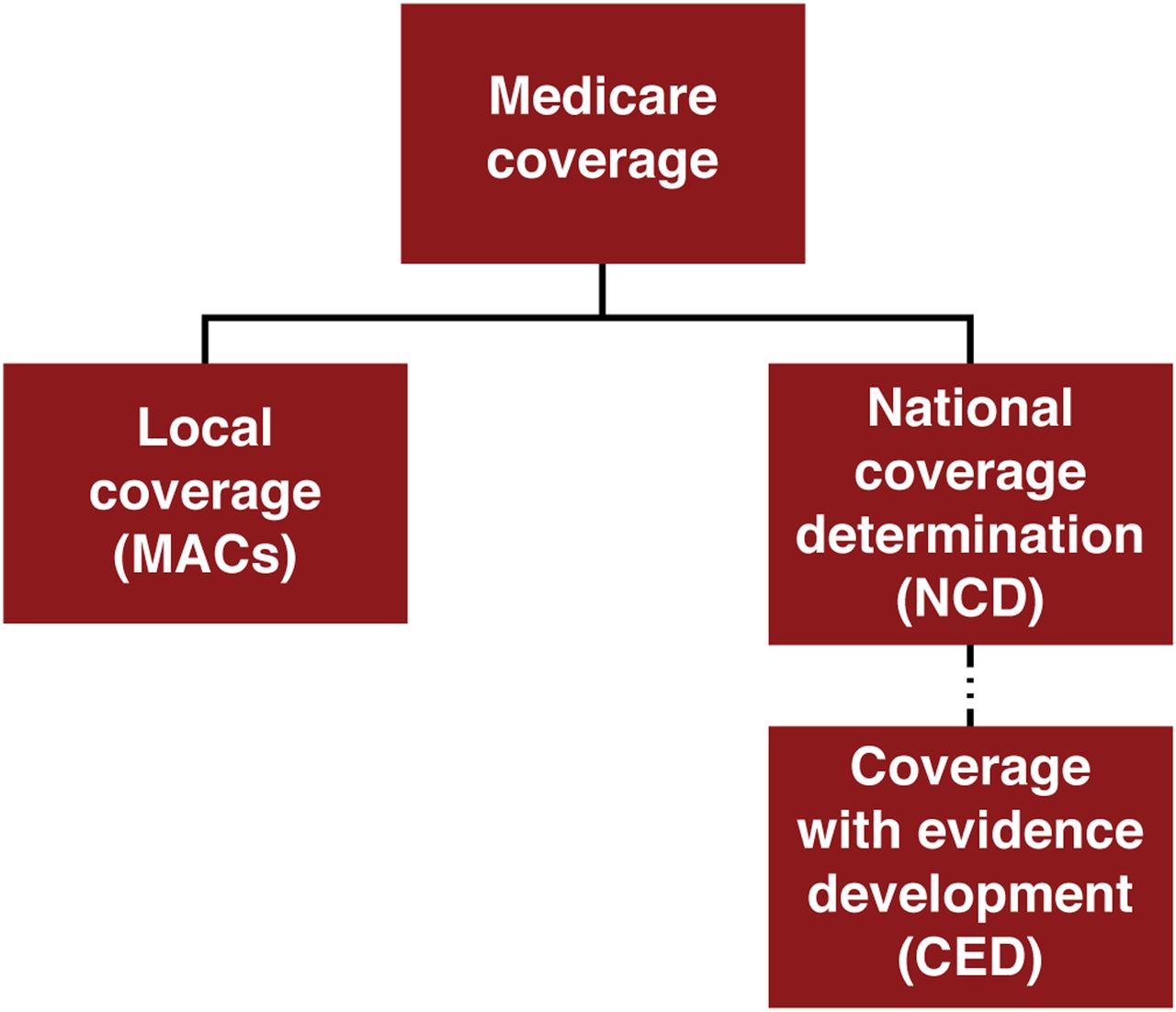
Medicare coverage components. Medicare coverage for new products or services occurs through 1 of 2 pathways: local coverage through MACs or national coverage through NCD.
Thus, most products and services do not go through the NCD process and are instead covered at the discretion of the local MACs. The MACs, in their coverage determination, may create either a Local Coverage Determination (LCD) (described in the supplemental materials available at http://jnm.snmjournals.org) (8–20) or a policy article defining the coverage criteria or the International Classification of Disease 10 codes (21) that can be used for the procedures. Sometimes they can be silent, that is, not subject to NCD, LCD, or coverage article but covered on the basis of medical necessity for that particular patient. The LCD process is typically reserved for a small portion of medical services covered by the MACs.
