


Abstract
A good-manufacturing-practices (GMP) 68Ge/68Ga generator that uses modified dodecyl-3,4,5-trihydroxybenzoate hydrophobically bound to a octadecyl silica resin (C-18) as an adsorbent has been developed that allows for dilute HCl (0.05N) to efficiently elute metal-impurity-free 68Ga3+ ready for peptide labeling. We characterized the performance of this generator system over a year in conjunction with the production of 68Ga-labeled DOTATOC and Glu-NH-CO-NH-Lys(Ahx)-HBED-CC (PSMA-HBED-CC) intended for clinical studies and established protocols for batch release. Methods: A 2,040-MBq self-shielded 68Ge/68Ga generator provided metal-free 68GaCl3 ready for peptide labeling in the fluidic labeling module after elution with 4 mL of 0.05N HCl. The compact system was readily housed in a laminar flow cabinet allowing an ISO class-5 environment. 68Ga labeling of peptides using GMP kits was performed in 15–20 min, and the total production time was 45–50 min. Batch release quality control specifications were established to meet investigational new drug submission and institutional review board approval standards. Results: Over a period of 12 mo, 68Ga elution yields from the generator averaged 80% (range, 72.0%–95.1%), and 68Ge breakthrough was less than 0.006%, initially decreasing with time to 0.001% (expressed as percentage of 68Ge activity present in the generator at the time of elution), a unique characteristic of this generator. The radiochemical purity of both 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC determined by high-performance liquid chromatography analysis was greater than 98%, with a minimum specific activity of 12.6 and 42 GBq/μmol, respectively. The radionuclidic (68Ge) impurity was 0.00001% or less (under the detection limit). Final sterile, pyrogen-free formulation was provided in physiologic saline with 5%–7% ethanol. Conclusion: The GMP-certified 68Ge/68Ga generator system was studied for a year. The generator system is contained within the fluidic labeling module, and it is compact, self-shielded, and easy to operate using simple manual techniques. The system provides radiolabeled peptides with high (>98%) radiochemical purity and greater than 80% radiochemical yield. The 68Ge levels in the final drug products were under the detection limits at all times. 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC investigational radiopharmaceuticals are currently being studied clinically under investigational new drug (IND) applications submitted to the U.S. Food and Drug Administration.
With the broad success of 18F-FDG, it is hard to imagine that 68Ga was the first short-lived, high-specific-activity positron emitter used for clinical investigation. 68Ga-ethylenediaminetetraacetic acid (EDTA) was used for brain tumor annihiscopy (1), a coincidence-based detection system used long before the advent of PET cameras. 68Ga use for clinical applications was only preceded by the long-lived 74As (17.5 d) and the carrier-added 64Cu (12.8 h, produced as 63Cu(n,γ)64Cu) (2). This first attempt of producing 68Ga (and specifically 68Ga-EDTA) was based in a liquid–liquid extraction, evidently not the most practical procedure (2). Follow-up was provided by Greene and Tucker in 1961, producing the first adsorbent-based 68Ge/68Ga generator, using a neutral alumina bed to retain the 68Ge while eluting the 68Ga as a 0.005 M EDTA complex in 25 mL of solution (pH 7) (3). Although an improvement over the solvent extraction system, the radionuclide was eluted in a large volume, necessitating preconcentration before use. The elution volume was reduced in a subsequent design by Yano and Anger in 1964, optimizing the original Greene and Tucker generator while eluting the 68Ga generator as 68Ga-EDTA (4). In some ways, it can be said that the 68Ge/68Ga generator is as old as its SPECT analog 99Mo/99mTc (5).
Despite the early endeavors in 68Ge/68Ga generator production, the surge faded because of the lack of a cationic gallium elution system that would allow for diverse drug development and the absence of a clinically viable positron imaging system. At the same time, the advent of the Anger camera in the 1960s (6) together with the reliable 99Mo/99mTc generator system (5) jumpstarted the use of 99mTc in nuclear imaging, increasing to this day. The invention of the first PET systems in the early 1970s combined with the successful labeling of glucose as 18F-FDG by Al Wolf and Joanna Fowler at Brookhaven National laboratories in 1978 drew attention further away from 68Ga (7).
The first reliable, durable, commercially available 68Ge/68Ga generator was produced by New England Nuclear and it was based on a β-SnO2 inorganic separation resin matrix, eluting ionic gallium and first described by Loch in 1980 (8). It was followed nearly 20 y later by the Obninsk 68Ge/68Ga generator (Russia), and it conveniently coincided with the surge of PET cameras for 18F-FDG metabolic imaging in the early 2000s (9). The Obninsk generator delivered cationic gallium in 5 mL of 0.1N HCl, which could be easily buffered for chelation (9). The Obninsk’s technology was further developed by Eckert and Ziegler into the first chemical-grade 68Ge/68Ga generator (IGG100), which recently received marketing authorization in the European Union and European Economic Area countries. This generator is based on a TiO2 matrix, which could potentially introduce metallic competitors during 68Ga chelation. Some prepurification of the 68Ga is necessary before labeling when high specific activity is required (10). The IGG100 specification sheet assures 65% 68Ga elution yield and a 68Ge breakthrough of 0.00003% when new to 0.001% after 200 elutions (expressed as percentage of 68Ge activity present in the generator at the time of elution).
