


Abstract
Prostate cancer (PCa) is the most common cancer in men worldwide. In general, PCa responds poorly to chemotherapy. Therefore, antibody–drug conjugates (ADCs) have been developed to specifically deliver highly cytotoxic drugs to the tumor. Because the prostate-specific membrane antigen (PSMA) is overexpressed in PCa, it represents a promising target for ADC-based therapies. The aim of this study was to evaluate the therapeutic efficacy of site-specifically conjugated duocarmycin- and monomethyl auristatin E (MMAE)–based anti-PSMA ADCs with drug-to-antibody ratios (DARs) of 2 and 4. Methods: The glycan group of the anti-PSMA antibody D2B was chemoenzymatically conjugated with duocarmycin or MMAE. Preservation of the immunoreactivity of the antibody on site-specific conjugation was investigated in vitro. Biodistribution and small-animal SPECT/CT imaging (18.5 ± 2.6 MBq) with 25 μg of 111In-labeled ADCs were performed on BALB/c nude mice with subcutaneous PSMA-positive LS174T-PSMA xenografts. Finally, the therapeutic efficacy of the 4 different ADCs was assessed in mice with LS174T-PSMA tumors. Results: The immunoreactivity of the anti-PSMA antibody was preserved on site-specific conjugation. Biodistribution revealed high tumor uptake of all agents. The highest tumor uptake was observed in mice administered with 111In-D2B-DAR2-MMAE, reaching 119.7 ± 37.4 percentage injected dose per gram at 3 d after injection. Tumors of mice injected with 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, and 111In-D2B-DAR4-MMAE could clearly be visualized with small-animal SPECT/CT. In contrast to unconjugated D2B or vehicle, treatment with either of the MMAE-based ADCs, but not with a duocarmycin-based ADC, significantly impaired tumor growth and prolonged median survival from 13 d (phosphate-buffered saline) to 20 and 29 d for DAR2 and DAR4 ADC, respectively. Tumor-doubling time increased from 3.5 ± 0.5 d to 5.2 ± 1.8 and 9.2 ± 2.1 d after treatment with D2B-DAR2-MMAE and D2B-DAR4-MMAE, respectively. Conclusion: The site-specifically conjugated anti-PSMA ADCs D2B-DAR2-MMAE and D2B-DAR4-MMAE efficiently targeted PSMA-expressing xenografts, effectively inhibited tumor growth of PSMA-expressing tumors, and significantly prolonged survival of mice.
- prostate cancer
- prostate-specific membrane antigen (PSMA)
- antibody–drug conjugate
- duocarmycin
- monomethyl auristatin E (MMAE)
Prostate cancer (PCa) is the most common cancer in men worldwide, leading to substantial morbidity and mortality (1). Despite improvements in conventional therapies such as radical prostatectomy with or without radiotherapy, the disease frequently recurs. Although recurrent disease can temporarily be managed with androgen ablation, PCa can progress to a hormone-refractory state, which frequently is accompanied by rapid progression of the disease (2). Hormone-refractory PCa usually is difficult to treat, and the treatment may include chemotherapeutic agents. Unfortunately, the chemosensitivity of PCa is limited and the responses are weak and accompanied by severe side effects (3).
In the past decade, therapy with monoclonal antibodies (mAbs) has attracted a tremendous amount of attention. However, most mAbs show limited antitumor activity in unmodified form. To overcome these limitations, mAbs can be covalently bound to cytotoxic drugs to form an antibody–drug conjugate (ADC) that selectively delivers the drug to tumor tissue while reducing toxicity to healthy tissues. In previous studies, it has been shown that the effectiveness of chemotherapeutic agents can be enhanced by conjugation to targeting molecules (4,5). Two main categories of cytotoxic drugs are used in ADC development: microtubule inhibitors and DNA-damaging drugs. At present, a variety of ADCs are available for clinical use, such as Kadcyla (ado-trastuzumab emtansine; Genentech), an ADC consisting of the anti-human epidermal growth factor receptor 2–directed mAb trastuzumab and the cytotoxic agent DM1. Another is Adcetris (brentuximab vedotin; Seattle Genetics), an ADC that contains monomethyl auristatin E (MMAE) and is a synthetic dolastatin 10 analog causing cytotoxicity by inhibition of tubulin polymerization (6) and the antibody brentuximab directed against CD30, which is overexpressed in Hodgkin lymphoma and other T-cell lymphomas.
In PCa, the prostate-specific membrane antigen (PSMA), a type II transmembrane glycoprotein, is abundantly and specifically expressed on PCa cells and internalized on binding, which renders it an ideal target for ADCs. PSMA has also been found to be expressed in the tumor-associated neovasculature of multiple solid tumor types; therefore, PSMA-targeted therapies might not be restricted to PCa but could also be applied to other malignancies in the future (7–9). So far, several anti-PSMA–based ADCs have been characterized. Ma et al. reported potent antitumor activity of an anti-PSMA mAb conjugated to MMAE in mice with PSMA-positive C4-2 xenografts (5). Kuroda et al. conjugated the anti-PSMA mAb hJ591 to the ribosome-inactivating protein toxin saporin and reported high antitumor activity in an LNCaP xenograft model (10), and the same antibody serves as scaffold for the ultra-potent DNA-crosslinking pyrrolobenzodiazepine dimer (ADCT401/MEDI3726), which most recently entered clinical trials (NCT02991911). An ADC consisting of the anti-PSMA antibody MLN591 and the antimicrotubule chemotherapeutic drug DM1 has been evaluated in patients with PCa in a phase I/II clinical trial (11). Although this study provided proof-of-principle evidence supporting anti-PSMA ADCs as potentially effective agents for patients with PCa, this particular ADC showed limited therapeutic efficacy and possessed an extremely narrow therapeutic window, with rapid deconjugation of the drug from the antibody causing dose-dependent peripheral neuropathy (11). Two other ADCs targeting PSMA are currently undergoing clinical evaluation, including one (which has completed phase II) containing the same linker technology and payload as present in Adcetris and another ADC (ADCT401/MEDI3726, which most recently entered clinical trials) based on antibody J591 and an ultra-potent DNA-crosslinking pyrrolobenzodiazepine dimer.
Frequently, ADCs are based on random conjugation to naturally available amino acid side chains (ε-amino group of lysines or thiol group of cysteines), resulting in stochastic distribution of the drug-to-antibody ratio (DAR) and leading to heterogeneous products containing a stochastic mixture of ADCs with different molar DARs and linked at different sites to the antibody. As a consequence, each component in the mixture may have different pharmacokinetics, immunoreactivity, charge, size, and stability (12). Random conjugation was shown to reduce the efficacy of ADCs, resulting in a low therapeutic index (amount of a therapeutic agent that causes the therapeutic effect in relation to the amount that causes toxicity) in which efficacious and maximum tolerated dose are similar (13). An important strategy to enhance the therapeutic index of a given mAb–drug combination is site-specific conjugation. One of the methods to achieve this is by engineering a specific amino acid into an antibody, which serves as an anchor for attachment of the drug (12,14). In the present study, site-specifically conjugated ADCs using the antibody’s glycan at asparagine-297 as an anchor were applied (12). A major advantage of this method is its applicability to any type of recombinant IgG isotype, irrespective of the mammalian expression system, and any linker−payload combination (12). ADCs produced by this method were found to be homogeneous and highly hydrolytically stable while displaying negligible aggregation (12).
The murine mAb D2B specifically targets PSMA and has been shown to efficiently accumulate in PSMA-expressing LNCaP xenografts (15).