When an NCD is required, it can be requested by any stakeholder, such as a manufacturer, a MAC, or a professional society. The CMS Coverage and Analysis Group (CAG) reviews NCD requests and determines whether to open an NCD. Once open, the CAG is responsible for reviewing all the data submitted by the requestor, as well as additional information submitted during the open public comment period. The final decision may be “covered,” “noncovered,” or “Coverage with Evidence Development” (CED); these possible responses can be thought of as “yes,” “no,” or “maybe.” All NCDs are maintained by the CAG in the NCD manual.
PET Coverage History and Background
Medicare began covering PET imaging in 1995 for studies using the cardiac perfusion agent 82Rb (22). From 1995 to 2005, for every new PET radiopharmaceutical developed and every new indication covered, a formal request was submitted to the CMS. After a few broad oncology coverage requests for 18F-FDG, the December 2000 NCD (23) included exclusionary language necessitating the many individual PET National Coverage Analyses (NCAs) that followed. The language said, “Our review of all evidence submitted and additional evidence gathered supports the conclusion that the request for broad coverage is denied.” Thus, all future new indications or new PET radiopharmaceuticals had to go through an NCA and, through 2005, this resulted in coverage for some cardiac indications, certain oncologic indications using 18F-FDG, evaluation of refractory epilepsy, and differential diagnosis of dementia, also using 18F-FDG. This process differs from the local coverage process via the MACs for most Medicare-covered products and services. Between 2005 and 2013, all requests submitted for PET resulted in the decision of CED, meaning that after the 9- to 12-mo NCA process had been completed, CMS required that additional data be collected through a CED study. The study then had to be developed, with the study design and endpoints needing approval by the CMS CAG—requiring additional time before Medicare beneficiaries can access the new product or indications. In 2013, another NCA request was submitted to the CMS asking that the exclusionary language be removed and that all new PET radiopharmaceuticals go to local MACs for coverage decisions, based on their FDA-approved indication. The final CMS decision allowed local MAC discretion to cover new FDA-approved PET oncologic tracers but required that the nononcologic PET tracers and indications remain within the NCD (24). This 2013 NCD represented a major change for oncologic PET radiopharmaceuticals.
PET Coverage Decisions
National Oncologic PET Registry (NOPR) Background and Current Status
The NOPR was developed in 2005 in response to the CMS decision to expand coverage for 18F-FDG PET through CED (25). Medicare reimbursement for all previously noncovered cancers and indications using 18F-FDG could be obtained if the patient’s referring physician and provider submitted data to a clinical registry to assess the impact of PET on patient management. The NOPR implemented this registry in order to collect the data CMS desired to make a coverage decision. 18F-NaF PET was added to the NOPR in 2010 for detection of osseous metastasis and was opened in 2011. The NOPR is sponsored by the World Molecular Imaging Society (formerly the Academy of Molecular Imaging) and managed by the American College of Radiology (26).
The 18F-FDG NOPR went into operation in May 2006. As of 2007, under the NCD process, the CMS had extended national coverage for 18F-FDG PET only in response to coverage requests that included clinical data sufficient to justify its use for a specific cancer type or indication. The CMS had issued its first coverage decision for 18F-FDG PET in January 1998, approving payment for the characterization of indeterminate solitary pulmonary nodules and for initial staging of suspected metastatic non–small cell lung cancer (27). In July 1999, the agency extended coverage to include restaging of suspected recurrent colorectal cancer, staging and restaging of lymphoma when used as an alternative to 68Ga scintigraphy, and evaluating the recurrence of melanoma before surgery when used as an alternative to 68Ga scintigraphy. In July 2001, in response to a request for broad coverage of 18F-FDG PET, the CMS began covering diagnosis, staging, and restaging for melanoma, lymphoma, and non–small cell lung, esophageal, colorectal, and head and neck cancers. In the same NCD, however, the CMS declined to cover 18F-FDG PET for many other requested indications, citing insufficient supporting clinical data. The CMS extended coverage to certain breast cancer indications in October 2002, to a specific thyroid cancer indication in October 2003, and to a specific cervical cancer indication in January 2005. Over the next 3 y, the NOPR investigators published several studies documenting the impact of 18F-FDG PET on management of patients with cancer (28–30), and in 2009, they submitted a request to the CMS to end the data collection requirements, thus opening an NCA. The result was positive coverage for cervical and ovarian cancers and multiple myeloma, allowing for initial and subsequent treatment strategy coverage for those indications. Positive coverage was also added for 1 initial treatment strategy scan (supplemental materials) for most other oncologic indications, and data collection by NOPR was continued for the subsequent treatment strategy of all remaining cancer indications. In 2012, after NOPR published further results (31), a final request was made to end the data collection requirement and to cover 18F-FDG PET for all oncologic indications; this NCA took an additional 9 mo and resulted in the ending of the NOPR for 18F-FDG PET and associated data collection requirements in 2013.
