


Abstract
The objective of the present PET study was to compare the effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on serotonergic neuronal systems and mitochondrial complex I (MC-I) activity with that of dopamine in conscious rhesus monkeys (Macaca mulatta). Methods: A Parkinson disease monkey model was prepared by repeated administration of MPTP. For the PET measurements, normal and MPTP-treated conscious monkeys received an intravenous injection of 11C-DASB for serotonin transporter, 18F-MPPF for serotonin 1A receptor, 11C-PE2I for dopamine transporter, 11C-6MemTyr for dopamine synthesis, 11C-raclopride for dopamine D2 receptor, or 18F-BCPP-EF for MC-I. Serotonin and dopamine parameters were calculated using time–activity curves in the cerebellum as the input function. The total distribution volume of 18F-BCPP-EF was assessed using Logan plot graphical analysis with metabolite-corrected plasma as the input function. Results: MPTP-induced diffuse reductions in MC-I activity were observed throughout the brain, except the cerebellum. Significant reductions in the presynaptic dopamine parameters—dopamine transporter and dopamine synthesis—were detected in the striatum and substantia nigra pars compacta of MPTP-treated monkeys, whereas no significant differences in postsynaptic dopamine D2 receptor binding were observed. Serotonin transporter binding was reduced by MPTP not only in striatal regions but also in extrastriatal regions. In contrast, serotonin 1A receptor binding was unaffected by MPTP anywhere in the brain. In the cortex, the reduction of serotonin transporter binding correlated with that of MC-I. Conclusion: The results obtained by multiparametric PET measurements in a Parkinson disease monkey model demonstrated that chronic MPTP treatment induced reductions not only in the dopaminergic system in the nigrostriatal pathway but also in serotonin transporter in the cortical and subcortical regions. These results suggest that the neurotoxicity of MPTP is not exclusive to the nigrostriatal pathway, as predicted from MC-I damage in the extrastriatal regions of the brain.
Parkinson disease is the second most common neurodegenerative disease; its prevalence is 1% of the population older than 55 y, and its etiology is unknown (1). A pathologic hallmark of idiopathic Parkinson disease is the presence of Lewy bodies and neurites, which are intracytoplasmic insoluble and aggregated proteins, including α-synuclein (2). Parkinson disease shows progressive degradation of nigrostriatal pathways, with selective loss of dopaminergic neurons in the substantia nigra pars compacta, resulting in movement disorders. These clinical symptoms of Parkinson disease are induced after 40%–50% of neurons in the substantia nigra pars compacta have been lost and dopamine in the striatum has been reduced to approximately 20% of normal levels (3).
Although the direct causes of this selective neurodegeneration in Parkinson disease remain unclear, mitochondrial dysfunction has emerged as a common aspect of its pathogenesis (4,5). Tissues with high energy demands, such as brain, kidney, and skeletal and cardiac muscles, contain large numbers of mitochondria. In mammalian cells, the electron transport chain in mitochondria consists of 5 complexes (I–V) that produce adenosine triphosphate, and mitochondrial complex I (MC-I) (nicotinamide adenine dinucleotide [reduced form]:ubiquinone oxidoreductase, Enzyme Commission number 1.6.5.3) forms the first and rate-limiting step of overall respiratory activity and oxidative phosphorylation. In addition, MC-I is a site of reactive oxygen species production and is particularly vulnerable to oxidative damage (6). The loss of MC-I catalytic activity in the electron transport chain was reported in multiple tissues from patients exhibiting sporadic Parkinson disease (4,5) with increased oxidative stress (7).
Exposure to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) causes a syndrome that mimics Parkinson disease symptoms and selective dopaminergic neurodegeneration in the substantia nigra pars compacta (8). 1-methyl-4-phenylpyridinium, a metabolite of MPTP by monoamine oxidase B in glial cells, selectively inhibits MC-I activity, leading to suppression of adenosine triphosphate synthesis and generation of reactive oxygen species (8). MC-I dysfunction has been identified in postmortem brain and biopsy tissue samples from Parkinson disease patients (4,5). However, to our knowledge, MC-I dysfunction had not been examined noninvasively until our recent PET study on the living brains of MPTP-treated cynomolgus monkeys (Macaca fascicularis) (9).
Abnormalities in the nigrostriatal dopamine pathway may be imaged in Parkinson disease patients using PET probes for the dopamine transporter and dopamine synthesis (10). Furthermore, we recently designed a novel PET probe, 2-tert-butyl-4-chrolo-5-{6-[2-(2-18F-fluoroethoxy)-ethoxy]-pyridin-3-ylmethoxy}-2H-pyridazin-3-one (18F-BCPP-EF), for quantitative PET imaging of MC-I in vivo and demonstrated its capability for diagnostic, prognostic, and therapeutic monitoring of neurodegenerative diseases using monkey models of stroke with middle cerebral artery occlusion and reperfusion (11), of Alzheimer disease with amyloid-β deposition (12), and of Parkinson disease prepared by MPTP (9), all of which are known to be related to impaired brain mitochondrial function in humans.
In our previous study on isoflurane-anesthetized cynomolgus monkeys, we observed MPTP-induced impairment of MC-I activity not only in the nigrostriatal pathway with abundant dopamine neurons (9) but also in extrastriatal nondopaminergic regions such as the cortex. Over the past 2 decades, Parkinson disease has been increasingly recognized as a disease not only with motor impairment but also with nonmotor symptoms such as depression and cognitive deficits, which are associated with changes in other monoamines (noradrenaline and serotonin) in extrastriatal regions (13). Previous findings from MPTP-treated rhesus monkeys suggested that the brain monoamine systems of dopamine, noradrenaline, and serotonin are susceptible to MPTP (14,15). In addition, despite some controversy, a deficiency in serotonin was reported in the cortical regions of postmortem brains from Parkinson disease patients (16). PET imaging has revealed diffuse reductions in serotonin transporter availability throughout the brain in nondepressed Parkinson disease patients (17), as well as reductions in serotonin 1A receptor binding in both nondepressed and depressed Parkinson disease patients (18). In contrast, other studies have argued that these alterations in the serotonergic system may be partly attributable to 3,4-dihydroxy-l-phenylalanine treatment and not to primary neuropathologic alterations (19).