The aim of this study was to compare the therapeutic efficacy of 4 different anti-PSMA ADCs. For this purpose, the mAb D2B was site-specifically conjugated with the cytotoxic drug MMAE, an irreversible inhibitor of microtubuli, or duocarmycin, a DNA-alkylating agent acting by binding to the minor groove of DNA in A-T–rich regions (16). To evaluate the relationship between DAR and ADC efficacy, 2 DAR variants were compared for both anti-PSMA ADCs, one with a DAR of 2 and one with a DAR of 4.
MATERIALS AND METHODS
Preparation of the Antibody
The anti-PSMA monoclonal antibody D2B (IgG1) was prepared as described before (17). Briefly, it was purified from a hybridoma culture supernatant by protein A affinity chromatography. The antibody-secreting cells were obtained according to the hybridoma technology from mice that were immunized with a lysate of membranes of LNCaP PCa cells.
Preparation of Acid-Cleavable Glycan-Conjugated ADCs
Glycan-conjugated ADCs were prepared as described previously (12). Briefly, anti-PSMA antibody D2B (15 mg/mL) was incubated with 1% (w/w) endoglycosidase in 20 mM Tris, pH 7.5, for 16 h at 37°C. Complete trimming was confirmed by mass spectral analysis on the Fc/2 domain. After deconvolution of peaks, 2 major products were observed (24,633 and 24,761 Da) for the trimmed anti-PSMA antibody D2B, resulting from core GlcNAc(Fuc) and GlcNAc(Fuc) without lysine (-Lys) anti-PSMA D2B. The starting material showed 3 major products (25,733, 25,859, and 26,022 Da), corresponding to G0F (-Lys), G0F, and G1F, respectively.
Subsequently, trimmed anti-PSMA D2B (10 mg/mL) was incubated with 5 mM F2-GalNAz (75 equivalents), 5% (w/w) glycosyltransferase (developed in house), and 10 mM MnCl2 in 20 mM Tris, pH 7.5, for 16 h at 30°C. Mass spectral analysis of anti-PSMA D2B antibody (F2-GalNAz)2 after FabULOUS (Genovis Inc.) and dithiothreitol treatment showed 2 major peaks corresponding to incorporated F2-GalNAz with dithiothreitol as an adduct, 25,000 Da (-Lys) and 25,129 Da. The dithiothreitol adduct is a result only of sample preparation for mass spectral analysis. The anti-PSMA D2B (F2-GalNAz)2 reaction mixture was loaded on a protein A column (endotoxin-free), and the antibody was eluted with 0.1 M glycine, pH 2.7, followed by dialysis to phosphate-buffered saline (PBS). The general protocol for conjugation of bicyclo[6.1.0]non-4-yne (BCN), modified with either 1 or 2 toxic payloads (MMAE or duocarmycin) via a cleavable linker (Val-Cit-PABC), to anti-PSMA D2B (F2-GalNAz)2 was as follows: 4 mg of anti-PSMA D2B (F2-GalNAz)2 (12 mg/mL) were conjugated with 5–10 equivalents of BCN-toxin in PBS and in the presence of 25% DMF. The ADCs were purified by size-exclusion chromatography in PBS.
Mouse Model
Animal experiments were conducted in accordance with the principles laid out by the revised Dutch Act on Animal Experimentation (2014) and approved by the institutional Animal Welfare Committee of the Radboud University Nijmegen. Male BALB/c nude mice (Janvier), 7–8 wk old, were housed in individually ventilated filter-topped cages (5 mice per cage) under nonsterile standard conditions with free access to standard animal chow and water. After being allowed 1 wk to adapt to laboratory conditions, the mice received a subcutaneous injection (right flank) of 3 × 106 PSMA-transfected LS174T cells (17) suspended in 200 μL of complete RPMI 1640 medium. LS174T-PSMA cells were grown in RPMI 1640 medium supplemented with 10% fetal calf serum (Life Technologies) and 2 mM glutamine in the presence of 0.3 mg/mL G418.
Radiolabeling
The ADCs were conjugated with diethylenetriaminepentaacetic acid (DTPA) as described previously (15). Both D2B-duocarmycin ADCs (108.2 μg) were radiolabeled with 120 MBq of 111In (Mallinckrodt Pharmaceuticals), whereas D2B and both D2B-MMAE ADCs (96.1 μg) were radiolabeled with 106.7 MBq of 111In in 0.1 M 2-(N-morpholino)ethanesulfonic acid buffer, pH 5.4 (3 times the volume of 111InCl3), and incubated for 20 min at room temperature under metal-free conditions. Subsequently, 50 mM ethylenediaminetetraacetic acid was added to the final concentration of 5 mM to chelate unincorporated 111In. The labeling efficiency was determined to be 85% by instant thin-layer chromatography using 0.15 M citrate buffer, pH 6.0, as the mobile phase. The 111In-labeled agents were purified by gel filtration on a PD-10 column (GE Healthcare), and the radiochemical purity of the agents exceeded 95%, as determined by instant thin-layer chromatography.
In Vitro Characterization
The immunoreactive fractions of 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, and 111In-D2B-DAR4-MMAE were determined essentially as described by Lindmo et al. (18). A serial dilution of LS174T-PSMA cells (3.3 × 106–5.2 × 107 cells/mL) in RPMI-1640 containing 0.5% bovine serum albumin was incubated with 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, or 111In-D2B-DAR4-MMAE. Nonspecific binding was determined by adding an excess of unlabeled D2B (5 μg) to a duplicate of the lowest cell concentration. After a 1-h incubation at 37°C, the cells were centrifuged and the activity in the cell pellet was measured in a γ-counter (Wizard 3″ 1480; LKB-Wallac, PerkinElmer). The inverse of the specific cell-bound activity was plotted against the inverse of the cell concentration, and the immunoreactive fraction was calculated from the y-axis intercept using GraphPad Prism (version 5.03 for Microsoft Windows).
LS174T-PSMA cells were cultured in 6-well plates and incubated with 1.9 kBq of 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, or 111In-D2B-DAR4-MMAE for 4, 24, and 48 h in 2 mL of binding buffer at 37°C in a humidified atmosphere with 5% CO2. Nonspecific binding and internalization were determined by coincubation with unlabeled D2B. After incubation, an acid wash buffer (0.1 M HAc, 0.15 M NaCl, pH 2.6) was added for 10 min to remove the membrane-bound fraction of the cell-associated 111In-labeled compounds. Subsequently, the cells were harvested from the 6-well plates, and the amount of membrane-bound and internalized activity was measured in a γ-counter.
Small-Animal SPECT/CT Imaging and Ex Vivo Biodistribution
Twenty male BALB/c nude mice (5 mice per agent) with PSMA-expressing LS174T-PSMA tumors (right flank) received a tail-vein injection of 25 μg of 111In-D2B-DAR2-duocarmycin (21.9 ± 0.6 MBq), 111In-D2B-DAR4-duocarmycin (21.8 ± 0.9 MBq), 111In-D2B-DAR2-MMAE (17.0 ± 0.7 MBq), or 111In-D2B-DAR4-MMAE (15.0 ± 0.4 MBq). Five additional mice received an intravenous injection of 25 μg of 111In-D2B (17.3 ± 0.6 MBq), serving as a reference group to determine the biodistribution of unmodified 111In-D2B. At 3 d after injection, the mice were euthanized with CO2/O2 asphyxiation. Of each group, 2 mice underwent SPECT/CT imaging (acquisition time 30 min) on a small-animal SPECT/CT scanner (U-SPECT II; MILabs) with a 1.0-mm-diameter pinhole mouse high-sensitivity collimator tube and CT parameters of 160-μm spatial resolution, 65 kV, and 615 μA. Scans were reconstructed with MILabs software, using ordered-subset expectation maximization, an energy window of 154–188 keV, 3 iterations, 16 subsets, a voxel size of 0.2 mm, and a gaussian filter of 0.4 mm. The SPECT/CT scans were analyzed, and maximum-intensity projections were created using the Inveon Research Workplace software (version 4.1). For each mouse, tissues of interest (LS174T-PSMA tumors, muscle, lung, spleen, kidney, liver, small intestine, salivary glands, and adrenals) were dissected and weighed, and the radioactivity was measured in a γ-counter. Blood samples were obtained by heart puncture. To calculate radioactivity uptake in tissue as a fraction of the injected dose, an aliquot of the injected dose was counted simultaneously.