For the 18F-NaF NOPR, ongoing since 2010, there was also a 1-y period between the coverage decision and patient enrollment. In 2015, a request to end the 18F-NaF NOPR data collection was made; the CMS decision was to extend CED to allow for additional data collection desired by the CAG. The 18F-NaF NOPR closed, and 18F-NaF PET coverage ended on December 14, 2017. Another NCA request was submitted to determine the future of coverage for 18F-NaF-PET; this request was denied in May 2018. Further discussions with the CMS need to occur to decide next steps.
IDEAS Background and Current Status
By providing access to amyloid imaging for more than 18,000 Medicare beneficiaries for whom there was ambiguity about the cause of their cognitive decline, the IDEAS (Imaging Dementia—Evidence for Amyloid Scanning) Study sought to demonstrate that amyloid PET can help clinicians determine the cause of cognitive impairment, provide the most appropriate treatments, and improve health outcomes for Medicare beneficiaries. It is anticipated that evidence obtained by the IDEAS Study will support reimbursement of amyloid imaging by Medicare and other third-party payers. The amyloid β PET imaging coverage request, submitted in 2012 after FDA approval, was the first initiated directly by the manufacturer. The coverage request was for the entire class of amyloid β products, rather than a single product, since additional amyloid PET agents were under review by the FDA at the time. The NCA request to cover amyloid β PET imaging resulted in a CED decision in September 2013. The IDEAS Study did not open to Medicare beneficiaries until February 2016, nearly 4 y after FDA approval of the first amyloid β product and about 3 y after the NCA request was submitted (supplemental materials). As of December, 2017, study enrollment ended. Currently, Medicare does not cover brain amyloid PET. Analysis of the IDEAS Study is under way, but the most important patient outcome results are not expected until sometime in late 2019 or early 2020.
Axumin Background and Current Status
Approved by the FDA in May 2016 after priority review, Axumin is the first FDA-approved 18F PET imaging agent indicated for use in patients with suspected recurrent prostate cancer. Immediately after FDA approval, Axumin was manufactured for clinical use in a few geographic areas. The manufacturer first submitted a request for coverage with MAC Cahaba Government Benefit Administrators, LLC (32) (jurisdiction J MAC to CMS covering Alabama, Georgia, and Tennessee at the time), which made the decision to cover the Axumin scans for the labeled indication in July 2016. As availability expanded, requests were sent to the other MACs around the United States and, as of January 1, 2017, Axumin PET was covered by all the MACs for the labeled indication, effectively achieving national coverage in just over 6 mo.
Requesting Coverage
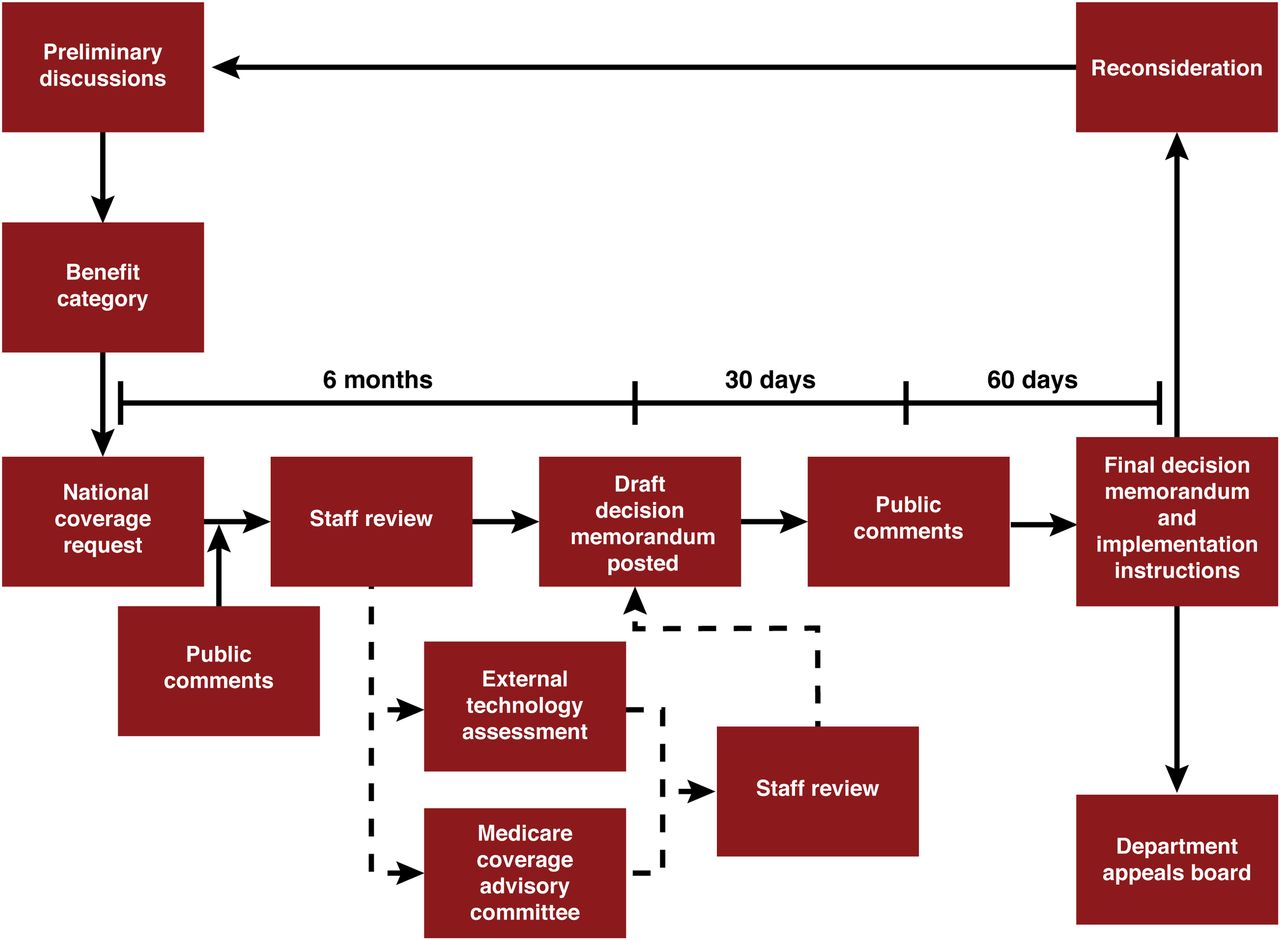
Typically, once a product is FDA-approved, the manufacturer can initiate the NCA request if it is necessary for the product. With the more recent inauguration of the “Program for Parallel Review of Medical Devices,” some formal discussion and analysis can begin before obtaining FDA approval (3). The typical NCA process, in its entirety, takes between 9 and 12 mo (Fig. 2). Thus, even when the coverage decision is positive, there is a significant time after FDA approval before Medicare beneficiaries have access to a new product. A CED decision is also coverage, but a lengthy process will ensue before Medicare beneficiaries have access to the covered product or service. A