The present study used a Parkinson disease monkey model prepared by repeated intravenous administration of MPTP over several months (9). We evaluated 11C-3-amino-4-(2-dimethylaminomethylphenylsulfanyl)-benzonitrile (11C-DASB) availability to serotonin transporter (20) and 4-(2′-methoxyphenyl)-1-[2′-(N-2″-pyridinyl)-p-18F-fluorobenzamido]ethylpiperazine (18F-MPPF) binding to serotonin 1A receptor (21) in parallel with dopaminergic parameters and MC-I activity in conscious monkeys.
MATERIALS AND METHODS
Animals and Drugs
Twelve young-adult male rhesus monkeys (Macaca mulatta: age range, 7.1–9.4 y old; weight range, 4.0–6.0 kg) were maintained and handled in accordance with the recommendations of the U.S. National Institutes of Health and the guidelines of the Central Research Laboratory, Hamamatsu Photonics K.K. The experiments were approved by the Ethical Committee of the Central Research Laboratory, Hamamatsu Photonics K.K. (approval HPK-2015-15).
MR images of the monkeys were obtained with a 3.0-T scanner (Signa Excite HDxt 3.0T; GE Healthcare Japan) using a 3-dimensional spoiled gradient echo sequence under pentobarbital anesthesia.
Kryptofix222 and K2CO3·1.5H2O were from Merck Milipore. Anhydrous CH3CN, dimethylformamide, dimethyl sulfoxide, silver trifluoromethanesulfonate, Tris(dibenzyldeneacetone)dipalladium(0), tri-o-tolylphosphine, and MPTP were from Sigma-Aldrich Japan. Precursors of 11C-DASB, 18F-MPPF, 6-11C-methyl-m-tyrosin (11C-6MemTyr), 11C-raclopride, 11C-N-(3-iodoprop-2E-enyl)-2β-carbomethoxy-3β-(4-methyl-phenyl) nortropane (11C-PE2I), 18F-BCPP-EF, and their corresponding standard compounds were from NARD Institute Ltd.
PET Ligand Syntheses
11C-DASB for serotonin transporter was labeled by 11C-N-methylation of its nor-compound with 11C-methyl triflate (20). Radiochemical purity was greater than 99%, and specific radioactivity was 47.6 ± 11.1 GBq/μmol. 18F-MPPF for serotonin 1A receptor was prepared by nucleophilic 18F-fluorination of its nitro precursor (21). Radiochemical purity was more than 99%, and specific radioactivity was 90.4 ± 25.6 GBq/μmol. 11C-PE2I for dopamine transporter was labeled by 11C-O-methylation of its nor-compound with 11C-methyl triflate. Radiochemical purity was greater than 98%, and specific radioactivity was 117.1 ± 42.9 GBq/μmol. 11C-6MemTyr for dopamine synthesis was labeled using a rapid palladium(0)-mediated cross-coupling reaction of 11C-methyl iodide and its corresponding boronate precursor (22,23). Radiochemical purity was greater than 99%, and specific radioactivity was 71.6 ± 37.4 GBq/μmol. 11C-raclopride for the postsynaptic dopamine D2 receptor was labeled by 11C-N-methylation of its nor-compound with 11C-methyl triflate. Radiochemical purity was greater than 98%, and specific radioactivity was 65.4 ± 15.8 GBq/μmol. 18F-BCPP-EF for MC-I was prepared by nucleophilic 18F-fluorination of the corresponding precursor as reported previously (9,12,13). Radiochemical purity was more than 99%, and specific radioactivity was 58.9 ± 7.9 GBq/μmol.
Parkinson Disease Model
Six of 12 young-adult male rhesus monkeys (M. mulatta) were used to prepare a Parkinson disease model according to our previous studies (9). To create bilateral striatal lesions, MPTP at doses ranging between 0.2 and 0.4 mg/kg of the free base in phosphate-buffered saline was injected intravenously over a 4-mo period until stable Parkinsonian syndrome was observed. The total doses of MPTP administered were between 8 and 14 mg/kg. To avoid the potential for spontaneous recovery and direct inhibition of 18F-BCPP-EF binding to MC-I by MPTP, PET measurements were started at least 2 mo after the last treatment with MPTP.
PET Measurements
Six normal and 6 MPTP-treated monkeys underwent 6 PET scans—using 11C-DASB, 18F-MPPF, 11C-PE2I, 11C-6MemTyr, 11C-raclopride, and 18F-BCPP-EF—while conscious. The order of scanning with each ligand was randomized. Scanning with arterial blood sampling was performed with 18F-BCPP-EF only, as reported previously (9,12,13). After an overnight fast, a venous cannula for ligand injection and an arterial cannula for blood sampling were inserted into both inferior limbs under temporal anesthesia using 2.0%–2.5% sevoflurane (1:2 O2:N2O). After the animal had completely recovered from the anesthesia, its head was rigidly fixed to the upper frame of a monkey chair using an acrylic head-restraint. The animal, sitting in the restraining chair, was placed in the gantry at a fixed position, with stereotactic coordinates aligned parallel to the orbitomeatal plane. Following a 60-min transmission scan using a 68Ge–68Ga rotation rod source, dynamic emission scanning with each probe was performed for 91 min after injection using a high-resolution animal PET scanner (SHR-38000; Hamamatsu Photonics K.K.). The data were reconstructed using a dynamic row-action maximum-likelihood algorithm with a 2.0-mm gaussian postprocessing filter (24), and attenuation was corrected using the transmission scan data. Individual PET and MR images were coregistered. Volumes of interest (VOIs) were drawn manually over brain regions on the MR images, in addition to regional information from BrainMaps.org (25), and the time–activity curve of each probe was measured for kinetic analyses.