The iThemba Labs 68Ge/68Ga generator was also introduced into the market in the 2000s, and it is an improved version of the one originally sold in the 1980s by New England Nuclear, also based in a β-SnO2 inorganic separation resin (8). Similarly to the IGG100 (TiO2-based), the 68Ge breakthrough and minor metallic impurities (zinc, iron, tin, titanium, copper, and aluminum) represent a potential problem because they can compete with 68Ga during complex formation (11,12). Stability studies of this generator showed 0.003% of 68Ge in the elution, increasing to 0.08% after 100 d (13). 68Ge did not pose a patient risk because it was completely eliminated after the DOTA precursor labeling by reversed-phase Sep-Pak (Waters) purification (0–11 Bq in the final composition). However, for practical purposes 68Ge should be kept below 0.01%, so that high volumes of long-lived liquid waste are not generated. The reported DOTA precursor labeling was also lower (60%–90% in 15 min at 95°C) than that obtained when using the TiO2-based IGG100 (>95% in 10–15 min at 95°C) (11).
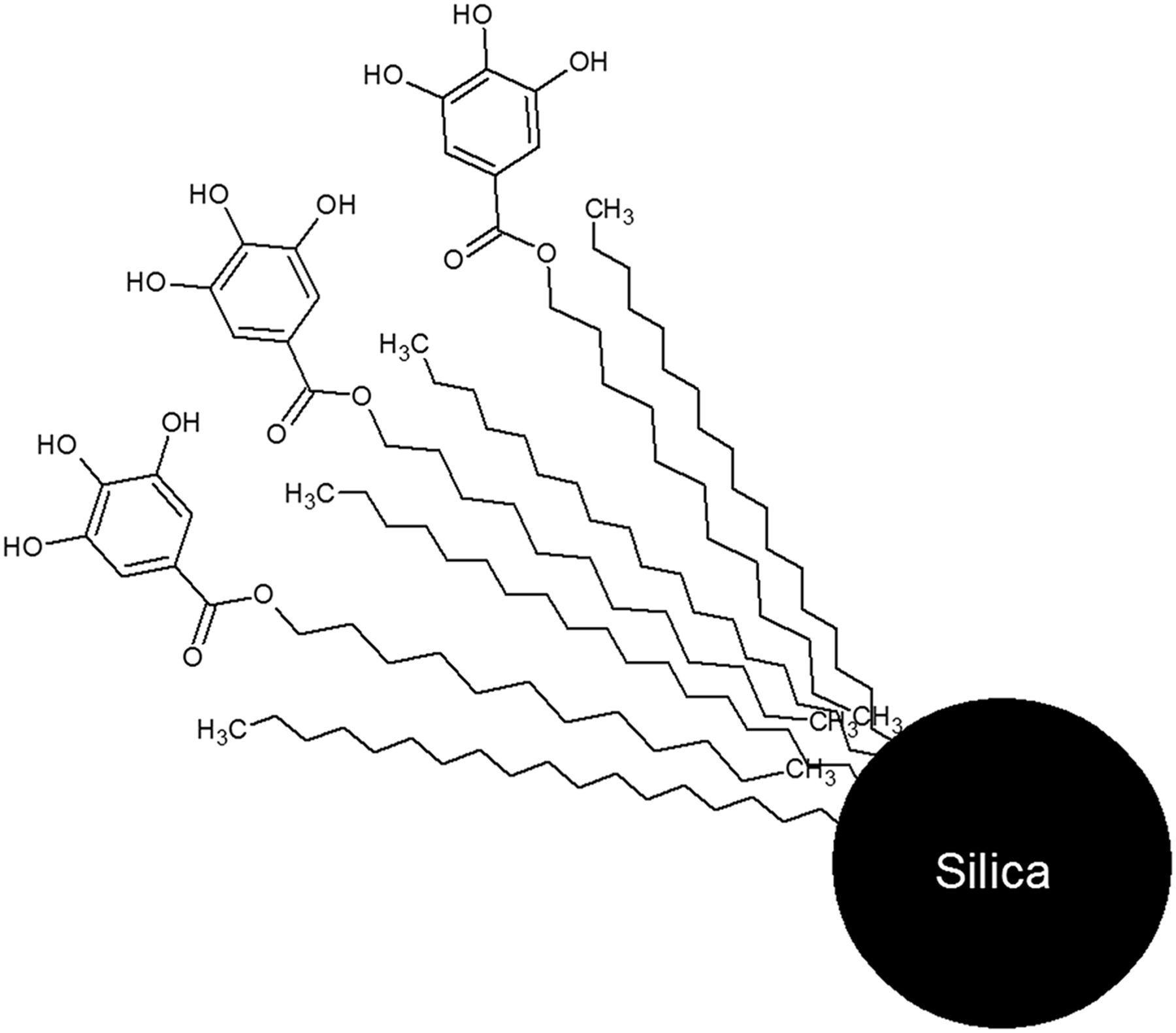
In both of these examples, prepurification of the eluent is needed to obtain high 68Ga labeling yields (>90%). A metal-free good-manufacturing-practices (GMP) generator was recently introduced into the market by Isotopen Technologies Garching GmbH (ITG GmbH)—the ITG 68Ge/68Ga generator, which is based on a modified dodecyl-3,4,5-trihydroxybenzoate hydrophobically bounded to an octadecyl-modified silica resin (C-18 resin) (Fig. 1). The generator is eluted with 0.05 M HCl and can be easily buffered for labeling. No prepurification of 68Ga is needed because of the metal-free column matrix; however, elution is recommended 24 h before labeling to eliminate excess 68Zn from 68Ga decay.