study will need to be designed and proposed. Some discussions on this can occur before the final decision to begin to conceptualize the study design, but the study would still need to be fully designed and formally proposed to the CAG. The CAG provides input on the study design and endpoints and ultimately approves the CED study. In cases of a positive coverage decision, including CED, CMS will provide instructions to the MACs on billing requirements. Implementation of a CED study can take some time—between 12 and 20 mo for the NOPR and IDEAS studies.

NCA process. Historically, once a manufacturer has FDA approval, it can initiate NCA request, if necessary.
When a product can be covered by the local MACs, the manufacturer may also initiate this process once the product has received FDA approval. The manufacturer will provide the MACs with information about the product, including clinical evidence supporting the use of the product in the patient population, the coding that would be used to bill for the product, the pricing compendia, and clinical articles supporting the use of the product. The MAC then will decide whether they will cover the product and whether to create an LCD or policy article or remain silent. When communicating with the MACs, most are responsive, with the typical wait between submitting information and receiving some sort of response or questions for additional information being up to 8 wk.
The differences between national and local coverage are highlighted in Figure 3.

NCD vs. local coverage. Differences between national and local coverage are highlighted.
Other Challenges
Other challenges are involved with coding, payment, and Medicare plan type (supplemental materials).
CMS CURRENT VIEW
Rosemarie Hakim, senior research advisor of CAG, CMS, presented as a representative of CMS.
The Medicare construct was established by the Social Security Act of 1965, title 18 (33). Everything done in CAG and CMS is governed by either a law, a statute, or a regulation (34).
CAG coverage determinations address our national coverage, assessing whether evidence is sufficient to conclude that the item or service will improve clinically meaningful health outcomes for the Medicare population. CAG performs in-house structured literature reviews. It considers the quality, strength, and totality of the evidence and, after 6 or 9–12 mo, comes to a decision.
Medicare Coverage of Clinical Studies
There are 3 ways for Medicare to cover clinical studies. The first is an investigational-device exemption. This is based on a regulation that allows coverage of FDA-approved premarket investigational-device exemption studies. In the past, all investigational-device exemptions were covered at the discretion of local MACs, resulting in different policies across the country. In 2015, this was consolidated into a centralized process, as described on the CMS website (https://www.cms.gov/Medicare/Coverage/IDE/). Sponsors submit their FDA-approved studies to the CMS. The CMS CAG performs a science review and decides whether the studies are adequate for coverage under CMS regulation. The second way for Medicare to cover clinical studies is clinical trial policy. This NCD allows coverage of many NCI-sponsored clinical trials. This coverage is for routine costs in clinical trials that are sponsored by the government. National Institutes of Health–sponsored trials, and anything that has an investigational-new-drug application by the FDA, is potentially coverable under this NCD. The third way is CED, which comprises individual CED studies that must go through the NCD process.