PET Data Analysis
For quantitative analysis of 18F-BCPP-EF, arterial blood samples were obtained every 8 s until 64 s after tracer injection and then at 90 and 150 s and at 4, 6, 10, 20, 30, 45, 60, 75, and 90 min. Blood samples were centrifuged to separate plasma and were weighed, and the radioactivity was measured using a γ-counter (1480 Wizard; Perkin Elmer). For metabolite analysis, ethanol was added to plasma samples (1/1 sample/ethanol [v/v]) obtained at 16, 40, and 64 s and then at 6, 10, 30, 45, 65, 75, and 90 min after injection, followed by centrifugation. The supernatants were developed using thin-layer chromatography plates (silica gel 60 F254; Merck) with ethyl acetate as the mobile phase. The ratio of unmetabolized fraction was assessed using a phosphor imaging plate and a bioimaging analyzer (FLA-7000; Fuji Film). The arterial input function for unmetabolized 18F-BCPP-EF was calculated using data obtained by a correction of the ratio of unmetabolized fraction to total radioactivity.
The kinetics of 18F-BCPP-EF were analyzed using Logan graphical analysis (26) with PMOD software (PMOD Technologies Ltd.), as reported previously (12,13). PMOD software was also used to calculate the nondisplaceable binding potential (BPND) of 11C-DASB, 18F-MPPF, and 11C-PE2I via a simplified reference tissue model (27), with the time–activity curve in the cerebellum applied as the indirect input function, as well as to perform multiple-time graphical analysis of 11C-6MemTyr (9,22,23,28,29), with the time–activity curve in the occipital cortex applied as the input function (30).
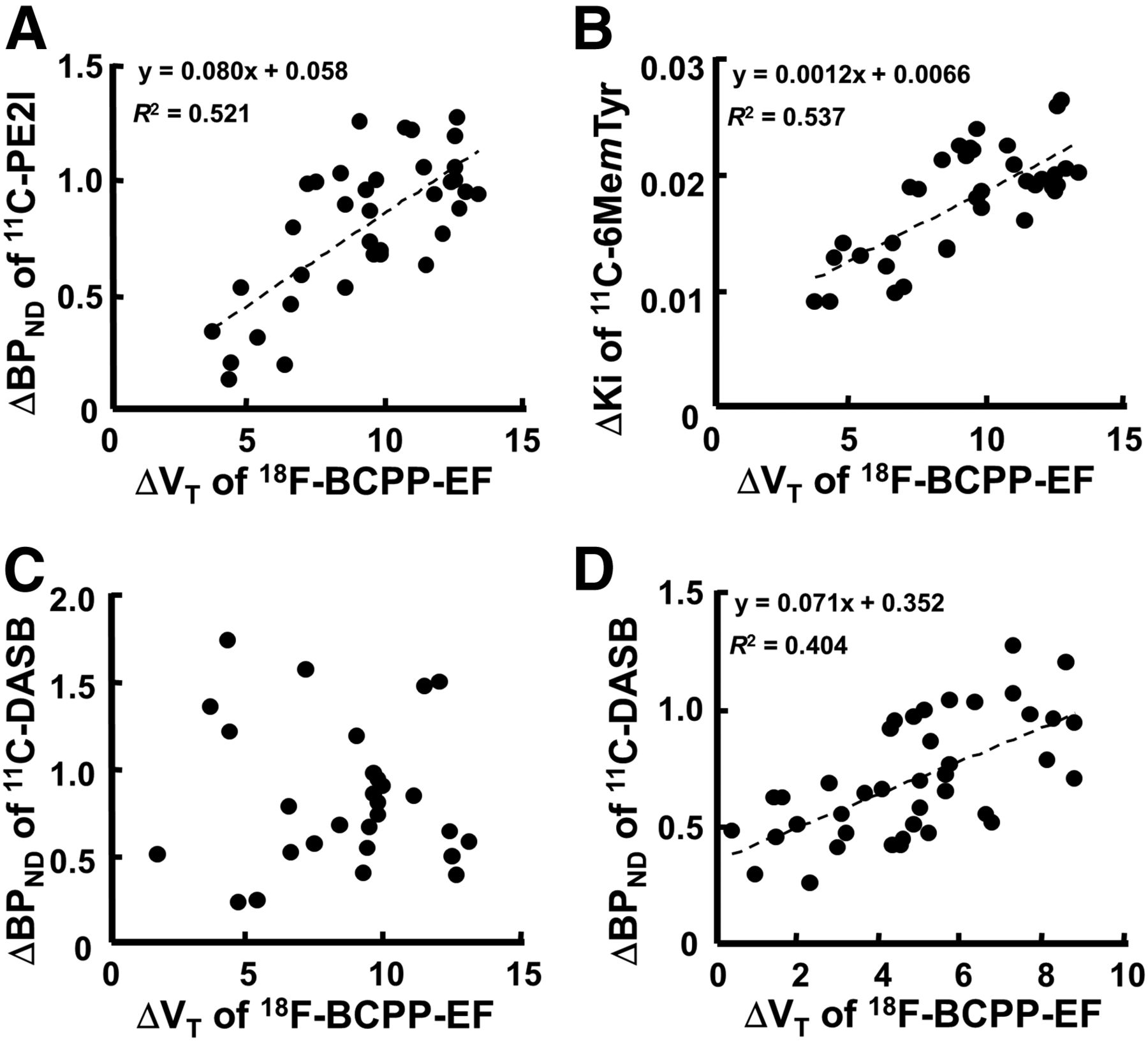
Correlation analysis was performed of the degree of MPTP-induced reduction in BPND for 11C-PE2I in nigrostriatal regions (caudate, putamen, and substantia nigra pars compacta), the degree of MPTP-induced reduction in inhibition constant (Ki) for 11C-6MemTyr in nigrostriatal regions, and the degree of MPTP-induced reduction in BPND for 11C-DASB in nigrostriatal and cortical regions (frontal, occipital, temporal, and parietal) against the degree of MPTP-induced reduction in distribution volume (VT) for 18F-BCPP-EF. The same VOIs, determined with the aid of individual MR images, were set on the parametric PET images of normal and MPTP-treated monkeys to obtain Ki for 11C-6MemTyr, BPND for 11C-DASB, and VT for 18F-BCPP-EF. Then, averaged Ki, BPND, and VT for each VOI of 6 normal monkeys were calculated and subtracted from each VOI of individual MPTP-treated monkeys to determine the degree of MPTP-induced reduction as ΔBPND for 11C-PE2I, ΔKi for 11C-6MemTyr, ΔBPND for 11C-DASB, and ΔVT for 18F-BCPP-EF. ΔKi and ΔBPND in each VOI were plotted against the corresponding ΔVT as reported previously (9).