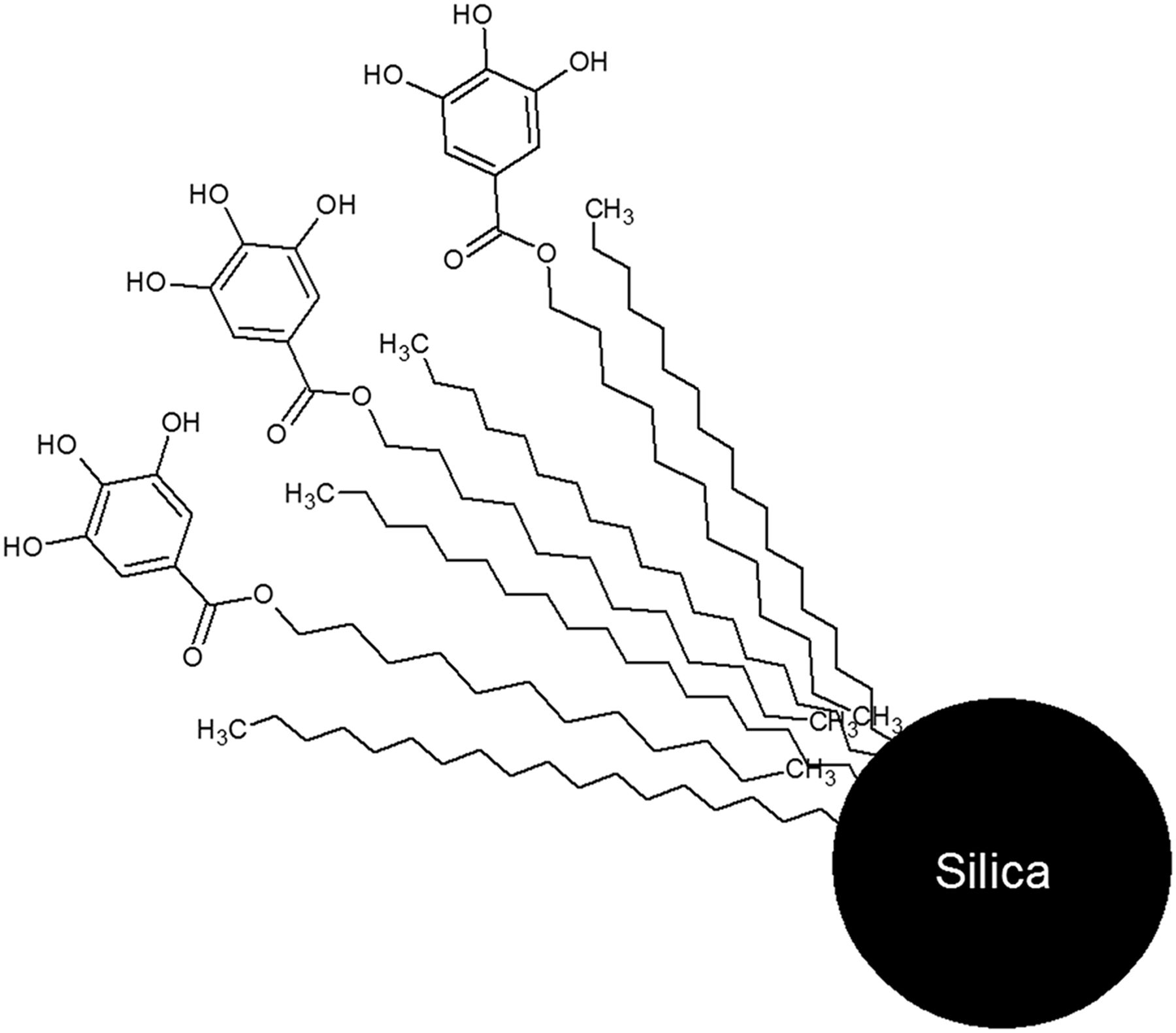
Active 68Ge trapping group in ITG 68Ge/68Ga generator.
Introducing an organic chelating group to retain the parent nuclide, 68Ge, in a long-lived generator poses potential radiolysis issues; thus, the generator was exhaustively tested. This article describes the characteristics of an ITG 68Ge/68Ga generator for a period of 1 y in routine use.