CASE STUDIES
PSMA-Targeted PET Probe for Prostate Cancer
68Ga-PSMA-11 has been frequently used outside the United States to stage patients with prostate cancer. This tracer has not been patented, and therefore no company or private entity will make the investment required to bring it to market. Accordingly, academic groups have taken the lead to collect the data necessary for submission of a new-drug application to the FDA, and academic institutions have initiated use of this tracer within expanded-access clinical trials. In head-to-head studies with 18F-fluorocholine PET, 68Ga-PSMA-11 PET has been shown to be superior (35,36). There are 2 main populations studied using 68Ga-PSMA-11 PET: preprostatectomy patients and biochemical recurrence patients.
Preprostatectomy Patients
There is a dramatic shift in patients undergoing prostatectomy in the United States, with a marked increase in the number of high-risk patients undergoing prostatectomy compared with prior years (37). In the high-risk prostatectomy population, the presence of nodal metastases is the worst prognostic factor for disease recurrence after surgery (38). Thus, the central issue that imaging can solve in primary preprostatectomy patients is the detection of nodal metastases before surgery. Additionally, the detection of distant metastases can dramatically change the management of patients who were being planned for prostatectomy. In collaboration with the SNMMI Clinical Trials Network, a preprostatectomy protocol (NCT02919111) has been developed to determine the sensitivity and specificity of 68Ga-PSMA-11 PET for detection of pelvic nodal metastases compared with histology at the time of radical prostatectomy. Initial staging of patients planned for curative-intent radiation therapy is of similar importance for treatment planning.
Biochemical Recurrence Patients
Biochemical recurrence is the most common indication for 68Ga-PSMA-11 PET outside the United States (39). The role in this setting is to determine the location of recurrent disease in patients with negative findings on conventional imaging who have a rising level of prostate-specific antigen. Particularly in patients with low prostate-specific antigen, that is, below 2.0 ng/mL, it is hoped that localizing solitary or oligometastatic disease may enable targeted treatment that could dramatically affect the patient outcome. The issue in this patient population is that PET-positive lesions are frequently subcentimeter-sized and particularly difficult to biopsy (small retroperitoneal nodes or osseous metastases). For this reason, it is difficult to define appropriate reference standards to determine whether positive scan findings are true-positive or false-positive.
Given the difficulty in developing endpoints in this population, approaches other than a pathologic reference have been pursued. For example, the University of California, San Francisco/University of California, Los Angeles, protocol (NCT03353740 and NCT02940262) developed a composite reference standard that involves both histology, when available, and conventional imaging follow-up to document true-positive sites of disease. The OSPREY trial, which is studying 18F-DCFPyL PET (NCT02981368), chose a different route requiring all patients to have a biopsy-capable lesion on conventional imaging before enrollment so that pathology can be used as a reference standard.
A separate approach would be to use a therapeutic outcome as the measure of the accuracy of the imaging study. Although not a registration study, this approach is being used in a study evaluating 18F-fluciclovine PET in radiation therapy planning (NCT01666808). In this study, patients are randomized to having radiation therapy planned with either conventional imaging or 18F-fluciclovine PET, with the primary outcome being biochemical recurrence-free survival at 3 y. This study is likely to provide valuable information because it will demonstrate the impact of imaging on patient outcome; however, it is not clear how traditional registration trial endpoints for imaging agents, such as sensitivity and specificity, would be incorporated into such a study.
Optical Probe for Image-Guided Surgery
Surgery remains the standard of care both for newly diagnosed oral cavity disease and for recurrent disease after medical management. Margins have a significant impact on overall survival. The positive margin rate ranges from 20% to 30% (40–44). The 5-y survival rate varies for those with positive versus close versus negative margins. Patients with positive margins have worse 2- and 5-y survival rates; this has been shown fairly consistently (41,45,46). Survival with negative margins is somewhere between 65% and 90%, but that with positive margins is less than 50%, roughly a 30% difference (40,41,44,45). This difference is seen for both early-stage and advanced-stage disease.
Challenge in Clinical Trial Design
There is limited standardization of how margins are managed and, even when there is consistency in surgical technique, there remains significant intersurgeon and intrasurgeon variability (supplemental materials).
Trial Design and Agent
In the study, the agent to be used is cetuximab, an antiepidermal growth factor receptor chimeric antibody used for therapy in several cancer types, with a known toxicity profile based on its use in thousands of patients. The fluorescent dye, IRDye800CW, is conjugated to the antibody, and the conjugate has been shown in nonhuman primate studies to have pharmacologic and toxicity properties similar to those of unlabeled cetuximab. The study proposes that cetuximab-IRDye800CW is indicated for intraoperative use in the detection of squamous cell carcinoma in patients undergoing curative resection. Cetuximab-IRDye800CW is used with a near-infrared imaging device as an aide to surgical decision making in conjunction with conventional techniques. Thus, the study proposes to demonstrate that, together with surgical judgment, cetuximab-IRDye800CW and the imaging device can be used to improve the completeness of surgical resection.