Statistical Analysis
Results are expressed as mean ± SD. Comparisons between conditions were made by unpaired 2-tailed Student t testing, and correlations were tested by a single regression analysis using KaleidaGraph (Synergy Software). A probability level of less than 5% (P < 0.05) was considered to indicate significance.
RESULTS
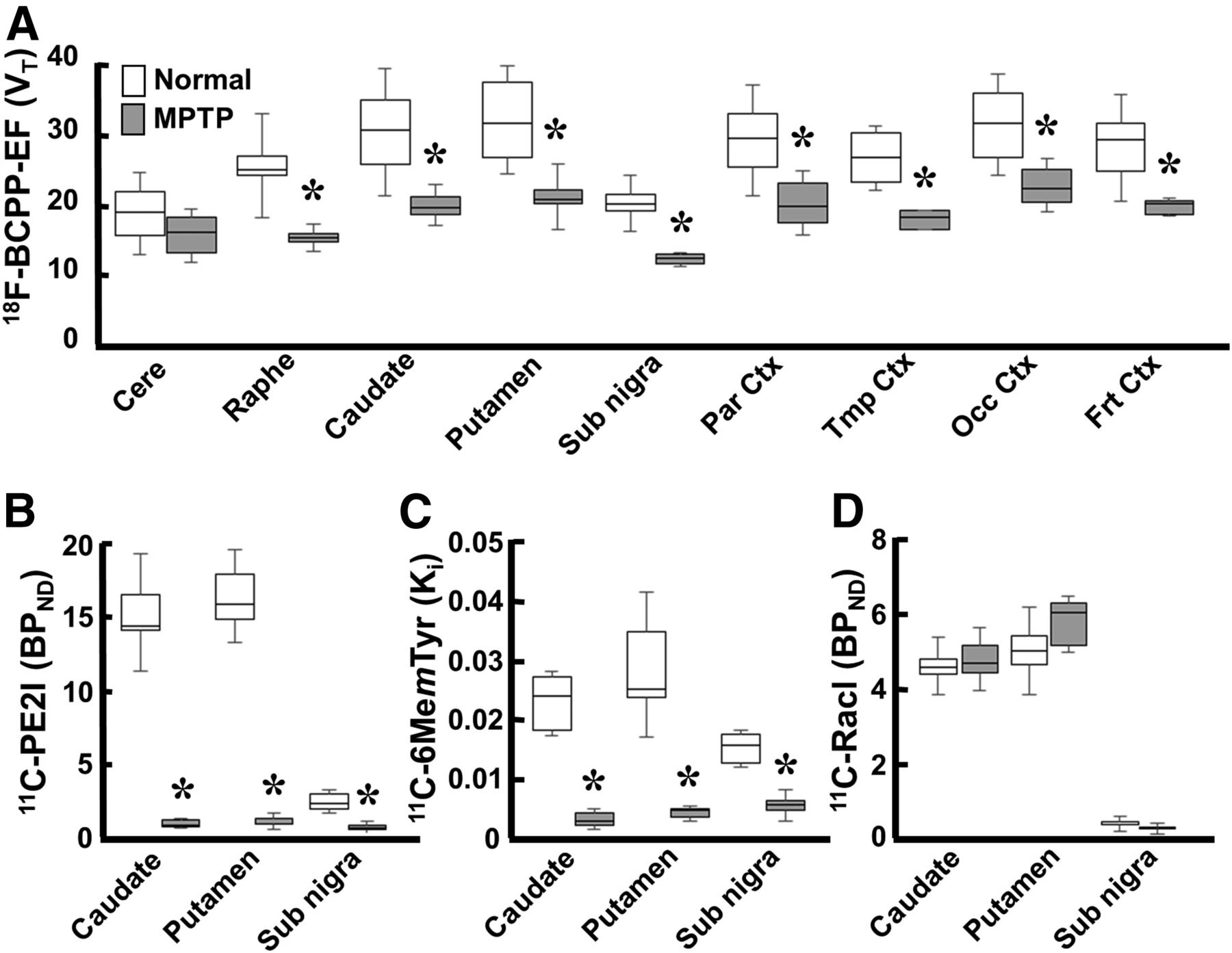
In MPTP-treated monkeys, VT for 18F-BCPP-EF (Figs. 1G and 2A) showed significant reductions to 68.9%, 72.3%, 67.7%, 66.4%, 68.1%, 65.2%, 60.9%, and 62.7% of normal levels (Figs. 1A and 2A) in the frontal, occipital, temporal, and parietal cortices; the putamen; the caudate; the raphe nucleus; and the substantia nigra pars compacta, respectively, and no significant decrease in the cerebellum.

