


Abstract
Disulfiram has been used for several decades in the treatment of alcoholism. It now shows promise as an anticancer drug and radiosensitizer. Proposed mechanisms of action include the induction of oxidative stress and inhibition of proteasome activity. Our purpose was to determine the potential of disulfiram to enhance the antitumor efficacy of external-beam γ-irradiation and 131I-metaiodobenzylguanidine (131I-MIBG), a radiopharmaceutical used for the therapy of neuroendocrine tumors. Methods: The role of copper in disulfiram-induced toxicity was investigated by clonogenic assay after treatment of human SK-N-BE(2c) neuroblastoma and UVW/noradrenaline transporter (NAT) glioma cells. The synergistic interaction between disulfiram and radiotherapy was evaluated by combination-index analysis. Tumor growth delay was determined in vitro using multicellular tumor spheroids and in vivo using human tumor xenografts in athymic mice. Results: Escalating the disulfiram dosage caused a biphasic reduction in the surviving fraction of clonogens. Clonogenic cell kill after treatment with disulfiram concentrations less than 4 μM was copper-dependent, whereas cytotoxicity at concentrations greater than 10 μM was caused by oxidative stress. The cytotoxic effect of disulfiram was maximal when administered with equimolar copper. Likewise, disulfiram radiosensitization of tumor cells was copper-dependent. Furthermore, disulfiram treatment enhanced the toxicity of 131I-MIBG to spheroids and xenografts expressing the noradrenaline transporter. Conclusion: The results demonstrate that the cytotoxicity of disulfiram was copper-dependent, the molar excess of disulfiram relative to copper resulted in attenuation of disulfiram-mediated cytotoxicity, copper was required for the radiosensitizing activity of disulfiram, and copper-complexed disulfiram enhanced the efficacy not only of external-beam radiation but also of targeted radionuclide therapy in the form of 131I-MIBG. Therefore, disulfiram may have anticancer potential in combination with radiotherapy.
Disulfiram, an inhibitor of aldehyde dehydrogenase (1), has been used for the treatment of alcoholism for several decades. This drug also has a variety of other actions that suggest that it has potential as an anticancer agent. These include the induction of oxidative stress (2,3), the generation of copper-dependent toxicity (2,4), proteasome inhibition (4–6), and nuclear factor-κB inhibition (7,8). Despite its diverse range of pharmacologic activities, prolonged treatment with disulfiram has negligible, reversible, adverse effects and is considered a safe drug (9). Disulfiram is currently undergoing clinical trials for the treatment of various cancers including melanoma and liver, lung, and prostate. To determine the potential application of this agent in cancer therapy, it is important to understand its mechanisms of action and its effects in combination with other therapeutic modalities.
Disulfiram-induced cytotoxicity has previously been reported to be mediated by oxidative stress (2,3), which may be enhanced by the presence of copper (2). Copper-binding drugs have been shown to inhibit proteasome activity (10) and generate reactive oxygen species (ROS) (11). Disulfiram chelates copper, and it may be the disulfiram–copper complex that is the toxic form of the drug (4). Many tumors contain elevated levels of copper (10), possibly rendering them selectively susceptible to disulfiram-induced toxicity.
A potentially significant mechanism of disulfiram-induced cell death involves inhibition of proteasome activity. The proteasome degrades misfolded, superfluous, or damaged proteins and controls many cellular processes involved in differentiation, proliferation, signal transduction, cell cycle progression, and apoptosis. Proteasome activity is increased in cancer cells, compared with normal cells (12), and the proteasome inhibitor bortezomib is being used in the treatment of multiple myeloma. Disulfiram has been reported to inhibit proteasome activity and induce apoptosis selectively in cancer cells but not normal cells (4). The copper-binding activity of disulfiram may also be involved in this mode of action because the formation of organic copper complexes appears to be responsible for proapoptotic proteasome inhibition (4). Other proteasome inhibitors (13), including the reduced form of disulfiram (diethyldithiocarbamate) (14), have radiosensitizing effects, and this may also apply to disulfiram, further increasing its potential as an anticancer agent.