Therapeutic Efficacy of ADCs
Sixty mice were inoculated subcutaneously with PSMA-transfected LS174T cells. When the tumors had grown to approximately 100 mm3, the mice were randomly assigned into 6 groups and received a single 5 mg/kg dose of ADC, unconjugated D2B, or PBS. Tumor size and body weight were measured 2 times per week by a technician who was masked to the treatment groups. Mice were removed from the experiment if one of the following endpoints was reached: a tumor volume of 500 mm3 or more, a body weight decrease of at least 20% compared with baseline or at least 15% in 2 d, ulceration, or invasive tumor growth.
Statistical Analysis
Differences in tumor uptake, blood concentration, tumor-to-blood ratios, and tumor-doubling time between 111In-DTPA-D2B, 111In-DTPA-D2B-DAR2-duocarmycin and 111In-DTPA-D2B-DAR4-duocarmycin or 111In-DTPA-D2B-DAR2-MMAE and 111In-DTPA-D2B-DAR4-MMAE were tested for significance via one-way analysis of variance and Bonferroni's multiple comparison post analysis using GraphPad Prism version 5.03. Survival was described as median, and survival curves were compared with the log-rank (Mantel–Cox) test using Prism, version 5.03. A P value of below 0.05 was considered significant.
RESULTS
111In-D2B-ADCs Specifically Bind to PSMA-Expressing Cells
The immunoreactive fractions of 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, and 111In-D2B-DAR4-MMAE were 69.3%, 86.5%, 75.5%, 68.4%, and 78.0%, respectively, indicating that the immunoreactivity of the D2B antibody was preserved during the conjugation procedures. Maximum binding to PSMA-expressing LS174T-PSMA cells reached 74%–80% for the reference compound and all 111In-D2B-based drug conjugates (Fig. 1).
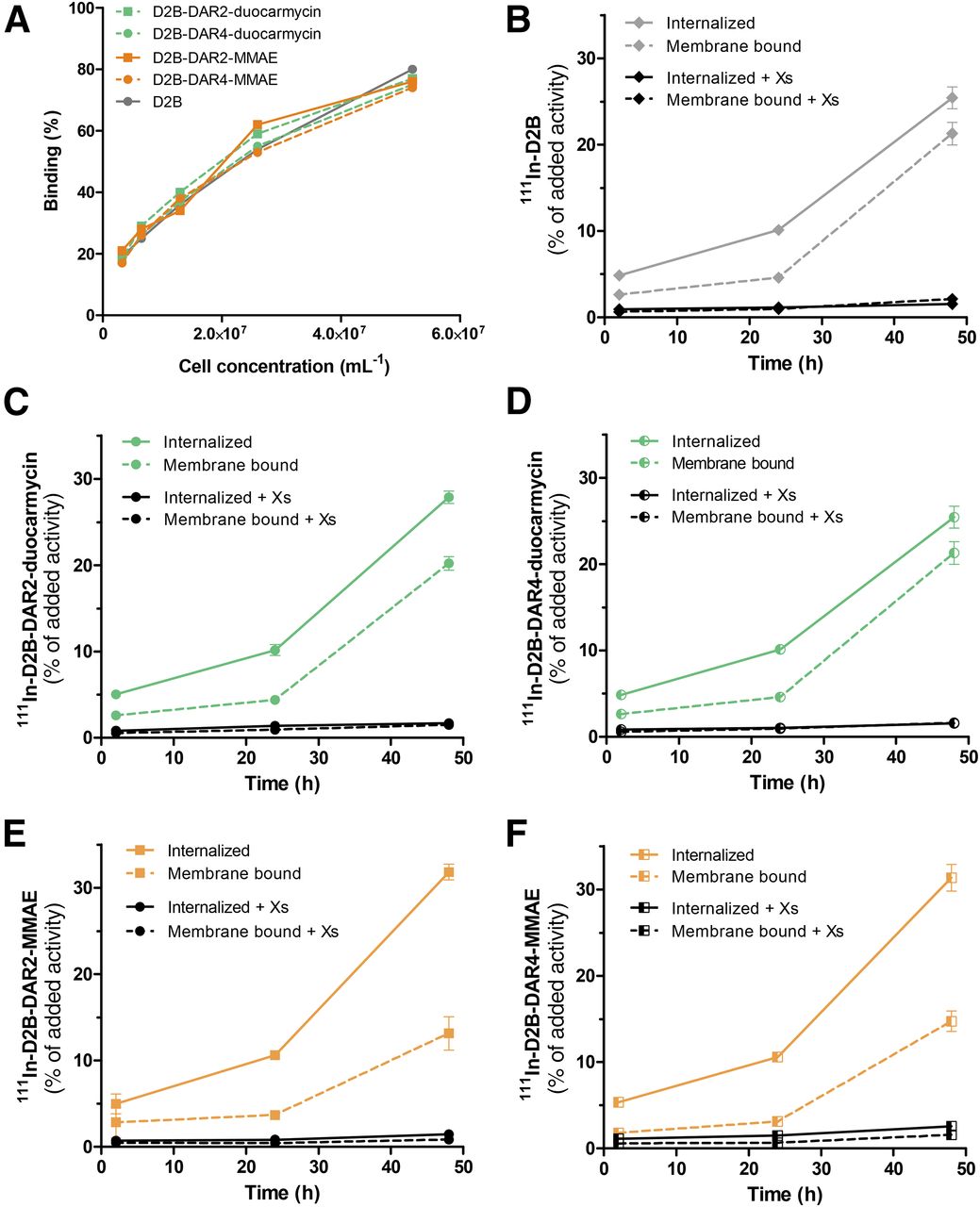
(A) Binding of 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, and 111In-D2B-DAR4-MMAE to PSMA-expressing LS174T-PSMA cells. No significant difference in immunoreactivity in any of the 5 agents was observed. (B–F) Internalization kinetics of 111In-D2B (B), 111In-D2B-DAR2-duocarmycin (C), 111In-D2B-DAR4-duocarmycin (D), 111In-D2B-DAR2-MMAE (E), and 111In-D2B-DAR4-MMAE (F) in LS174T-PSMA cells. Binding and internalization are presented as percentage of added activity after 48 h of incubation (mean ± SD).
111In-D2B-ADCs Gradually Internalize into PSMA-Expressing Cells
All radiolabeled D2B-ADCs and native D2B gradually and specifically internalized into PSMA-expressing LS174T-PSMA cells, although absolute binding and internalization were higher for 111In-D2B-DAR2-MMAE and 111In-D2B-DAR4-MMAE than for native 111In-D2B, 111In-D2B-DAR2-duocarmycin, and 111In-D2B-DAR4-duocarmycin. During the first 24 h of incubation, all compounds were mainly membrane-bound. The internalized fraction gradually increased until 48 h of incubation, reaching 25.4% ± 1.3%, 27.9% ± 0.7%, 25.4% ± 1.3%, 31.8% ± 0.9%, and 31.4% ± 1.5% of the added activity of 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, and 111In-D2B-DAR4-MMAE, respectively.