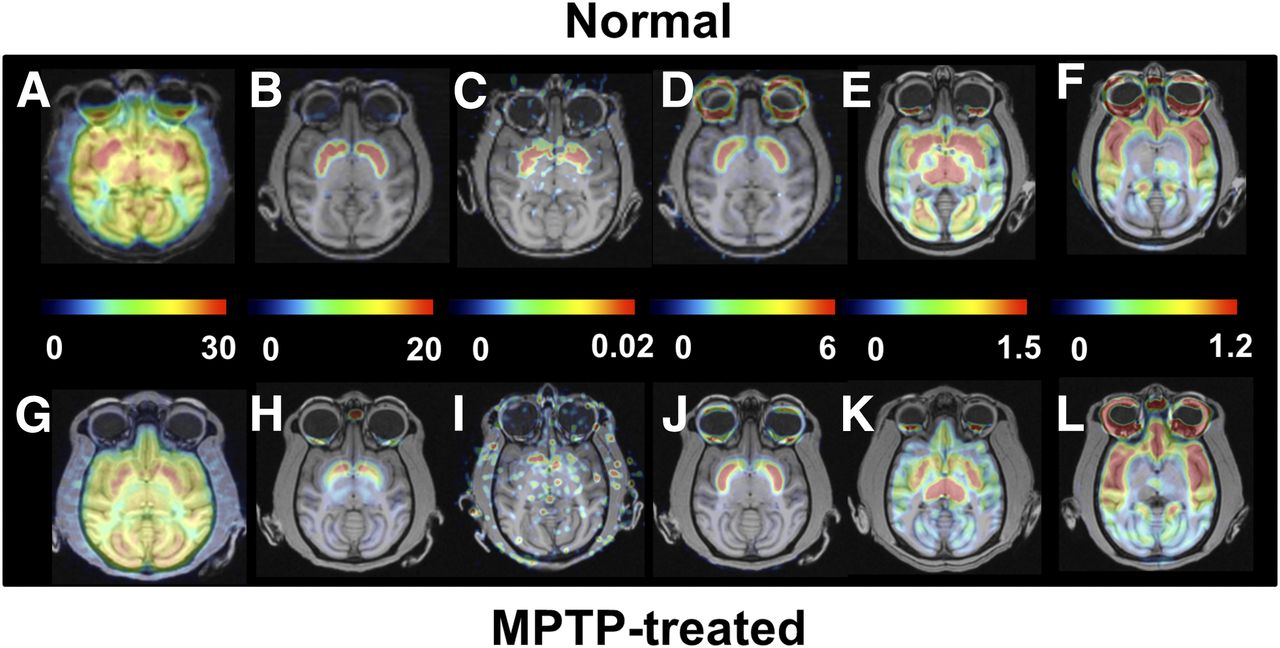
Effect of MPTP treatment on PET images of MC-I, dopamine, and serotonin in conscious monkeys. Scanning was performed for 91 min with 18F-BCPP-EF (MC-I) (A and G), 11C-PE2I (dopamine transporter) (B and H), 11C-DASB (serotonin transporter) (E and K), and 18F-MPPF (serotonin 1A receptor) (F and L) and for 60 min with 11C-6MemTyr (dopamine synthesis) (C and I) and 11C-raclopride (dopamine D2 receptor) (D and J). VT images of 18F-BCPP-EF were created using Logan graphical plot analysis with metabolite-corrected plasma input function. BPND images of 11C-PE2I, 11C-raclopride, 11C-DASB, and 18F-MPPF and multiple-time graphical analysis Ki images of 11C-6MemTyr were created using the corresponding time–activity curves in cerebellum as input function.
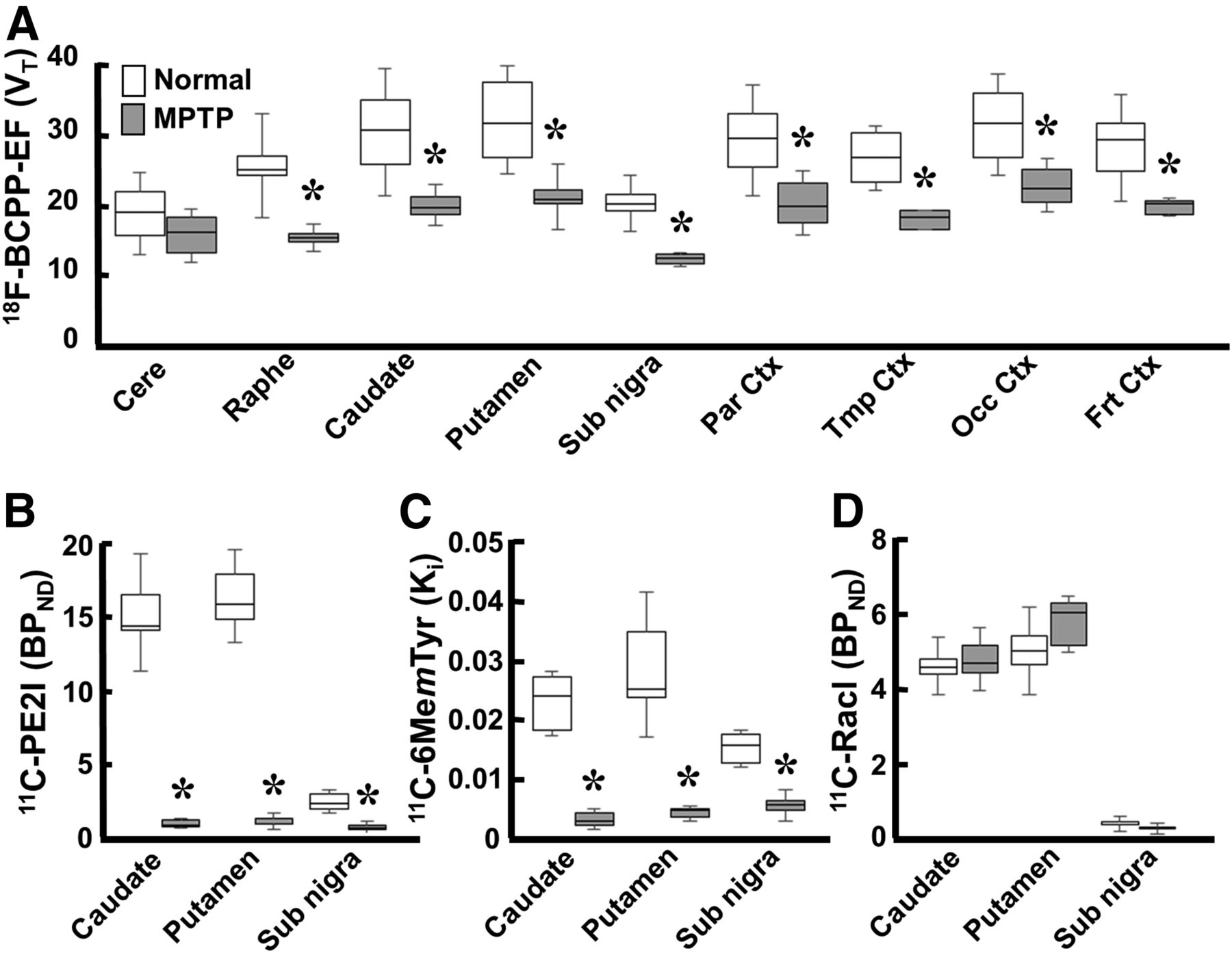
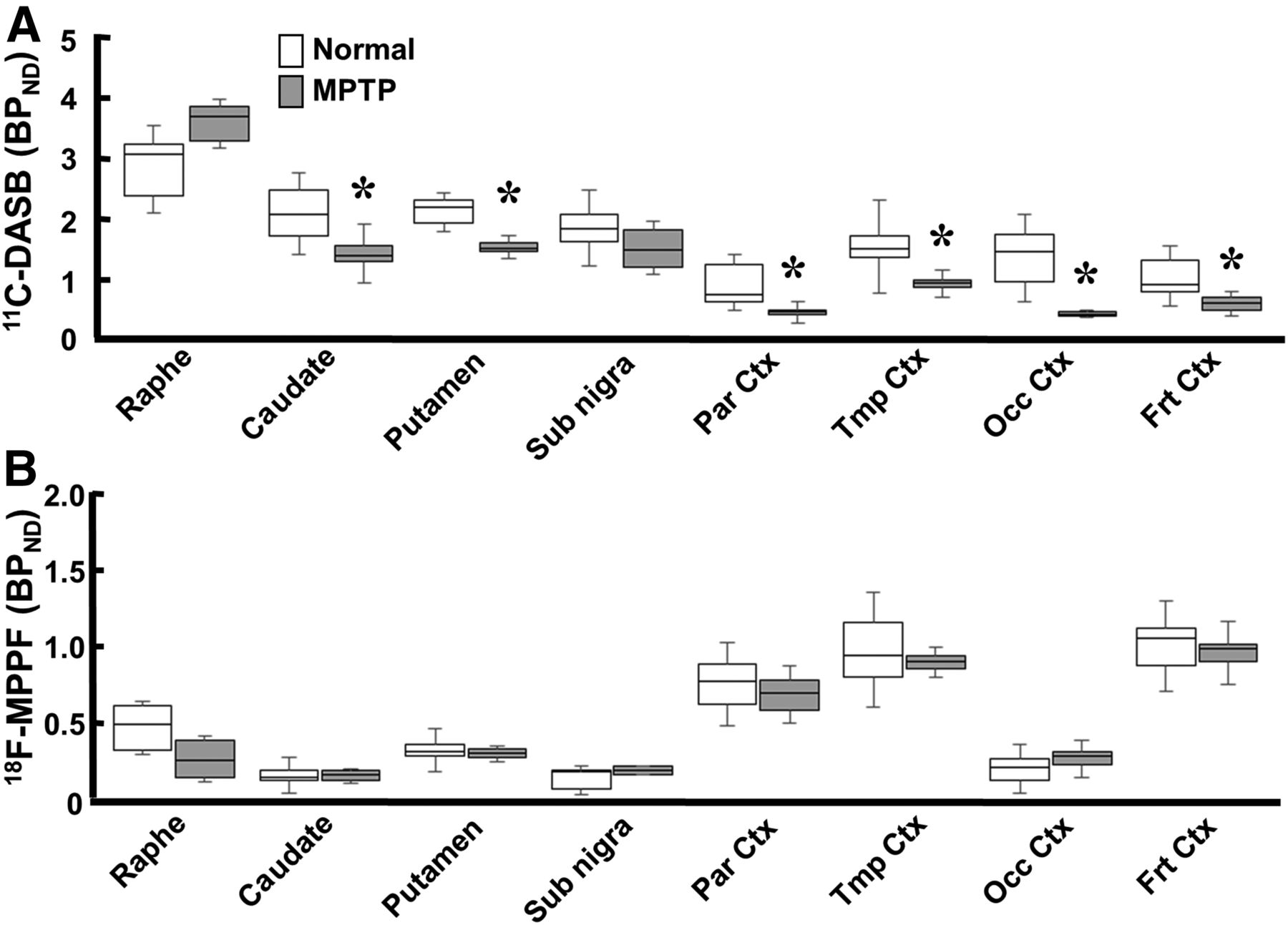
Effect of MPTP treatment on MC-I and pre- and postsynaptic dopamine parameters in conscious monkeys. (A) Binding of 18F-BCPP-EF to MC-I was calculated using Logan graphical plot analysis. (B and D) 11C-PE2I availability to dopamine transporter (B) and 11C-raclopride binding to dopamine D2 receptor (D) were calculated using BPND analysis. (C) 11C-6MemTyr for dopamine synthesis was calculated using multiple-time graphical analysis. Data are expressed as box plots showing median, minimum, and maximum. *P < 0.05 vs. normal.