MATERIALS AND METHODS
All reagents were at least metal-trace grade. Hydrochloric acid (Sigma-Aldrich), 99.999% trace metals basis (100 mL), was used to elute the ITG 68Ge/68Ge generator. MilliQ (18.2 MΩ) water was obtained from a Direct Q system (Millipore). DOTATOC (GMP) and DKFZ-PSMA-11 (GMP) were obtained from ABX Pharmaceuticals. Ultrapure anhydrous sodium acetate (NaOAc) was used to buffer the generator elution (Sigma-Aldrich). Labeling kits and fluidic cassettes were obtained directly from ITG GmbH.
The 68Ge/68Ga generator was eluted with 4 mL of 0.05 M HCl for labeling. For testing, the generator was eluted with 6 mL of 0.05 M HCl, with the collection of 1-mL fractions to determine 68Ga activity (CRC-15 PET; Capintec), elution yield, and profile. The dose calibrator setting was verified with a 68Ge/68Ga National Institute of Standards and Technology (NIST)–traceable volumetric source. Fractions were saved for over 24 h and tested for 68Ge content (Wallac Wizard 3″ 1480; Perkin-Elmer). A NIST-traceable 68Ge/68Ga 12 × 75 mm (1-mL bottom) source containing 4.54 kBq (122.72 nCi; Bench/Mark) at calibration was used to determine well-counter detection efficiency and quantify the total 68Ge activity in the eluent. The dose calibrator accuracy was tested using another NIST-traceable volumetric source (syringe and vial geometry) containing 20.017 MBq (0.541 mCi; Bench/Mark) at the time of calibration.
The metallic content of the generator eluent was determined by inductively coupled plasma mass spectrometry (ICP-MS) (7700x ICP-MS; Agilent Technologies). Metallic impurities might interfere with 68Ga during the labeling process and thus lower the radiochemical yield. Trace metals of interest were iron, nickel, copper, zinc, niobium, and lead. To determine the content of these metals, calibration standards containing these elements in the following concentration were prepared to obtain a calibration curve: 10, 20, 30, 40, 50, and 100 μg/L. The generator eluent was diluted by a factor of 10 with 0.05 M HCl. The 0.05 M HCl was further measured as the blank sample.
Labeling was performed using the iQS 68Ga Fluidic Labeling Module (ITG) and the 68Ga Peptide Radiolabeling Kit (ITG) at 95°C for 5 min for both 68Ga-DOTATOC and 68Ga-Glu-NH-CO-NH-Lys(Ahx)-HBED-CC (PSMA-HBED-CC) (14). For 68Ga-DOTATOC labeling, 100 μL of a solution (250 μg/mL; 25 μg) of DOTATOC (GMP) were predissolved in 1 mL of buffer solution (part of the labeling kit), whereas only 5 μL of a PSMA-HBED-CC (1 mg/mL; 5 μg) solution were used in similar fashion. The labeled molecule was purified in-line with the labeling module using a reversed-phase C-18 Sep-Pak Lite (Waters) and filtered through a Cathivex-GV 0.22-μm filter (Merck Millipore) for sterilization. Radiochemical purity was assessed by high-performance liquid chromatography using a Waters NovaPak C18 4.6 × 150 mm column in a Varian binary solvent dual UV system (Agilent) and radiodetector (Eckert & Ziegler). All preparations were compared with known stable Ga-DOTATOC and Ga-PSMA-HBED-CC standards (ABX). Radionuclidic purity was evaluated using a multichannel analyzer (Canberra) and in a high-efficiency, low-background well-counter after 68Ga decay (>24 h after labeling). Sterility was tested by aliquoting 0.1 mL of the drug product into trypticase soy broth and thioglycollate medium and incubating for 15 d to assess bacterial growth. Endotoxin content was measured using an automated Endosafe system (Charles Rivers). Filter integrity (0.22-μm filter used for sterilization) was tested at 50 ψ with a bubble-point method. Ethanol content of the final product was confirmed to be under the 10% limit using gas chromatography (SRI Instruments).