For tumor types that are both radiosensitive and chemosensitive, such as neuroblastoma, combined chemotherapy and radiotherapy may offer the best prospects for a positive outcome to treatment. A feature of neuroblastoma is expression of the noradrenaline transporter (NAT), allowing targeted radiotherapy using a radiolabeled structural analog of noradrenaline, 131I-metaiodobenzylguanidine (MIBG). This radiopharmaceutical has proven to be an excellent palliative, and long-term remissions have been achieved after single-agent treatment (15). However, the optimal way to use 131I-MIBG has yet to be defined, and increasingly it is administered in combination with other treatments in attempts to exploit the expression of several different targets in the rapidly evolving cells of tumors. Future advances in 131I-MIBG therapy are expected to derive from concurrent administration of radiosensitizers. Drug resistance is also an important cause of failure in the treatment of neuroblastoma, possibly caused by increased P-glycoprotein expression after chemotherapy (16). Disulfiram inhibits P-glycoprotein–mediated drug resistance (17), thereby increasing sensitivity to chemotherapeutic agents, and this approach has been used successfully for treating pediatric tumors, including retinoblastoma and neuroblastoma (18). Having previously demonstrated synergy between the topoisomerase I inhibitor topotecan and 131I-MIBG (19,20), we now describe the use of a similar methodology to test the potential synergistic effect of disulfiram with 131I-MIBG treatment.
We report an investigation of the extent to which disulfiram’s ability to induce cell death occurred through copper- or ROS-dependent mechanisms. In the SK-N-BE(2c) and UVW/NAT cancer cell lines, clonogenic survival was decreased by disulfiram treatment, and the presence of copper was necessary for tumor cell kill at low, clinically achievable, concentrations of disulfiram. Moreover, the molar ratio of disulfiram to copper was critical for cytotoxicity. Finally, a potent radiosensitizing effect of disulfiram was demonstrated in vitro and in vivo for the first time, to our knowledge, and this also was copper-dependent.
MATERIALS AND METHODS
Reagents
Disulfiram (tetraethylthiuram disulfide) was purchased from Sigma-Aldrich and Antabuse obtained from Actavis. Stock solutions of both agents were prepared in dimethyl sulfoxide, and then diluted in culture medium (maximum dimethyl sulfoxide concentration, 0.1% v/v). All other drugs were dissolved in culture medium. All cell culture medium and supplements were purchased from Invitrogen, and all other chemicals were from Sigma-Aldrich. No-carrier-added 131I-MIBG was prepared as described previously (20).
Cell Culture
Human neuroblastoma–derived SK-N-BE(2c) cells were purchased from the American Type Culture Collection. The UVW cell line was derived from a human glioblastoma (21). Cell lines were authenticated in-house using the AmpF/STR Identifiler kit (Applied BioSystems). SK-N-BE(2c) cells were maintained in Dulbecco modified Eagle medium containing 15% (v/v) fetal calf serum. UVW cells were transfected to express the NAT gene as previously described (22) and were maintained in minimum essential medium containing 10% (v/v) fetal calf serum and geneticin (1 mg/mL) at 37°C in a 5% CO2 atmosphere.
Clonogenic Survival Assay
Cells were seeded in 25-cm2 flasks at 105 cells per flask. When cultures were in the exponential growth phase, medium was removed and replaced with fresh medium containing disulfiram; the cell-impermeable copper chelator bathocuproine disulfonic acid (BCPD) (300 μM), the antioxidant N-acetyl-L-cysteine (NAC) (1 mM), copper (II) chloride, or various combinations of these agents were added simultaneously. Cells were incubated with drugs for 24 h. Cells were γ-irradiated using an Alcyon II 60Co-cobalt source at a dose rate of 1 Gy/min, then incubated for 24 h. For experiments conducted in serum-free conditions, cells were washed twice with phosphate-buffered saline before the serum-free medium with or without drugs was added. After drug treatment, cells were seeded for clonogenic assay as previously described (19,20).