No significant changes in 11C-raclopride binding to dopamine D2 receptor were observed in the striatum or substantia nigra pars compacta between MPTP-treated monkeys (Figs. 1J and 2D) and normal animals (Figs. 1D and 2D). When dopamine transporter availability, a presynaptic parameter, was assessed with 11C-PE2I, availability in the caudate, putamen, and substantia nigra pars compacta was found to be significantly lower in MPTP monkeys (4.0%, 4.9%, and 18.6% of the normal level) (Figs. 1H and 2B) than in normal animals (Figs. 1B and 2B). When dopamine synthesis, another presynaptic parameter, was assessed using 11C-6MemTyr, synthesis in the striatum and substantia nigra pars compacta was also found to be markedly lower in MPTP monkeys (13.6%, 12.3%, and 41.1% of the normal level) (Figs. 1I and 2C) than in normal animals (Figs. 1C and 2C).
Quantitative analysis of 11C-DASB revealed that MPTP-treated monkeys had reduced serotonin transporter availability in all brain regions except the raphe nucleus (Figs. 1K and 3A). The reduction was greatest in the occipital cortex (19.7% of normal); was lower in the frontal, parietal, and temporal cortices (50.0%, 40.7%, and 51.6% of normal, respectively); was lower still in the caudate (60.3% of normal); and was least in the putamen and substantia nigra pars compacta (66.0% and 67.7% of normal, respectively) (Figs. 1E and 3A). In contrast, binding of 18F-MPPF to serotonin 1A receptor did not significantly change in any brain region of MPTP monkeys (Figs. 1L and 3B) compared with normal animals (Figs. 1L and 3B).