RESULTS
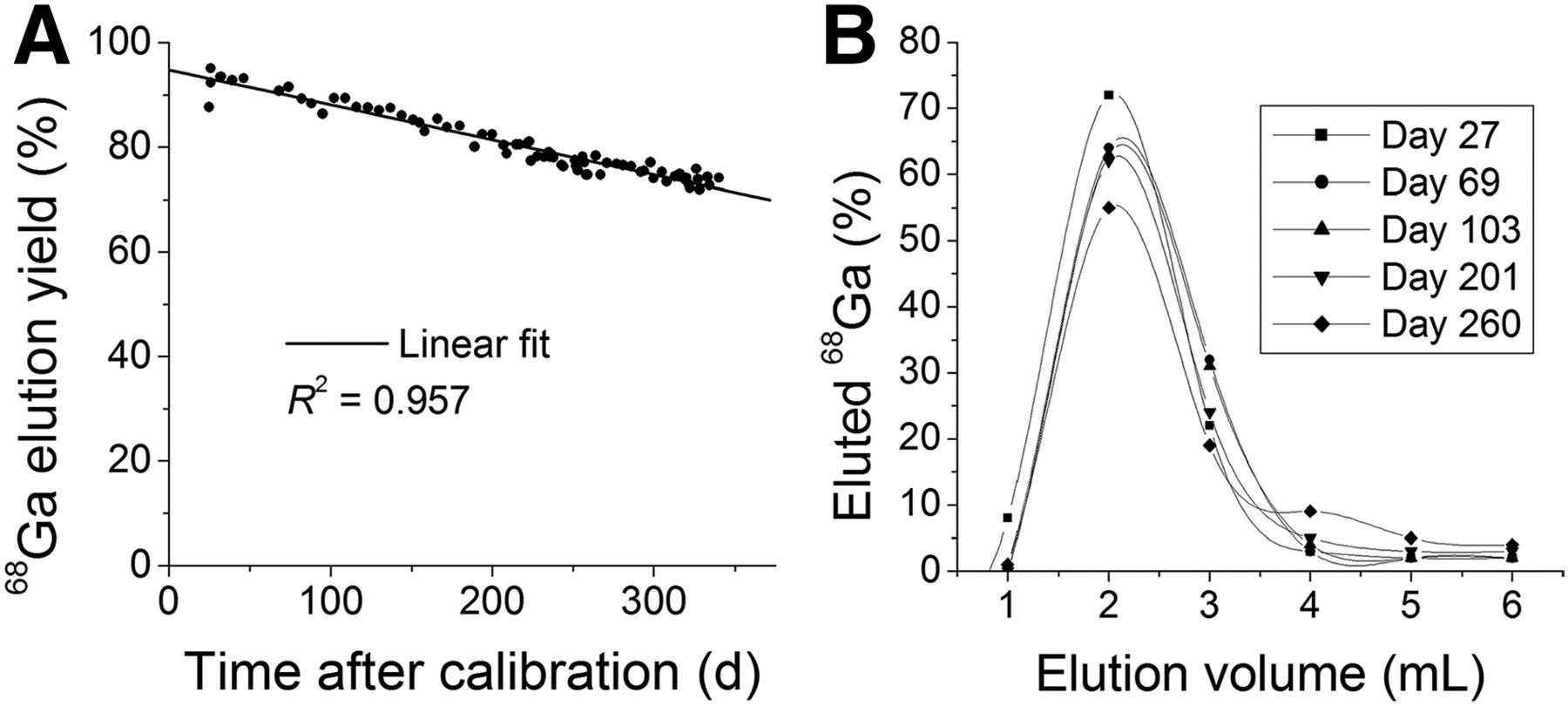
The GMP ITG 68Ge/68Ga generator contained 2,040 MBq (55.14 mCi) of 68Ge at calibration date. The elution yield of the ITG 68Ge/68Ga generator during a 1-y period was an average of 80% and never went below 70%. The highest yield obtained was 95.1% at day 26 after calibration, whereas the lowest obtained was 72.0% on day 328 after calibration. The elution yield decreased with a linear trend (R2 = 0.957, Fig. 2) over the period studied. This behavior was also described during the characterization of the IGG100 and the iThemba labs 68Ge/68Ga generators. In the case of the IGG100, the decrease was attributed to rearrangements that take place in the matrix in which suspension/precipitation of the TiO2 resin forms layers that cover the adsorbed 68Ge, and once it decays the resulting 68Ga is no longer accessible for elution because of physical trapping. No feasible theory was provided for this effect during the iThemba Labs generator characterization, but it can be safely assumed that a similar process is likely to occur as well for both the iThemba and the ITG generator.