Spheroid Growth Assay
Multicellular spheroids were used for the evaluation of 131I-MIBG treatment because β-particle cross-fire irradiation makes a considerable contribution to its efficacy. This component of tumor cell kill would be underrepresented in cellular monolayers (23). SK-N-BE(2c) and UVW/NAT cells were cultured as multicellular tumor spheroids using the liquid overlay technique (24). Briefly, spheroids were initiated by inoculating cells into an agar-coated flask. After 3–4 d, spheroids were retrieved by centrifugation and resuspended in fresh serum-free culture medium containing disulfiram or copper for 24 h. Simultaneously, 131I-MIBG was added to the medium at a final concentration of 1 MBq/mL for SK-N-BE(2c) and 0.5 MBq/mL for UVW/NAT spheroids. After treatment, spheroids were washed twice and those of approximately 100 μm in diameter were transferred individually into agar-coated wells of 24-well plates. Individual spheroid growth was monitored twice per week using an inverted phase-contrast microscope connected to an image acquisition system. Two perpendicular diameters, dmax and dmin, were measured using image analysis software (ImageJ) and the volume, V (μm3), was calculated using the formula V = π × dmax × dmin2/6 (25).
Combination Treatments
The cytotoxic interaction between disulfiram and radiation was examined using a clonogenic assay, according to the method of Chou and Talalay (26). Initially, exponentially growing cells were treated with each agent alone to determine effective doses. Cells were subsequently treated with a range of doses of disulfiram and γ-radiation, administered simultaneously, using a fixed dose ratio of disulfiram to γ-radiation, so that the proportional contribution of each agent in the mixtures would be the same at all treatment intensities. The fixed dose ratio was 0.1 μM disulfiram to 0.9 Gy of γ-radiation, based on inhibitory concentration of 50% values of 0.34 μM and 3 Gy, for disulfiram and γ-radiation, respectively. The effectiveness of combinations of disulfiram and γ-radiation was quantified by determining a combination index (CI) at various levels of cytotoxicity. Because the method of interaction was unknown, CI values were calculated assuming both mutual exclusivity (where the drugs have similar mechanisms of action) and mutual nonexclusivity (where the drugs have dissimilar mechanisms of action). CIs less than, equal to, and greater than 1 indicate synergism, additivity, and antagonism, respectively.
Tumor Xenografts
Six-week-old female, congenitally athymic nude mice of strain CD1 nu/nu were obtained from Charles River plc. In vivo experiments were performed in accordance with the Animals (Scientific Procedures) Act 1986. Tumors in athymic mice formed from SK-N-BE(2c) and UVW/NAT cells express the NAT, enabling active uptake of 131I-MIBG. Subcutaneous tumor growth was established as previously described (19). Mice were used for experimental therapy when the SK-N-BE(2c) tumor volumes had reached approximately 500 mm3 or when UVW/NAT tumors had grown to approximately 60 mm3. To monitor potential toxicity, experimental animals were examined daily for signs of distress and weighed weekly. Mice were randomized into treatment groups, each consisting of 6 animals that received phosphate-buffered saline solution (intraperitoneal injection), disulfiram solution (200 mg/kg; suspension in water, by oral gavage), 10 MBq of 131I-MIBG (intraperitoneal injection), 5 Gy of γ-radiation or simultaneous administration of disulfiram and 131I-MIBG, or disulfiram and γ-radiation. The activity of 131I-MIBG given to the mice was shown previously by us to induce a significant delay of growth but incomplete sterilization of UVW/NAT xenografts (19). Tumors were measured with calipers immediately before treatment and twice weekly thereafter. On the assumption of ellipsoidal geometry, diameter measurements were converted to an approximate tumor volume by multiplying half the longest diameter by the square of the mean of the 2 shorter diameters. Mice whose xenograft volume reached 1,900 mm3 were euthanized.
Statistics
Data are presented as mean ± SEM, unless otherwise stated, with the number of independent repetitions provided in the legend to each figure. Statistical significance was determined using a Student t test. A P value of less than 0.05 was considered to be statistically significant and less than 0.01 highly significant.