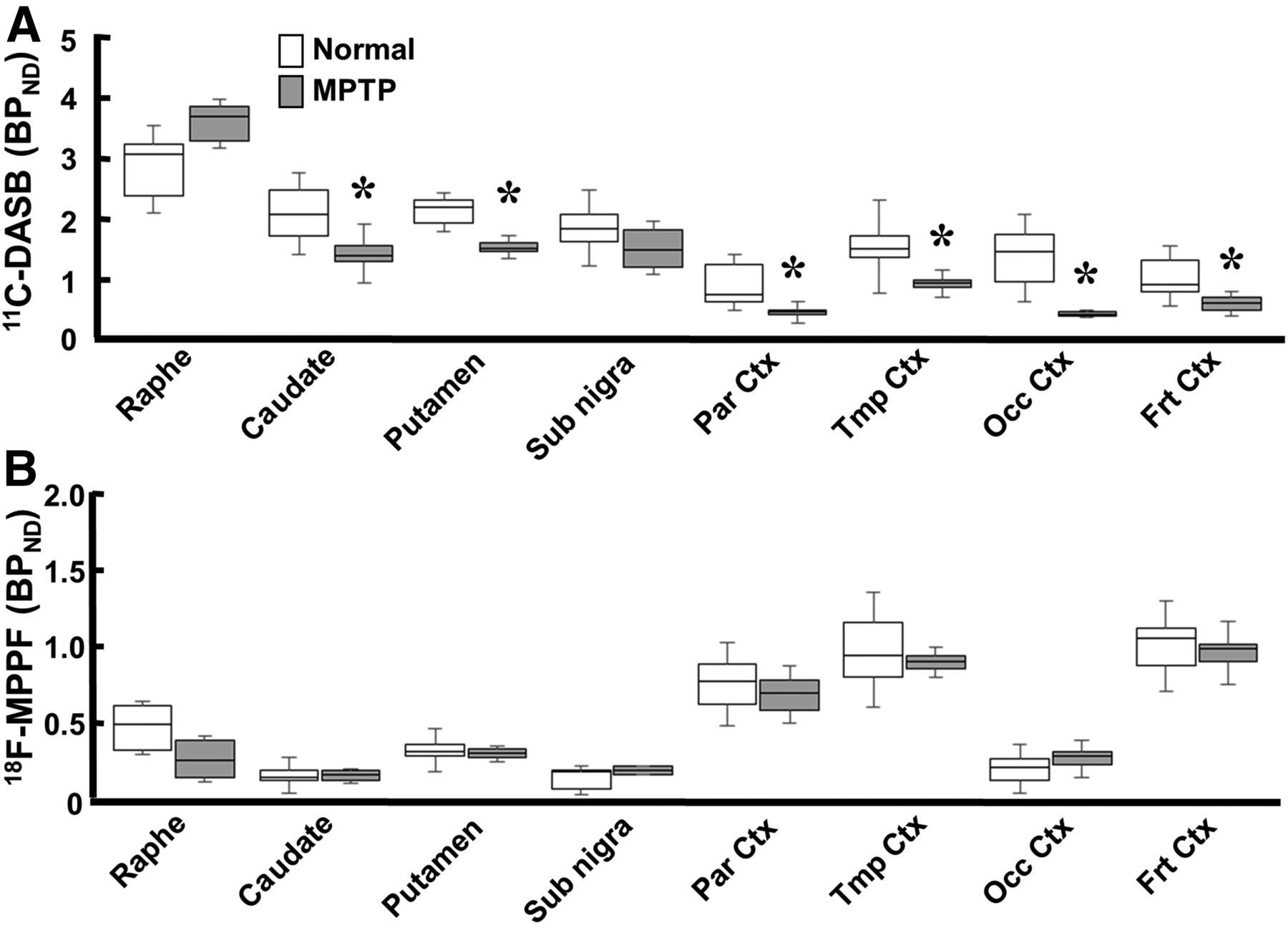
Effect of MPTP treatment on serotonin transporter and serotonin 1A receptor in conscious monkeys. 11C-DASB availability to serotonin transporter (A) and 18F-MPPF binding to serotonin 1A receptor (B) were measured using BPND analysis. Data are expressed as box plot showing median, minimum, and maximum. *P < 0.05 vs. normal.
As shown in Figure 4, when ΔBPND for 11C-PE2I, ΔKi for 11C-6MemTyr, and ΔBPND for 11C-DASB in each VOI were plotted against the corresponding ΔVT for 18F-BCPP-EF, significant positive correlations were observed between ΔBPND for 11C-PE2I and ΔVT for 18F-BCPP-EF (r2 = 0.521, P < 0.01) (Fig. 4A), between ΔKi for 11C-6MemTyr and ΔVT for 18F-BCPP-EF in nigrostriatal regions (r2 = 0.537, P < 0.01) (Fig. 4B), and between ΔBPND for 11C-DASB and ΔVT for 18F-BCPP-EF in cortical regions (r2 = 0.403, P < 0.01) (Fig. 4D). In contrast, no significant correlation between ΔBPND for 11C-DASB and ΔVT for 18F-BCPP-EF was detected in nigrostriatal regions (Fig. 4C).

Effect of MPTP-induced MC-I impairment on dopamine transporter availability (A), dopamine synthesis in nigrostriatal region (B), and serotonin transporter in nigrostriatal (C) and cortical regions (D) of conscious monkeys. Averaged BPND for 11C-PE2I, Ki for 11C-6MemTyr, BPND for 11C-DASB, and VT for 18F-BCPP-EF in each VOI of normal monkeys were calculated and subtracted from each VOI of individual MPTP-treated monkeys to determine degree of MPTP-induced reduction. Then, ΔKi or ΔBPND in each VOI was plotted against the corresponding ΔVT.