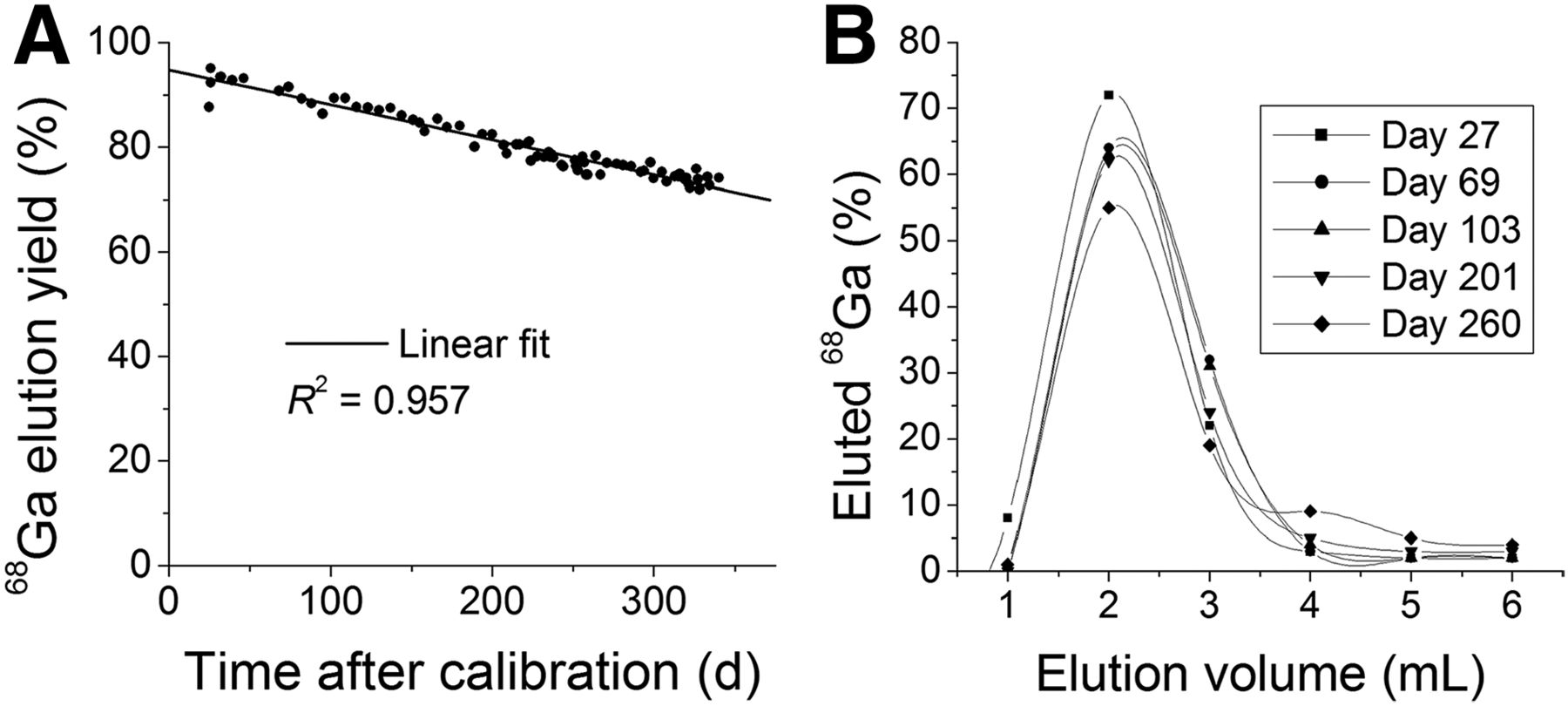
ITG 68Ge/68Ga generator long-term elution yield (A) (linear fit provided for reference) and elution profile study (365 d) (B).
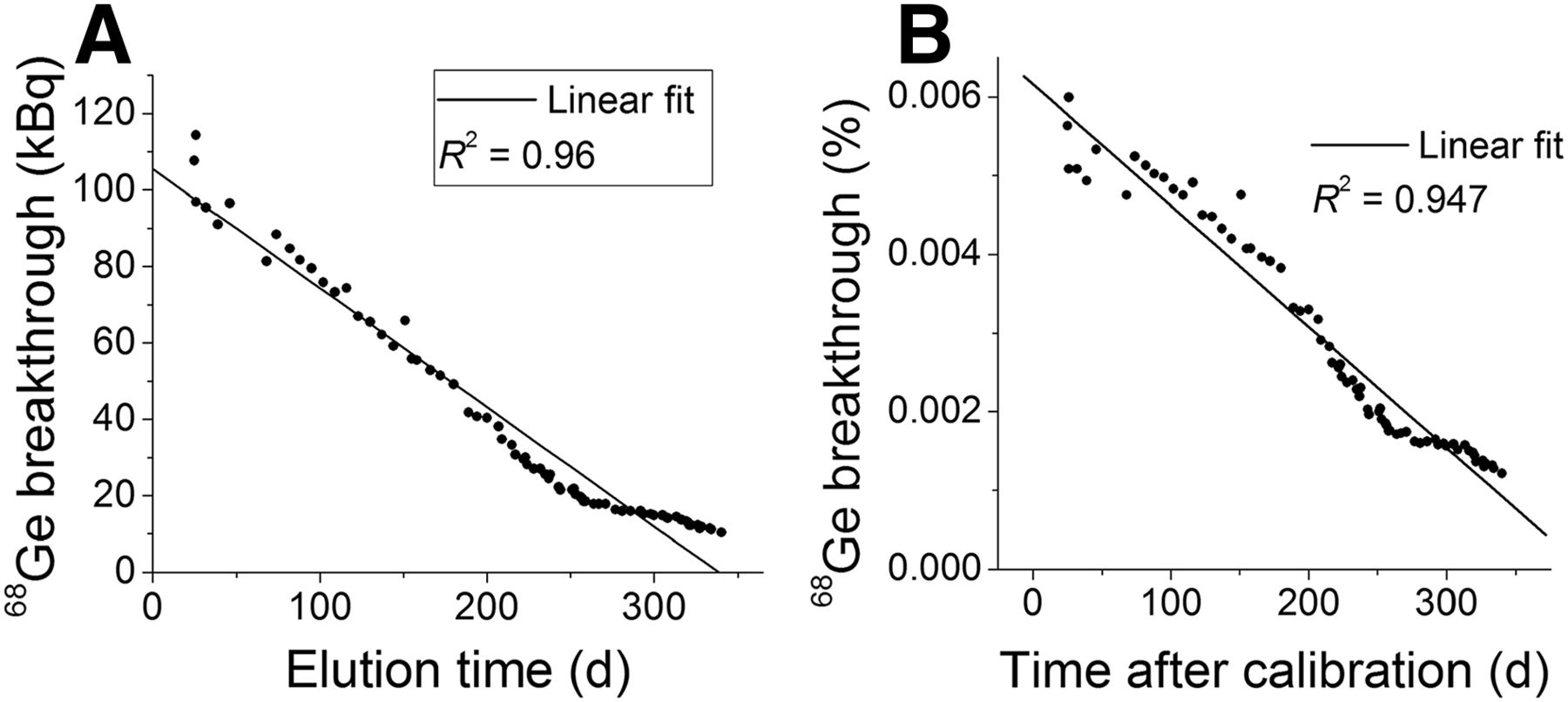
Most of the 68Ga-eluted activity (>95%) was eluted within the first 3 mL of 0.05 M HCl elution within the first 200 d of use. However, the elution profile changed (Fig. 2, elution profiles) after this period, eluting only 85% of the total eluted activity in the first 3 mL and 10%–12% in the last mL. In contrast, the same 68Ge concentration was found in every fraction, making the total eluted activity dependent on the total elution volume. The initial eluted 68Ge activity was just under 120 kBq (≈3 μCi), or 0.006% of the 68Ge activity present in the generator at the time of elution (Fig. 3).

ITG generator’s 68Ge breakthrough as neat eluted activity (A) and percentage of eluted germanium with respect to total 68Ge activity present at time of elution (B). Linear fit provided for reference.
In contrast with previous results obtained while testing the IGG100 and the iThemba Labs generators, the amounts of 68Ge in the eluent decreased to 10.4 kBq (0.3 μCi) after 340 d for the ITG generator. The decrease was observed not only for the absolute activity, but also in the 68Ge percentage (down to 0.0012%). This characteristic is unique for the ITG generator and could be attributed to the overall decrease in radiolysis and activity concentration with the decrease of total activity; nevertheless, no controlled experiment has been designed to date to test these conditions. In any case, there was no evidence of significant radiolysis because no ultraviolet absorbent chemical impurities were found in any of the chromatograms (given that trihydroxybenzoate will absorb in the same wavelengths used for the drug quality control, 220 and 280 nm) performed during the quality control. The metallic content of the eluate was found to be extremely low and is given in Table 1.
Concentration of Metallic Impurities of ITG 68Ge/68Ga Generator in 4 Milliliters of Eluate at Time of First Elution (1,850 MBq of 68Ga)