RESULTS
Cytotoxicity of Disulfiram
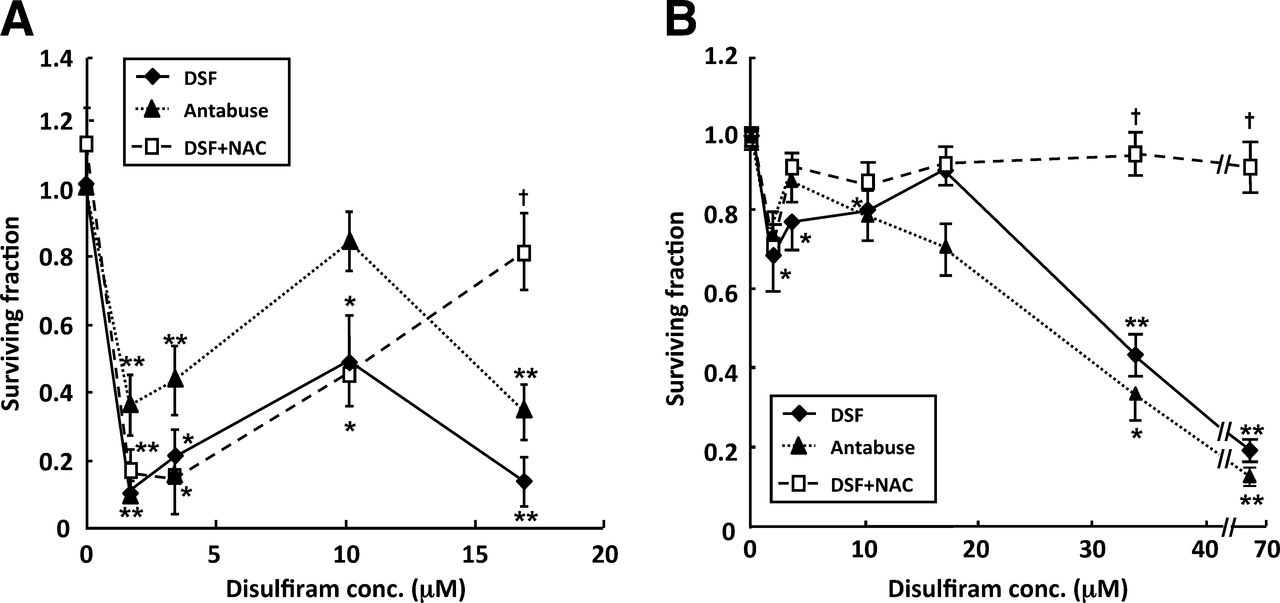
Escalating concentrations of disulfiram caused a biphasic toxicity profile in SK-N-BE(2c) cells (Fig. 1). Initial clonogenic kill was maximal after the administration of 1.7 μM disulfiram. Increasing disulfiram beyond this concentration resulted in partial reversal of cytotoxicity, up to 10 μM disulfiram. Thereafter, cytotoxicity increased, and after the administration of 17 μM disulfiram, clonogenic survival was 10% of untreated control levels (Fig. 1A). The effect on clonogenic survival of the clinical formulation of disulfiram used in animal studies, Antabuse, was also evaluated. A concentration-dependency profile similar to that generated by disulfiram was observed (Fig. 1A).
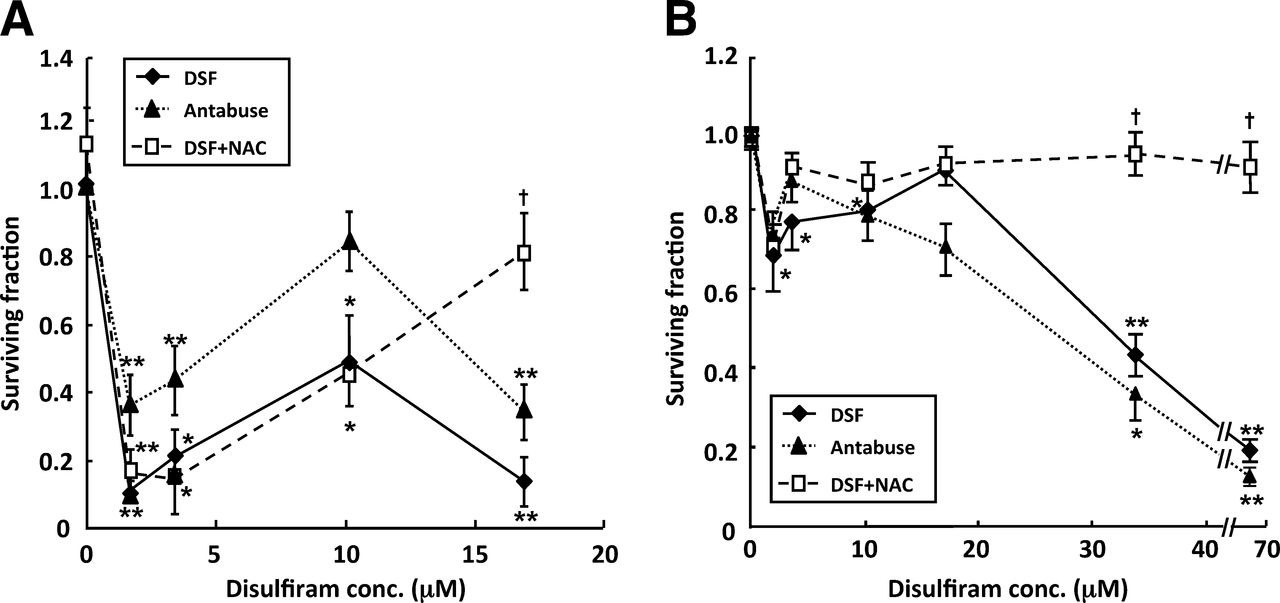
Cytotoxicity of disulfiram is biphasic with respect to dose. Clonogenic survival after exposure of SK-N-BE(2c) cells (A) or UVW/NAT cells (B) to disulfiram for 24 h in absence or presence of NAC or to clinical formulation of disulfiram (Antabuse); n = 4. Data are mean ± SEM. Significance of differences: *P < 0.05. **P < 0.01 from untreated control. †P < 0.05 from disulfiram alone. conc. = concentration; DSF = disulfiram.
A glioma cell line transfected with the NAT gene, UVW/NAT, responded in an analogous manner with respect to clonogenic cell kill in response to a range of concentrations of disulfiram (Fig. 1B). Again, the initial clonogenic survival nadir was observed after treatment with 1.7 μM disulfiram. A similar biphasic dose–toxicity profile was also observed when UVW/NAT cells were exposed to Antabuse (Fig. 1B).