One endpoint will be the number of patients who have a positive margin identified on near-infrared light that was not recognized by white light, and a second endpoint will be the sensitivity and specificity of this imaging modality by comparison to permanent histologic sections as the reference standard. The optimal dose is based on a tumor-to-background ratio in a dose-escalation study (47) in a limited number of patients (n = 12).
Assumptions
Important assumptions are that the frequency of positive margins is between 20% and 30%, that imaging can identify additional positive margins in about 50% of patients (the hypothesis), and that negative margins are associated with 30% better survival. The calculations indicate that about 100–120 patients would be needed for the study to have an α-value of 0.05 and a power of 80%. The expectation for this protocol is to reduce the number of patients with positive margins from 20% to 10%.
Discussion
The difference between the positive margin detection rate is an appropriate endpoint. However, it will eventually need to be considered whether detection of the additional 10% of patients with positive margins under near-infrared light translates into improved clinical outcomes. It is known that the positive margin rate results in a reduction of survival by about 30%, depending on the cancer type. Because of the multimodality treatment provided in head and neck cancer and the variation in tumor location, a clinical trial in a specific head and neck subsite, for tumors likely to recur (stage III or IV), receiving otherwise identical treatment (chemotherapy/radiation/surgery), would require a large number of patients to determine benefit. It is possible that, on the basis of the strong correlation of margin status and outcome, increased numbers of positive margins using near-infrared light should allow for a more complete resection, in turn improving survival.
The CMS would require stronger clinical outcome data to demonstrate value. For example, if 20% fewer cases return to the operating room and the need for reoperation is minimized, that would be a desirable clinical outcome. For the proposed case, CMS would like to see a link back to long-term follow-up for outcome (overall survival) in these patients. Local Medicare contractors would be looking for some divergence in patient outcomes by taking the different pathways based on the evidence.
Ultrasound with Targeted Microbubbles
Targeted microbubbles have only recently been used in human studies. Some of these agents have been produced by Bracco, a company that has also funded related clinical trial work.
Ovarian Cancer
Early detection of ovarian cancer could potentially result in a very large difference in outcomes. The 5-y relative survival rate for late-detected ovarian cancer (distant spread at diagnosis) is 27.6%, whereas for localized disease it can be as high as 93.5%. However, only 15% of ovarian cancers are diagnosed as localized disease because of the mostly later presentation with vague clinical symptoms. Many women with ovarian cancer, having different genetic mutations such as BRCA1 and BRCA2, and the associated data around them (48,49), need to be followed with new strategies to detect these cancers early.
Ovarian Cancer Imaging
Current first-line imaging is typically transvaginal sonography followed by additional imaging as needed. A large study done in the United Kingdom (UKCTOCS) randomized subjects to 3 arms (annual screening using a multimodal screening strategy, an ultrasound screening strategy, or no screening) and included transvaginal sonography following the CA-125 blood biomarker study as part of the multimodal screening strategy. It was unclear if the ovarian cancer screening cost-effectively reduced mortality. Currently, the goal would be to make transvaginal sonography much more specific while keeping the sensitivity high to enable successful screening detection of patients with ovarian cancer.
Ovarian Cancer Screening
Most early detection work suggests that a new test should begin with a high-risk population that has a greater pretest likelihood of disease. After succeeding there, one can move to a population-based screening approach where the pretest likelihood is lower. For ovarian cancer, there are numerous reasons why a population-based approach is problematic: most women show no or nonspecific symptoms; the natural history of the disease is largely unknown; the prevalence of the disease is relatively low, requiring high specificity; most cases occur without identifiable risk factors; and a definitive evaluation for a positive screen includes surgery. Thus, studying high-risk women is likely more practical. However, current screening methods lack the sensitivity and specificity for both population-based and high-risk patient screening. Molecular imaging demonstrates clear potential to improve screening by offering the needed sensitivity and specificity and the ability to show change over time in a panel of biomarkers. Because CA-125 is not specific enough, new panels are needed. However, achieving the needed test performance remains difficult. If one accepts a 10% positive predictive value (1/10 patients found to have tumor at surgery), the levels of accuracy needed, at given sensitivities and specificities, are very high (50).
Decision Modeling for Ovarian Cancer Screening
Decision models using stochastic microsimulation have been built that show what would happen to a large cohort of individuals moving down one of several screening test combination arms (51). Screening combinations involving CA-125, transvaginal sonography, a panel of blood biomarkers that may be more accurate, and a next-generation imaging modality were evaluated in modeling the mortality reduction achieved and the cost per year of life saved using different screening intervals with repeat blood testing and/or imaging (6, 12, 18, and 24 mo). This allows for modeling of the performance needed for a given test. On the basis of this modeling, the performance characteristics needed for the imaging test, blood test, and the two to be coupled together in a new screening test are known. Generally, the imaging test needs to have about 95% sensitivity and specificity, with a cost not exceeding $750 per imaging session.