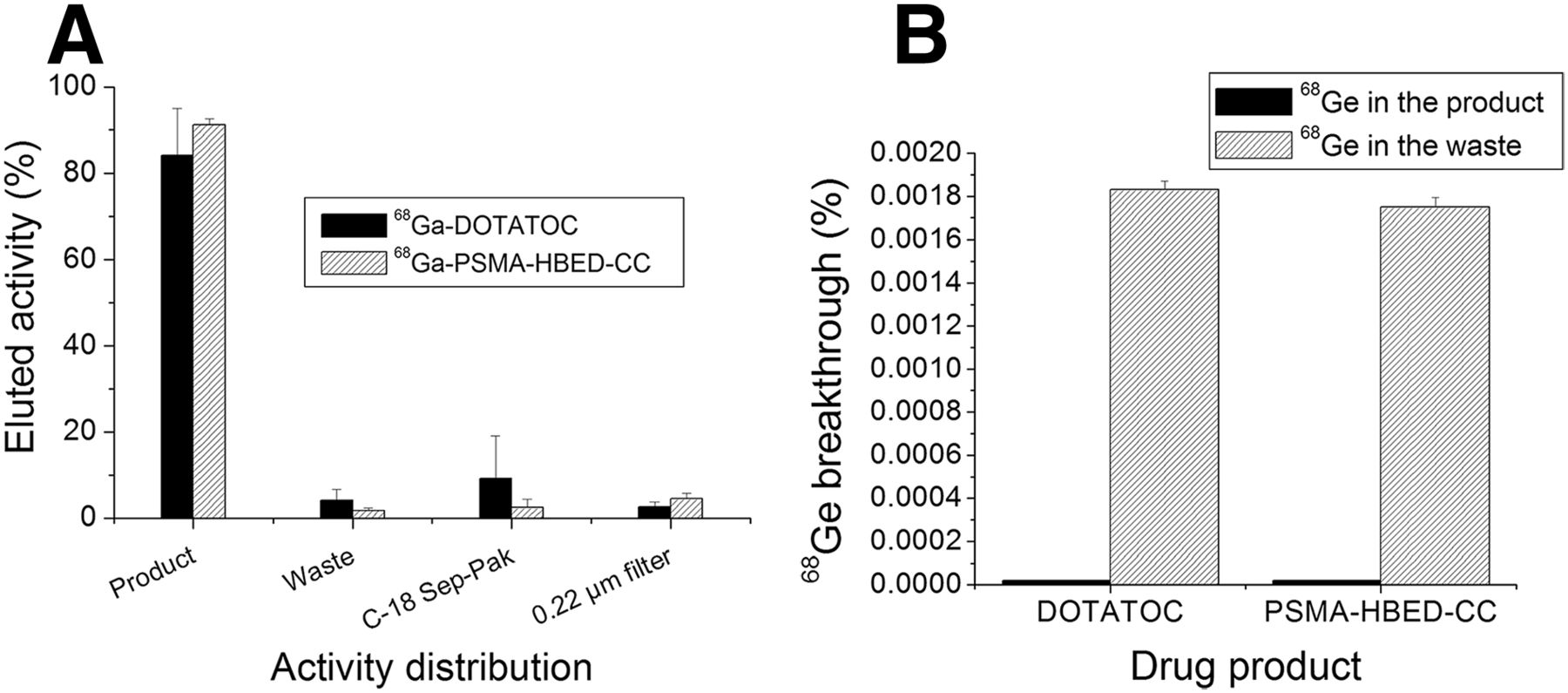
Radiolabeling of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC was performed using the same standard operating procedure (supplemental materials [available at http://jnm.snmjournals.org]). The HBED chelate is capable of complexing gallium at room temperature (25°C); however, the labeling was performed at 95°C for 5 min and the higher temperature provided consistency and better mixing and did not diminish the radiochemical purity of the final product. Having the same procedure and the same labeling set up for both imaging agents (68Ga-DOTATOC and 68Ga-PSMA-HBED-CC) of clinical interest simplifies the overall operation. The contents of the product vial, waste vial, C-18 Sep-Pak, and 0.22-μm filter were measured in a dose calibrator to assess labeling yield and efficiency of trapping/elution (Fig. 4).

Percentage of activity measured of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC in product vial, waste vial, C-18 Sep-Pak, and 0.22-μm filter (A) and 68Ge in product and waste vial after 24 h (as percentage of 68Ge activity in generator at time of elution) (B).
DISCUSSION
Higher radiochemical yield and smaller variability were found for the labeling of 68Ga-PSMA-HBED-CC (91.3% ± 1.3%) than for 68Ga-DOTATOC (84.1% ± 10.1%). Nevertheless, the main variability was found during the elution from the C-18 Sep-Pack, whereas the activity in the waste vial (presumably free 68Ga) was under 5% (1.7 ± 0.6 and 4.1% ± 2.5%, respectively). Around 5% of the produced 68Ga-PSMA-HBED-CC activity (4.5% ± 1.2%) was retained in the filter whereas only 2.5% ± 1.3% of the labeled 68Ga-DOTATOC was. These results demonstrate the high 68Ga labeling yields (≥95%) obtained in short labeling times (5 min) while using the ITG 68Ge/68Ga generator eluate, presumably because of the absence of metal impurities in the eluent. Invariably the eluted 68Ge radionuclidic impurity was successfully eliminated during the C-18 purification stage, because it was found only in the waste vial whereas the product vial