111In-D2B-ADCs Efficiently Accumulate in PSMA-Expressing Tumors
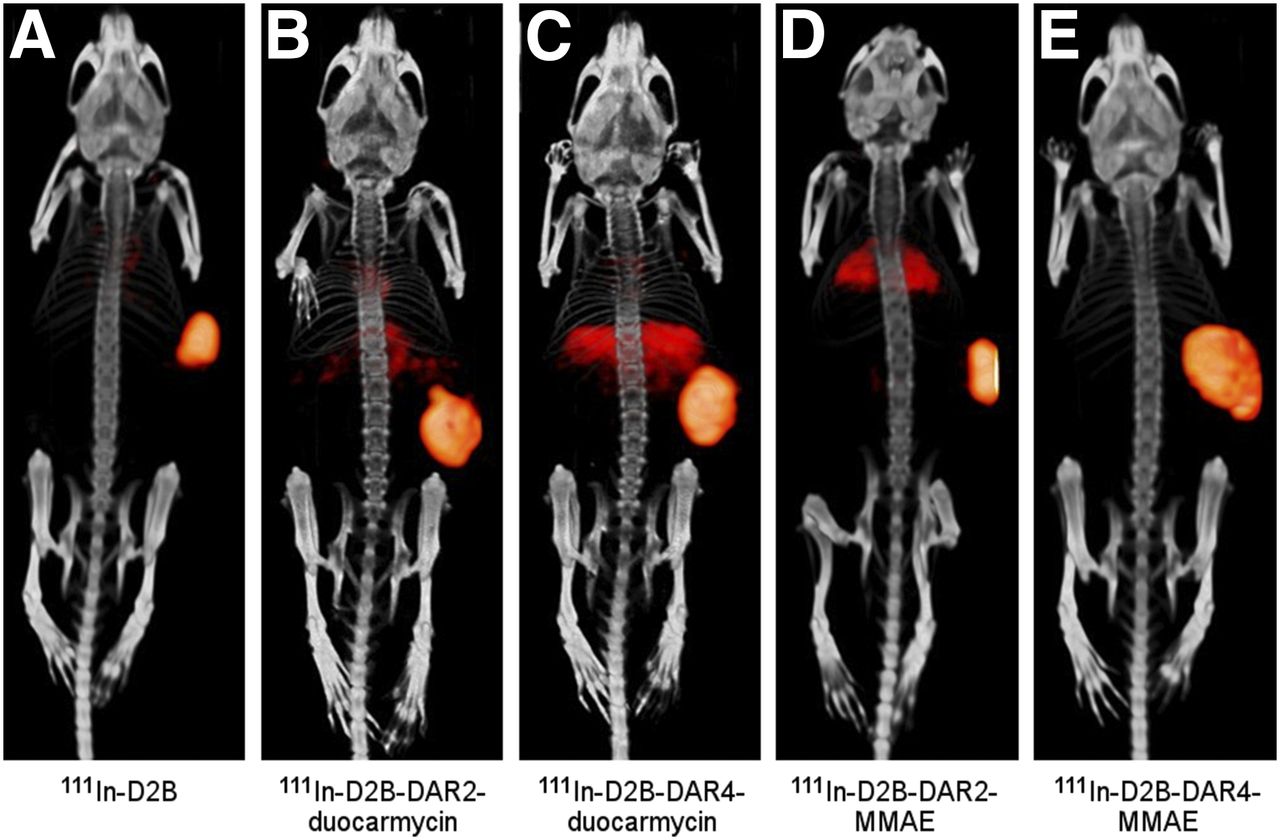
Ten BALB/c nude mice with subcutaneous LS174T-PSMA tumors were imaged with the small-animal SPECT/CT scanner at 3 d after injection of 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, 111In-D2B-DAR4-MMAE, or 111In-D2B. A typical set of SPECT/CT acquisitions is shown in Figure 2. LS174T-PSMA tumors were clearly visualized, with homogenous uptake for all 4 111In-labeled ADCs. Liver uptake seemed to be higher for the duocarmycin variants than for the MMAE variants.

Representative small-animal SPECT/CT images of mice with subcutaneous LS174T-PSMA xenografts on right flank. Images were acquired 3 d after injection of 111In-D2B (17.8 MBq) (A), 111In-D2B-DAR2-duocarmycin (22.1 MBq) (B), 111In-D2B-DAR4-duocarmycin (20.8 MBq) (C), 111In-D2B-DAR2-MMAE (17.2 MBq) (D), and 111In-D2B-DAR4-MMAE (15.2 MBq) (E).
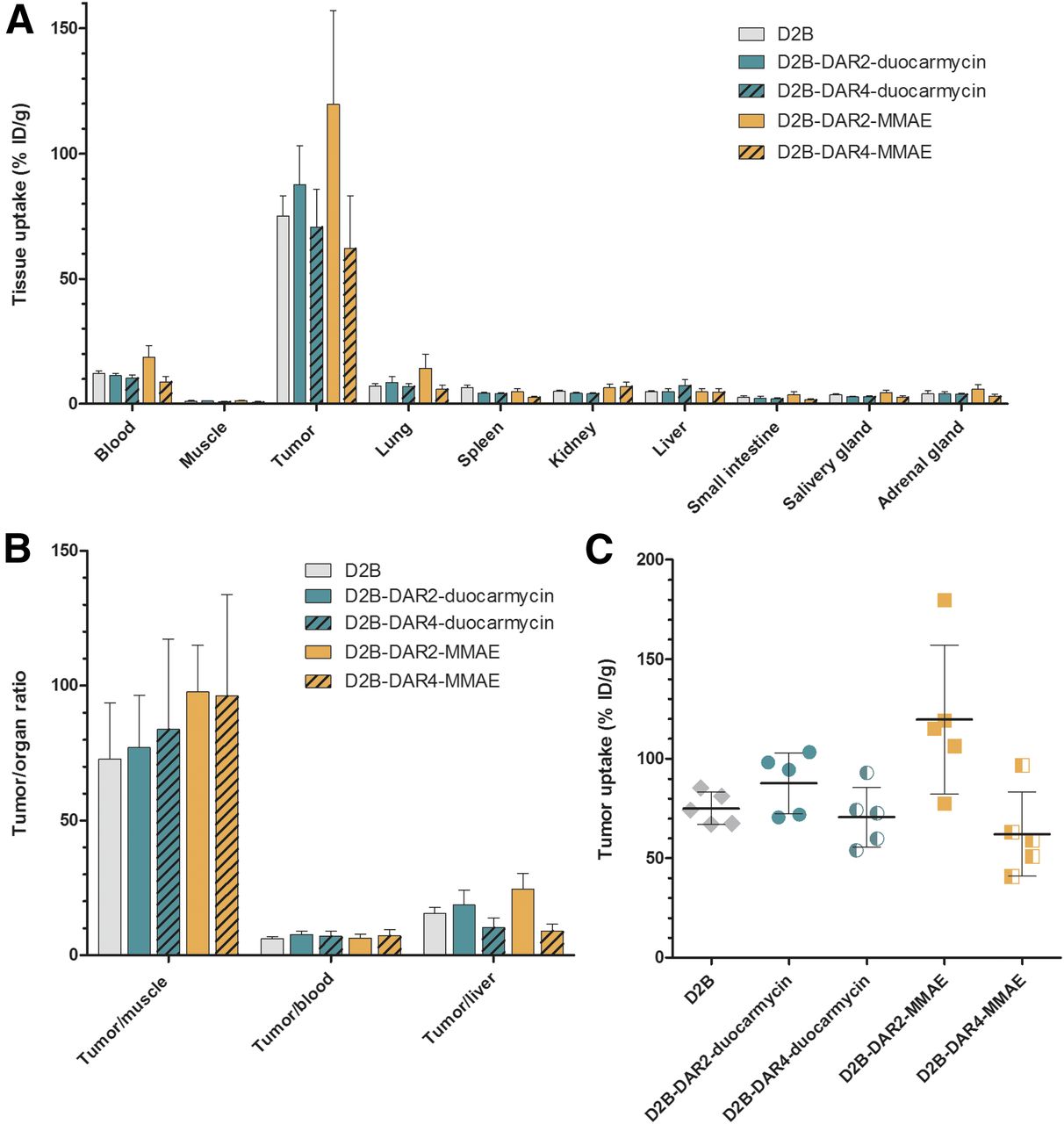
Ex vivo biodistribution revealed that all ADCs showed high accumulation in PSMA-positive LS174T-PSMA tumors (Table 1). Tumor uptake 3 d after injection was significantly higher for 111In-D2B-DAR2-MMAE (119.7 ± 37.4 percentage injected dose per gram [%ID/g]) than for the reference compound, 111In-D2B (75.1 ± 8.1 %ID/g, P < 0.05). In addition, tumor uptake of 111In-D2B-DAR2-MMAE (119.7 ± 37.4 %ID/g) was significantly higher than that of 111In-D2B-DAR4-MMAE (62.1 ± 21.1 %ID/g) or 111In-D2B-DAR4-duocarmycin (70.7 ± 15.1 %ID/g) at 3 d after injection (P < 0.01 and P < 0.05, respectively). No significant difference in tumor uptake was observed between the reference compound, 111In-D2B (75.1 ± 8.1 %ID/g), and 111In-D2B-DAR2-duocarmycin (87.7 ± 15.4 %ID/g) or 111In-D2B-DAR4-duocarmycin (70.7 ± 15.1 %ID/g) (Fig. 3).