BR55: Targeted Microbubble Probe
Here, the target is vascular endothelial growth factor receptor type 2, also known as the kinase insert domain receptor. BR55, one of several bubble types that could be useful, was chosen because the target kinase insert domain receptor is present on the vasculature of many different kinds of tumors, making it useful beyond ovarian cancer, and it was one of the furthest along in the Bracco development pipeline in toxicity testing, thus facilitating filing of an investigational-new-drug application. With targets on the vascular endothelium, such as the kinase insert domain receptor, ultrasound after microbubble administration provides a means to image the tumor microvasculature and neoangiogenesis. Ultrasound can then be used to locate the bubbles and destroy them if needed.
First-in-Women Targeted Microbubble Ultrasound Clinical Study
Initial results of these agents were obtained through preclinical studies followed by studies done in Europe and in the United States. The agents were initially applied in women with breast tumors or ovarian cancer and in men with prostate cancer. The first-in-women study (52) was designed to assess whether BR55 was safe and allowed assessment of kinase insert domain receptor expression using immunohistochemistry as the reference standard and to determine the optimal contrast agent dose in patients with cancer. The results of this study were recently published (52). Overall, the microbubbles were found to be safe.
Clinical Trial
On the basis of the pilot results, Amelie Lutz from Stanford submitted a grant to the NCI, funded in 2017, to conduct a clinical trial that will characterize the behavior of these targeted microbubbles in women at high risk for ovarian cancer. Ovarian cancer was chosen for this initial efficacy trial because of the potential large impact given that there is no competing modality that performs well. High risk is defined as familial or hereditary ovarian cancer, BRCA1/2 mutations, Lynch syndrome, and Li-Fraumeni syndrome.
The hypothesis for this current trial is that BR55-targeted microbubble ultrasound has a higher sensitivity and specificity for ovarian cancer detection in high-risk women than does traditional non–contrast-based transvaginal sonography (the current default strategy). The new strategy must be found to do better if it is to proceed to further trials. For the trial, 2 patient groups will be recruited in parallel: patients with suspected ovarian cancer (to evaluate performance of the new imaging method in the ovarian tumor setting with histopathologic correlation) and patients at high risk for ovarian cancer. The objectives are to determine the performance and specificity of targeted microbubble contrast-enhanced ultrasound for in vivo imaging of the ovarian cancer vascular network at the molecular level. Additional exploratory objectives in the 2 groups will be pursued (supplemental materials). The primary endpoint in this initial study is when all 60 patients with suspected ovarian cancer and all 50 menopausal patients undergoing prophylactic surgery are imaged. The goal is to have real tissue for validating what is seen on the images to collect accurate sensitivity and specificity. This trial, now starting up, is anticipated to take 2 y.
MAJOR FINDINGS: KEY TAKEAWAYS
FDA Review Optimization
For NETSPOT, a priority review product with orphan drug status, the FDA points out the missed opportunity for collecting patient outcome data, which could have been implemented at relatively low additional cost and possibly used to enhance the sponsor’s claim. It might have been determined how often patients were downstaged or upstaged on the basis of imaging findings (53). Additionally, the primary endpoint data were limited. These included agreement, sensitivity, and specificity evaluations by comparison to approved imaging agents, with follow-up imaging and/or pathology as a reference standard. Overall, FDA approval was achieved without clinical outcome data, and the agent is now reimbursed.
Commercialization Issues
FDA approval is only the first step in commercialization; significant effort also is needed to address planning for coverage, coding, and payment level by Medicare, Medicaid, and commercial insurers. Obtaining Medicare coverage for new PET radiopharmaceuticals and examinations via interactions with local MACs has allowed for significantly faster access than is possible via the NCD process (in months as opposed to years).
Parallel Review
Recent efforts by the FDA and CMS have made parallel review available for devices (3). A sponsor developing a new technology can ask the CMS and the FDA to evaluate the proposed premarket study jointly. This is important, as the CMS sometimes does not approve a new technology based on evidence that was satisfactory for FDA approval because of slightly different statutory mandates. Parallel review allows a sponsor to design a study that, if successful, should satisfy both the FDA and the CMS mandates, thus speeding the process toward coverage. Much of this information is outlined in a guidance document (54) on the process of NCDs and LCDs, and coverage in general.
PSMA-Targeted PET Probe for Prostate Cancer
68Ga-PSMA-11 PET has an important role in initial staging of patients before definitive therapy (prostatectomy or curative-intent radiation therapy) and in restaging of patients with biochemical recurrence. Trials have been completed, and data will subsequently be submitted to the FDA for approval of this agent, through a collaboration between the University of California, San Francisco, and the University of California, Los Angeles. Outcome trials have different endpoints from accuracy trials and cannot be easily performed simultaneously.