contained amount below the detection limit (0.00001% of the 68Ge activity present at the time of elution). A high product radiochemical purity (>98%) was found for all preparations (supplemental materials) quantified by radio–high-performance liquid chromatography. We established the release criteria for 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC for routine production using the methods described in this article (Table 2).
Release Criteria for 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC
CONCLUSION
The GMP-certified ITG 68Ge/68Ga generator system was studied for a year. An average 80% 68Ga elution yield, and always a more than 70% yield, was found. The generator system is contained within the iQS Fluidic Labeling Module, and it is compact, self-shielded, and easy to operate, using simple manual techniques. The system provides radiolabeled peptides with high (>98%) radiochemical purity and greater than 80% noncorrected radiochemical yield. The amounts of 68Ge present in the generator elution decreases with time of use, a unique characteristic of this generator. The 68Ge levels in the drug products were under the detection limits at all times.
Currently at our institution, clinical studies using these radiopharmaceuticals have been initiated for imaging neuroendocrine tumors (68Ga-DOTATOC) and prostate cancer (68Ga-PSMA-HBED-CC) under separate investigational new drug (IND) applications submitted to the U.S. Food and Drug Administration. Our institution’s institutional review board approved these studies, and all subjects signed a written informed consent form. The ability to successfully prepare high-purity pharmaceutically acceptable injectable solutions of 68Ga-DOTATOC and 68Ga-PSMA-HBED-CC on a routine basis has facilitated the introduction of these important imaging agents into the clinic.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Apr. 21, 2016.
- © 2016 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication January 8, 2016.
- Accepted for publication March 14, 2016.





{kind=link}
{kind=link}
{kind=link}
{kind=link}