Biodistribution at 3 Days After Injection
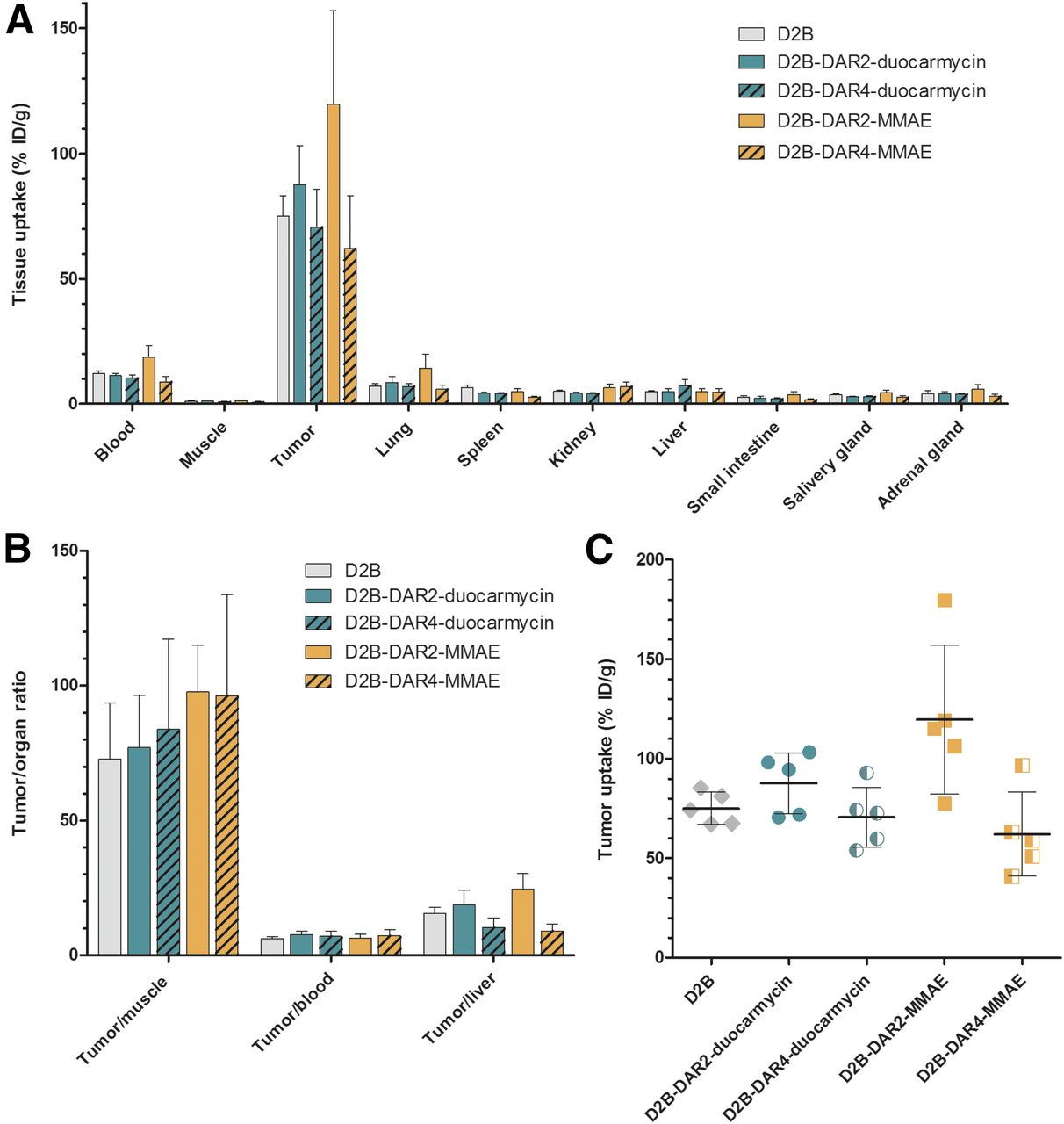
(A) Biodistribution of 111In-D2B, 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, and 111In-D2B-DAR4-MMAE in PSMA-expressing LS174T-PSMA tumors and healthy organs at 3 d after injection (n = 5 mice per group). (B) Tumor-to-organ ratios of all 4 agents and control agent 111In-D2B. (C) Tumor uptake per mouse and targeting agent.
The uptake of 111In-D2B-DAR2-duocarmycin and 111In-D2B-DAR4-duocarmycin remained low in most normal tissues; hepatic uptake reached 5.0 ± 1.2 %ID/g and 7.4 ± 2.4 %ID/g, respectively, at 3 d after injection, whereas kidney uptake reached 4.2 ± 0.5 %ID/g and 4.1 ± 0.4 %ID/g, respectively. For 111In-D2B-DAR2-MMAE and 111In-D2B-DAR4-MMAE, hepatic uptake was 4.9 ± 1.3 %ID/g and 4.8 ± 1.3 %ID/g, respectively (n = 5), whereas kidney uptake was 6.5 ± 1.5 %ID/g and 7.0 ± 1.7 %ID/g, respectively. Hepatic uptake did not significantly differ between the reference compound, 111In-D2B, and 111In-D2B-DAR2-duocarmycin, 111In-D2B-DAR4-duocarmycin, 111In-D2B-DAR2-MMAE, or 111In-D2B-DAR4-MMAE.
111In-D2B-DAR2-MMAE showed significantly higher blood levels at 3 d after injection (18.6 ± 4.7 %ID/g) than did the reference compound, 111In-D2B (12.2 ± 1.1 %ID/g, P < 0.01), or 111In-D2B-DAR4-MMAE (8.8 ± 2.2 %ID/g, P < 0.001). In addition, 111In-D2B-DAR2-MMAE showed significantly higher blood levels than did 111In-D2B-DAR2-duocarmycin (11.4 ± 0.9 %ID/g, P < 0.01) or 111In-D2B-DAR4-duocarmycin (10.3 ± 1.5 %ID/g, P < 0.001). No significant difference in tracer concentration in the blood was observed between the reference compound, 111In-D2B, and 111In-D2B-DAR4-MMAE, 111In-D2B-DAR2-duocarmycin, or 111In-D2B-DAR4-duocarmycin.
However, tumor-to-blood ratios did not significantly differ among 111In-D2B-DAR2-duocarmycin (6.5 ± 1.3), 111In-D2B-DAR4-duocarmycin (7.3 ± 2.3), 111In-D2B-DAR2-MMAE (7.7 ± 1.2), and 111In-D2B-DAR4-MMAE (7.0 ± 1.9) at 3 d after injection or between these compounds and the reference compound, 111In-D2B (6.2 ± 0.7).