Biphasic Dose–Response of Disulfiram
Treatment with 1 mM of the antioxidant NAC alone had no significant effect but prevented the reduction in clonogenic survival of SK-N-BE(2c) cells and UVW/NAT cells induced by concentrations of disulfiram greater than 10 μM, whereas NAC had no significant effect on disulfiram-induced toxicity at concentration of 10 μM or less (Fig. 1). Therefore, a ROS-independent mechanism of cell kill predominated at low concentrations of disulfiram (≤10 μM).
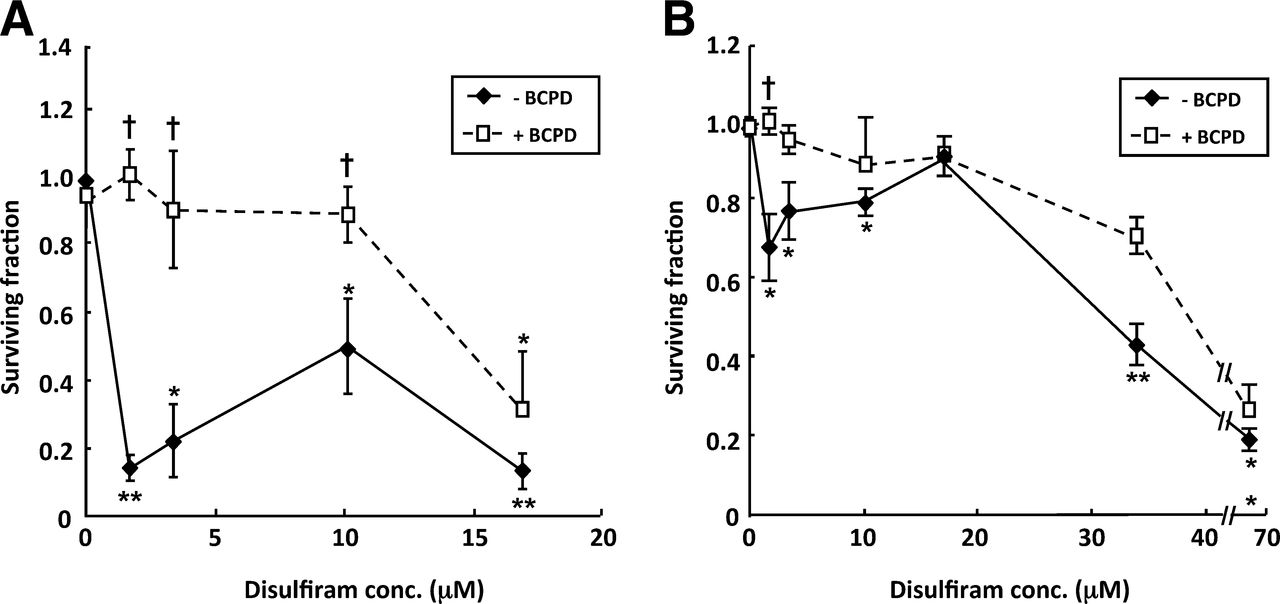
Although BCPD alone had no significant effect, the disulfiram-induced reduction in clonogenic survival of SK-N-BE(2c) cells was prevented by BCPD, at 300 μM, in the first phase of the dose–response, up to and including 10 μM disulfiram, suggesting a role for copper in disulfiram-induced cytoxicity at low concentrations. Although UVW/NAT cells were more resistant to disulfiram, the initial, maximal, clonogenic cell kill observed after treatment with 1.7 μM disulfiram was also prevented by BCPD (Fig. 2B).

Copper is necessary for toxicity of disulfiram at concentrations < 4 μM. Clonogenic survival after exposure of SK-N-BE(2c) cells (A) or UVW/NAT cells (B) to disulfiram for 24 h in absence or presence of BCPD; n = 5. Data are mean ± SEM. Significance of differences: *P < 0.05. **P < 0.01 from untreated control. †P < 0.05 from disulfiram alone. conc. = concentration.