DISCUSSION
The present study demonstrated that MPTP-induced damage detected as MC-I impairment was not exclusive to the dopaminergic nigrostriatal pathway and that damage in the extrastriatal regions was partly attributable to serotonergic neuronal damage.
Administration of MPTP causes slow, progressive loss of dopamine neurons over several months, and the decrease in nigrostriatal dopamine levels is considered responsible for the motor symptoms of MPTP-treated monkeys. Impaired electron transport and oxidative phosphorylation due to an MC-I deficiency may account for neuronal cell death in Parkinson disease. Alternatively, the MC-I deficiency in Parkinson disease may be secondary to mitochondrial damage due to oxidative stress. MC-I, a site at which reactive oxygen species are produced, is particularly vulnerable to oxidative damage (6). We previously demonstrated the potential of 18F-BCPP-EF to detect MPTP-induced impairment of MC-I activity, which strongly correlated with damage in presynaptic dopamine neurons assessed as dopamine transporter availability or dopamine synthesis in a Parkinson cynomolgus model under isoflurane anesthesia (9).
Measurement of MC-I activity using 18F-BCPP-EF is not affected by isoflurane anesthesia (12). Dopamine D2 receptor binding (31) and dopamine transporter availability (32), however, have been reported to be affected—thus our intention to assess all PET parameters in conscious animals in the present study. The present study also confirmed that MPTP treatment induced an impairment of MC-I activity as measured using 18F-BCPP-EF, and this finding correlated well with reduced dopamine transporter availability as measured using 11C-PE2I and with presynaptic dopamine synthesis as measured using 11C-6MemTyr (9,22,23).
Our preliminary observations (9) suggested that impairment of MC-I activity is also induced in the nondopaminergic brain regions of MPTP-treated cynomolgus monkeys, and the present study with rhesus monkeys confirmed that the toxic effect of MPTP occurred not only in the dopamine-rich regions of the substantia nigra pars compacta and striatum but also in brain regions with very few dopaminergic neurons. Thus, in this conscious monkey model, 18F-BCPP-EF detects MPTP-induced impairment of MC-I activity in extrastriatal brain regions such as the cortex, mid brain, and limbic system, and this impairment is associated with decreased binding of 11C-DASB to serotonin transporter. This result is inconsistent with previous findings showing, in postmortem Parkinson disease brain, that MPTP induces neuronal damage (8) and MC-I deficiency (33) exclusively in dopaminergic neurons in the nigrostriatal regions.
Two transport systems, uptake 1 and uptake 2, are responsible for terminating monoaminergic neurotransmission by clearance from extracellular space. Uptake 1 is an Na+- and Cl−-dependent high-affinity transporter system and includes dopamine transporter, serotonin transporter, and norepinephrine transporter. These are expressed on the nerve endings of monoaminergic neurons, and their clearance is from the synaptic cleft. Uptake 2 was originally characterized as an Na+- and Cl−-independent low-affinity, high-capacity transport system in peripheral organs; however, this transporter was suggested to be involved in monoamine signaling pathways in the brain (34,35). Uptake 2 consists of multiple organic cation transporters; in particular, organic cation transporters are prominent not only for endogenous monoamines but also for prototypical organic cations such as 1-methyl-4-phenylpyridinium (35,36). Thus, it is not difficult to imagine that 1-methyl-4-phenylpyridinium can be gradually transported into monoaminergic cells, including serotonergic neurons, through the uptake 2 system during repeated MPTP administration. 1-methyl-4-phenylpyridinium enters mitochondria and selectively binds to and inhibits the MC-I contained within all kinds of monoaminergic cells, leading to inhibition of adenosine triphosphate synthesis and of reactive oxygen species production (33). A small loss (∼25%) in MC-I activity is sufficient to decrease adenosine triphosphate synthesis in neuronal cells, resulting in neurodegenerative damage in monoaminergic neurons. In fact, previous studies on MPTP-treated rhesus monkeys showed that the brain monoamine systems for not only dopamine but also noradrenaline and serotonin are susceptible to MPTP (14).
There is increasing evidence that in addition to causing motor symptoms, Parkinson disease is associated with nonmotor symptoms such as depression and cognitive deficits, which may be related to changes in other monoamines (noradrenaline and serotonin) in extrastriatal regions (13). Lewy body and neurite deposition occurs within the raphe nucleus, which contains the serotoninergic neurons of the caudal brain stem (33), before it occurs in the midbrain, which contains dopaminergic neurons (37,38). Decreases in serotonin concentration have been reported postmortem in the cortical brain regions of Parkinson disease patients (15). PET imaging of nondepressed Parkinson disease patients has revealed diffuse reductions in 11C-DASB binding to serotonin transporter throughout the brain, suggesting a diminished density of serotonergic fibers (16,17). In contrast, other studies have indicated an increase in 11C-DASB binding to serotonin transporter in the limbic structures of depressed Parkinson disease patients (39,40), suggesting compensatory upregulation of serotonin transporter leading to a reduction in serotonergic neurotransmission.
Although the present study demonstrated no significant MPTP-induced changes in 18F-MPPF binding to serotonin 1A receptor in monkeys, clinical PET research has reported significantly weaker serotonin 1A receptor binding in nondepressed and depressed Parkinson disease patients than in controls (18). Because some studies have refutably demonstrated that these alterations in the serotonergic system are partly attributable to 3,4-dihydroxy-l-phenylalanine treatment (19), these controversial findings do not appear to reflect the primary neuropathologic alterations in Parkinson disease.
CONCLUSION
The present results suggest that 18F-BCPP-EF has the potential to become a useful PET probe for quantitative imaging of MPTP-induced degeneration in not only dopaminergic but also nondopaminergic neurons, as shown by impaired MC-I activity in conscious monkeys.
DISCLOSURE
No potential conflict of interest relevant to this article was reported.
Acknowledgments
We gratefully acknowledge Norihiro Harada, Dai Fukumoto, and Aiko Iwazaki for their technical assistance.
Footnotes
Published online Mar. 9, 2017.
- © 2017 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication December 26, 2016.
- Accepted for publication February 17, 2017.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.