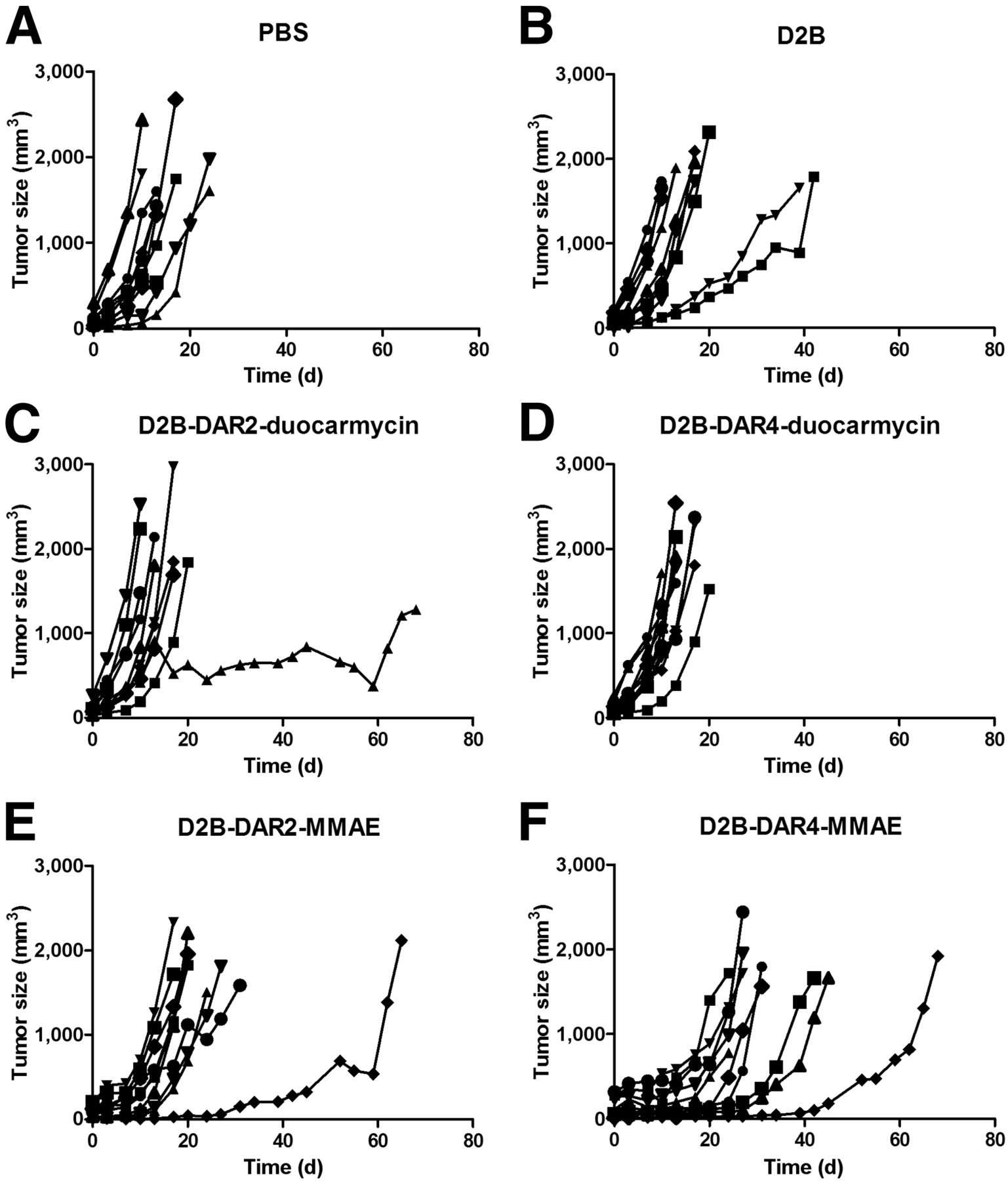
Tumor Growth Is Inhibited by 111In-D2B-ADCs
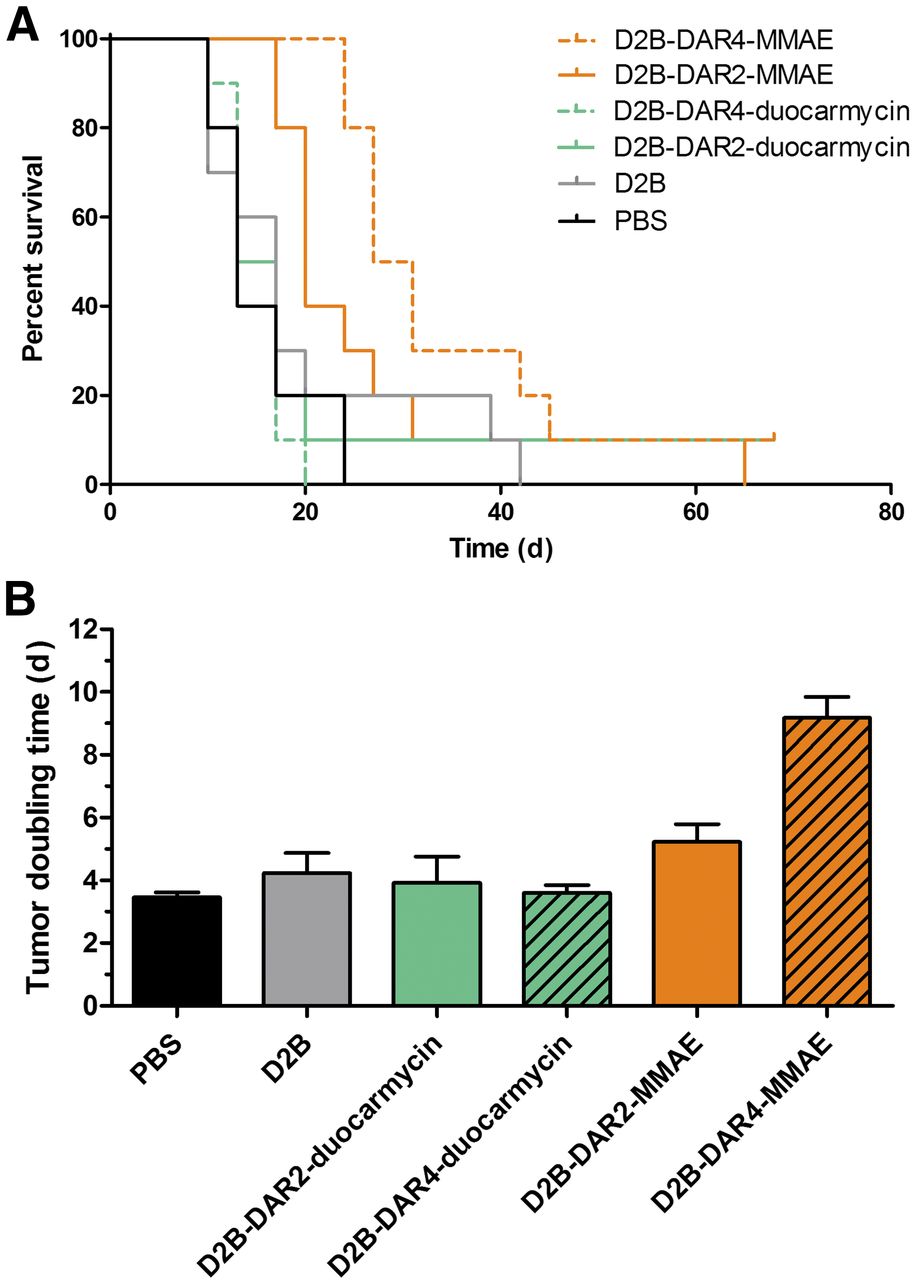
Body weight and tumor size did not significantly differ among the treatment groups at the beginning of treatment. Tumor growth after treatment with PBS, unmodified D2B, D2B-DAR2-duocarmycin, D2B-DAR4-duocarmycin, D2B-DAR2-MMAE, and D2B-DAR4-MMAE in individual mice is depicted in Figure 4. The longest tumor-doubling time was observed after treatment with D2B-DAR4-MMAE (9.2 ± 2.1 d), and was significantly longer than that in the PBS control mice (3.5 ± 0.5 d, P < 0.001) or in the mice treated with unmodified D2B (4.2 ± 2.0 d, P < 0.001), D2B-DAR2-MMAE (5.2 ± 1.8 d, P < 0.001), D2B-DAR2-duocarmycin (3.9 ± 2.7 d, P < 0.001), or D2B-DAR4-duocarmycin (3.6 ± 0.8 d, P < 0.001) (Fig. 5B). Tumor-doubling time did not significantly differ among mice that received PBS only, unmodified D2B, D2B-DAR2-MMAE, D2B-DAR2-duocarmycin, or D2B-DAR4-duocarmycin.