Effect of Copper on Cytotoxicity of Disulfiram
In serum-free medium and in the absence of copper, disulfiram, at concentrations up to 10 μM, induced no significant reduction in survival of either SK-N-BE(2c) or UVW/NAT clonogens (Fig. 3), whereas the inclusion of copper chloride in the incubation medium resulted in dose-dependent clonogenic cell kill. This was maximal when disulfiram and copper concentrations were equimolar (Fig. 3). These observations suggest not only that the presence of copper is important for the cytotoxicity of disulfiram, but also that the concentration of copper relative to disulfiram is a major determinant of disulfiram’s potency.

Clonogenic cell kill is dependent on relative concentrations of disulfiram and copper. Clonogenic survival after exposure of SK-N-BE(2c) cells (A) or UVW/NAT cells (B) to disulfiram for 24 h in absence or presence of CuCl2 in serum-free medium; n = 4. Data are mean ± SEM. Significance of differences from untreated controls: *P < 0.05. **P < 0.01. conc. = concentration.
Copper Dependence of Disulfiram Radiosensitization
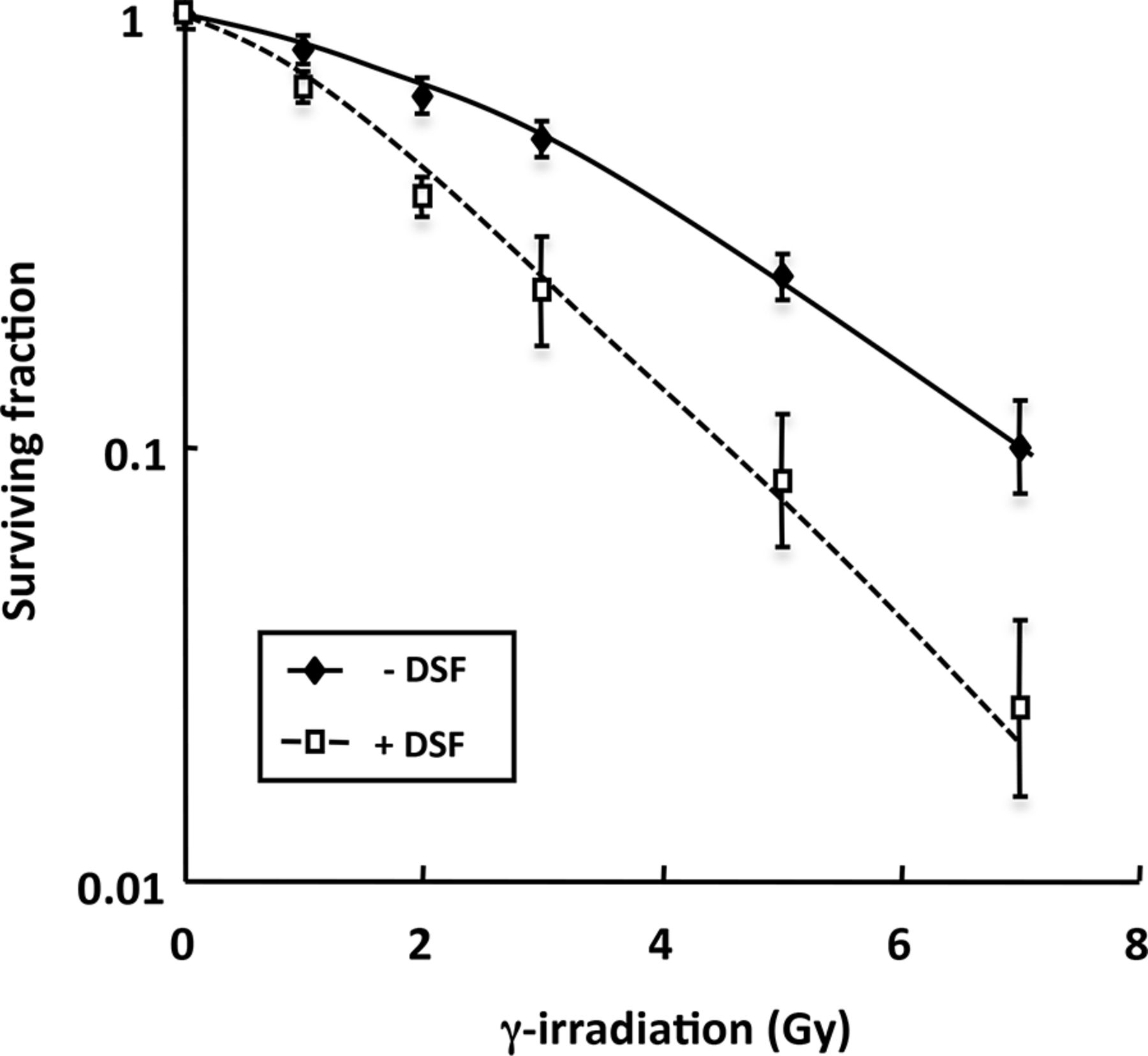
To investigate the potential for disulfiram to act as a radiosensitizer, SK-N-BE(2c) cells were γ-irradiated alone or in combination with disulfiram. A radiation dose response was clearly demonstrated with enhancement of the radiation-induced clonogenic kill by disulfiram at all radiation doses (Fig. 