Optical Probe for Image-Guided Surgery
Surgery remains the standard-of-care curative-intent treatment for most solid tumor types. Margins have a significant impact on overall survival. The enhanced ability to visualize tumor must be demonstrated by clinical outcome. Phase III trials must be designed using strategies that minimize bias and maximize standardization. The Cysview (hexaminolevulinate HCl; Photocure ASA) study is an appropriate starting point for this trial design (supplemental materials). Paramount endpoints should demonstrate that the use of imaging improves the accuracy of positive margin identification.
Ultrasound with Targeted Microbubbles
Targeted microbubbles hold promise as a novel strategy for detecting various malignancies by using imaging agents that target vasculature-specific biomarkers such as VEFGR2. Initial results in humans are encouraging; more clinical trials on various cancers, including ovarian, are now under way. Investigators planning clinical trials of novel diagnostic tests need to consider greater use of decision models. Initial use of 18F-FDG PET in clinical trials applied decision models examining lung cancer, melanoma, and colorectal cancer, and these results were presented to the CMS in lieu of pending clinical trial results (55–57). Given an observed PET accuracy in a particular application, the models predicted, for example, how many patients would be upstaged or downstaged, as validated later by studies assessing outcomes in terms of management changes.
DISCUSSION: RECOMMENDATIONS FROM MEETING STAKEHOLDERS FOR NEXT STEPS
The research community calls for an end-to-end continuum map outlining the entire process of a new molecular imaging agent and/or device, moving from development through the process of obtaining FDA approval and eventual CMS coverage.
Research investigators would like further incorporation of theranostics, particularly in coupling diagnostic imaging with targeted radiotherapy, to motivate sponsors to support imaging development, as in the case of antibodies for therapy.
Future meetings should include more European investigators to help understand how advances in molecular imaging have evolved more rapidly in Europe.
A helpful topic for future meetings is the role and existing frameworks of academia, industry, networks, and cooperative groups in fostering the types of trial needed for registration and coverage decisions for molecular imaging agents. Another helpful topic is other applications of molecular imaging, such as response assessment and therapeutic target measurement. These topics are mentioned in the supplemental materials, which also provide other recommended steps, background detail, and a summary of key points from audience questions and panel discussion.
DISCLOSURE
Thomas Hope is on the JNM editorial board. Barry A. Siegel is a JNM consulting editor; an advisory board member for Blue Earth Diagnostics and GE Healthcare; and a consultant for Avid Radiopharmaceuticals, Inc., BTG Management Services, Capella Imaging, LLC, Curium Pharma, ImaginAb, Inc., Merrimack Pharmaceuticals, Inc., and Progenics Pharmaceuticals, Inc. He also receives clinical trial support from Blue Earth Diagnostics and Progenics Pharmaceuticals, Inc. Sanjiv S. Gambhir has active imaging grants from Sanofi-Aventis, US, Inc., Biogen Inc., Novocure, Philips Medical, and Pliant Therapeutics, Inc. He is a cofounder of CellSight Technologies, Endra Life Sciences, and Earli Inc. He is on the advisory boards of Centella Therapeutics, Inc., Click Diagnostics, Verily, ImaginAb, MagArray Inc., Konica-Minolta Inc., Vave Inc., Nines Inc., NinePoint Medical, GE Healthcare (ad hoc), Alpha Source Inc., RefleXion Medical, Spectrum Dynamics, Vor Biopharma, TauMark Inc., Piramal MI, Life Molecular Imaging, Infinitus Inc., NuvOx Inc., Bracco Inc., Grail Inc., and VisualSonics/Fuji Inc. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Peter Gardiner (chief medical officer) and Priscilla Harlan (vice president, Corporate Communications) of Blue Earth Diagnostics for their editing; and James Strommer (Stanford Radiology) for figure preparation.
Footnotes
Published online Mar. 8, 2019.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication September 4, 2018.
- Accepted for publication November 28, 2018.





{kind=link}
{kind=link}
{kind=link}
Jump to section
- Article
- FDA CURRENT VIEW: REGULATORY INNOVATIONS IN CANCER IMAGING DEVELOPMENT
- PET AND MOLECULAR IMAGING AGENT APPROVAL PROCESSES: PAST AND PRESENT
- CMS CURRENT VIEW
- CASE STUDIES
- MAJOR FINDINGS: KEY TAKEAWAYS
- DISCUSSION: RECOMMENDATIONS FROM MEETING STAKEHOLDERS FOR NEXT STEPS
- DISCLOSURE
- Acknowledgments
- Footnotes
- REFERENCES
- Figures & Data
- Supplemental
- Info & Metrics
Related Articles
Cited By...
- No citing articles found.