Tumor size of individual mice in each group after treatment with PBS (A), D2B (B), D2B-DAR2-duocarmycin (C), D2B-DAR4-duocarmycin (D), D2B-DAR2-MMAE (E), or D2B-DAR4-MMAE (F) (5 mg/kg).
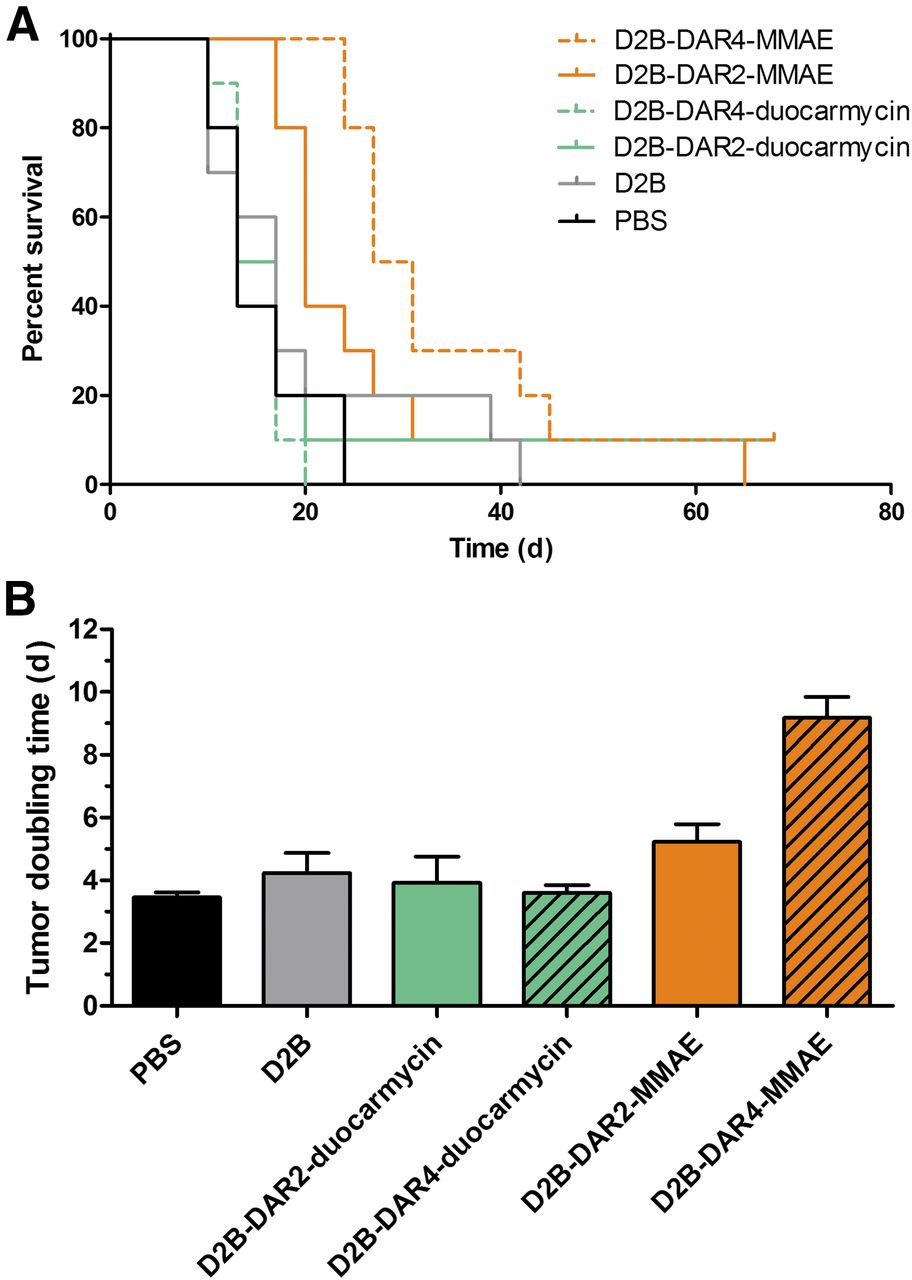
(A) Kaplan–Meier survival plot of mice with subcutaneous LS174T-PSMA tumors treated with PBS, D2B, D2B-DAR2-duocarmycin, D2B-DAR4-duocarmycin, D2B-DAR2-MMAE, or D2B-DAR4-MMAE. (B) Tumor-doubling time.
In control mice that received PBS, median survival was 13 d. In mice treated with D2B in its unmodified form, median survival (17 d) was not significantly different, indicating no therapeutic effect. Treatment with D2B-DAR2-duocarmycin (median survival 15 d) and D2B-DAR4-duocarmycin (median survival 13 d) did not prolong median survival significantly compared with treatment with PBS or D2B alone (Mantel–Cox test, P = 0.8884) (Fig. 5A). Treatment with D2B-DAR4-MMAE and D2B-DAR2-MMAE significantly improved median survival from 13 to 29 d (P < 0.001) and 20 d (P < 0.01), respectively (Fig. 5A). No significant difference in median survival was observed between treatment with D2B-DAR4-MMAE and treatment with D2B-DAR2-MMAE. Treatment with D2B-DAR4-MMAE resulted in significantly prolonged median survival compared with treatment with D2B-DAR2-duocarmycin (P < 0.05) or D2B-DAR4-duocarmycin (P < 0.001). Moreover, treatment with D2B-DAR2-MMAE resulted in significantly prolonged median survival compared with treatment with D2B-DAR2-duocarmycin (P < 0.01) or D2B-DAR4-duocarmycin (P < 0.001).
DISCUSSION
This report describes the in vitro and in vivo characterization of 4 novel site-specifically conjugated anti-PSMA ADCs consisting of the anti-PSMA mAb 111In-D2B conjugated to either 2 or 4 molecules of duocarmycin or MMAE.
So far, most of the tested anti-PSMA ADCs have been based on random conjugation of the drug to the antibody, resulting in mixtures of ADCs with different molar DARs, linked at different sites to the antibody and with different pharmacokinetics, immunoreactivity, charge, size, and stability (13). In addition, in randomly conjugated ADCs, DAR0 species can occur, which do not contribute to drug-mediated antitumor activity and compete for antigen-binding sites with higher DAR species (19). Several strategies have been developed to improve the therapeutic efficacy of ADCs, among which is the site-specific conjugation of antibodies to cytotoxic drugs (13,20). This strategy is based on providing an anchor point in the antibody molecule for attachment of the drug, allowing specific conjugation to a certain point in the molecular structure and preventing random binding. It was shown previously that site-specific conjugation minimizes the heterogeneity of ADCs, rendering the properties of ADCs more predictable and allowing consistent conjugate production (20). In addition, it was shown that engineered site-specific ADC conjugates were better tolerated than traditional ADC conjugates in both rat and cynomolgus monkey toxicity models (13,20,21), presumably because the engineered ADCs do not disintegrate in vivo as much as the traditional ADCs.
Recently, van Geel et al. developed a method to site-specifically conjugate ADCs using the antibody’s glycan at asparagine-297 as an anchor (GlycoConnect method; Synaffix BV) (12). Major advantages of this method are that it is applicable to any type of recombinant IgG isotype, irrespective of mammalian expression system and linker–payload combination (12). ADCs produced by this method were found to be homogeneous and hydrolytically stable while displaying negligible aggregation (12). In addition, immunoreactivity is preserved entirely, as the drug is conjugated to the antibody at a site not affecting antigen binding or affinity. Glycan-remodeled trastuzumab showed significantly better antitumor activity than the clinically available ADC Kadcyla, demonstrating the superiority of GlycoConnect ADC over a randomly conjugated ADC based on the same components (12).
In addition to site-specific conjugation, it has been shown that the antitumor effect of ADCs can be modified by varying the drug loading per antibody (22). Hamblett et al. demonstrate that a single dose of an ADC with a DAR of 2 can be less effective than treatment with an ADC with a DAR of 4. In contrast, it was shown that ADCs containing higher-DAR species (DAR6 and DAR8) are generally less stable and clear more rapidly than ADCs containing lower-DAR species, depending on the conjugation method (22). Therefore, in the present study, we concentrated on evaluating the antitumor activity of DAR2 and DAR4 species.
Previously, the number of drug molecules that can be conjugated to an antibody molecule was shown to be limited, as it can reduce the antigen-binding capacity by blocking binding sites (23). In the present study, in vitro, the immunoreactivity of the anti-PSMA mAb 111In-D2B was preserved on site-specific conjugation with MMAE or duocarmycin, and all 4 ADC conjugates showed binding to PSMA-expressing cells comparable to that of 111In-D2B IgG, thereby demonstrating that site-specific conjugation does not interfere with antigen binding. The binding capacity to PSMA-expressing tumor cells did not significantly differ among the various anti-PSMA ADCs, nor was it significantly different from that of the unmodified 111In-D2B antibody.
In vivo, small-animal SPECT/CT images allowed high-contrast visualization of PSMA-expressing tumors for all 4 ADC variants. Tumor visualization was comparable to that using the reference compound 111In-D2B, confirming that in vivo tumor targeting remains intact on conjugation to the ADCs. 111In-labeled anti-PSMA ADCs showed a biodistribution profile comparable to that of 111In-D2B, with high tumor uptake and relatively low uptake in healthy organs. The highest tumor uptake, however, was reached in the 111In-D2B-DAR2-MMAE variant, which had significantly higher uptake than unmodified 111In-D2B or the 111In-D2B-DAR4 variant, as might be explained by altered pharmacokinetics in the different DAR variants in vivo. Compared with the other variants, the 111In-D2B-DAR2-MMAE variant showed significantly prolonged blood circulation, which may have resulted in increased tumor uptake. Differences in blood circulation have been described previously for ADCs with different DARs. ADCs with higher DARs, especially MMAE ADCs, clear more rapidly from the circulation than ADCs with lower DARs (22, 24). The explanation for this observation is most likely the enhanced liver clearance of the higher conjugated species due to the hydrophobic nature of the payload, leading to alterations in pharmacokinetics.
Subsequently, the antitumor activity of MMAE- and duocarmycin-based anti-PSMA ADCs in terms of median survival and tumor-doubling time was compared with that of mice that received PBS or D2B without the addition of a cytotoxic drug. Both MMAE-based anti-PSMA ADCs significantly prolonged median survival. Because the DAR4 variant carries more-cytotoxic drugs than the DAR2 variant, the DAR4 variant was expected to have higher antitumor activity than the DAR2 variant (22). Indeed, the tumor-doubling time of D2B-DAR4-MMAE was significantly higher than that of D2B-DAR2-MMAE. However, no significant difference in prolongation of median survival was observed between D2B-DAR2-MMAE and D2B-DAR4-MMAE. An explanation for this finding could be the prolonged circulation time and increased tumor uptake of the D2B-DAR2 variant, leading effectively to relatively more intracellular free toxin and thereby compensating for the absolute lower number of cytotoxic drugs carried.
In contrast to the D2B-DAR2-MMAE and D2B-DAR4-MMAE variants, no significant difference between the DAR2 and DAR4 variants of the duocarmycin-based anti-PSMA ADC was observed—neither a difference leading to prolonged tumor-doubling time nor a difference in median survival. Although MMAE inhibits microtubule assembly, ultimately precluding mitosis and causing apoptosis in dividing tumor cells, duocarmycin acts in both dividing and nondividing cells because it alkylates DNA, resulting in DNA damage, mitochondrial stress, impaired DNA transcription, apoptosis, and ultimately cell death. On the basis of these mechanisms, both duocarmycin and MMAE are expected to have high antitumor activity. The lack of antitumor activity in the duocarmycin-based ADCs might be explained by the relatively slower onset (in rapidly dividing cells such as those in the present study) of DNA-damaging agents in duocarmycin-based ADCs than in micotubuli inhibitors. Moreover, the lower antitumor activity of duocarmycin might stem from this drug’s need to reach the nucleus before becoming active whereas MMAE acts in the cytoplasm. In addition, structural changes due to conjugation of the drug to the antibody might cause reduced internalization of the ADC or diminished lysosomal degradation (25). In the present study, mice received a single 5 mg/kg dose of the ADC. Although no maximum tolerable dose was determined here, Owonikoko et al. reported that side effects such as neuropathy occur at 15 mg/kg doses of BMS936561, an ADC consisting of an anti-CD70 antibody covalently linked to duocarmycin (26). In that study, the authors reported 8 mg/kg to be the best-tolerated dose (26), suggesting that upscaling of the dose of the duocarmycin-based ADCs evaluated here might be well tolerated. The therapeutic effect of these duocarmycin-based ADCs should be evaluated at higher doses. Aggressive growth of PSMA-expressing LS174T-PSMA tumor cells may have masked less pronounced treatment effects. Therefore, in future therapy studies, a tumor model that shows a less aggressive growth pattern should be evaluated as well. Finally, it is important to point out that although we treated the tumor-bearing mice with only 1 dose of our D2B-DAR4-MMAE ADC, we obtained a doubling of the survival time in our fast-growing tumor model. Because it is well known that therapeutic strategies to destroy tumor cells are based on repeated administration of the drugs to improve the antitumor efficacy, in future the efficacy of this new treatment should be evaluated with repeated ADC administration cycles to increase the antitumor response.
CONCLUSION
In this study, site-specifically conjugated MMAE- and duocarmycin-based anti-PSMA ADCs with different DARs have been characterized. Although the duocarmycin-based anti-PSMA ADCs did not show antitumor activity at the dose used in this study, the site-specifically conjugated anti-PSMA ADCs D2B-DAR2-MMAE and D2B-DAR4-MMAE effectively inhibited growth of PSMA-expressing tumors and prolonged survival of the mice. The highest antitumor activity was achieved with the MMAE-based anti-PSMA ADC containing 4 molecules of MMAE per antibody molecule.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Bianca Lemmers-van de Weem, Iris Lamers-Elemans, Kitty Lemmens-Hermans, and Karin de Haas-Cremers for technical assistance with the animal experiments. Furthermore, we thank Synaffix for making the conjugation technology available.
Footnotes
Published online Nov. 16, 2017.
- © 2018 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication May 18, 2017.
- Accepted for publication September 11, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}