4). Inhibitory concentration of 50% values of 3.23 or 1.88 Gy were observed in the absence or presence of disulfiram, respectively. The effective doses of disulfiram and radiation were estimated from dose responses of each agent alone. A constant dose ratio of 1:9 of disulfiram (μM) to γ-irradiation (Gy) was used in the combination experiments. The CI values were determined according to the study of Chou and Talalay (26) and are shown in Table 1. Because the manner of the interaction between the agents was uncertain, the formula was solved in 2 different ways, corresponding to modes of action of the 2 agents, which are similar or distinct. Synergistic kill of SK-N-BE(2c) clonogens after treatment with combined disulfiram and γ-irradiation was manifested by CI values less than 1 for all dose intensities examined. For example, at the ED30 (dose required to kill 30% of clonogens), CI values were 0.45 ± 0.12 or 0.68 ± 0.19, assuming mutually exclusive or independent modes of action, respectively. Corresponding CI values at the ED50 (dose required to kill 50% of clonogens) were 0.62 ± 0.14 and 0.70 ± 0.19, respectively, and at the ED90 (dose required to kill 90% of clonogens) values were 0.70 ± 0.15 and 0.80 ± 0.2, respectively. Increasing CI values suggest decreasing synergism at higher dose intensities. Nonetheless, the observation of CI values less than 1 throughout the toxicity range of the experimental combination therapy indicated synergy at all strengths of treatment.
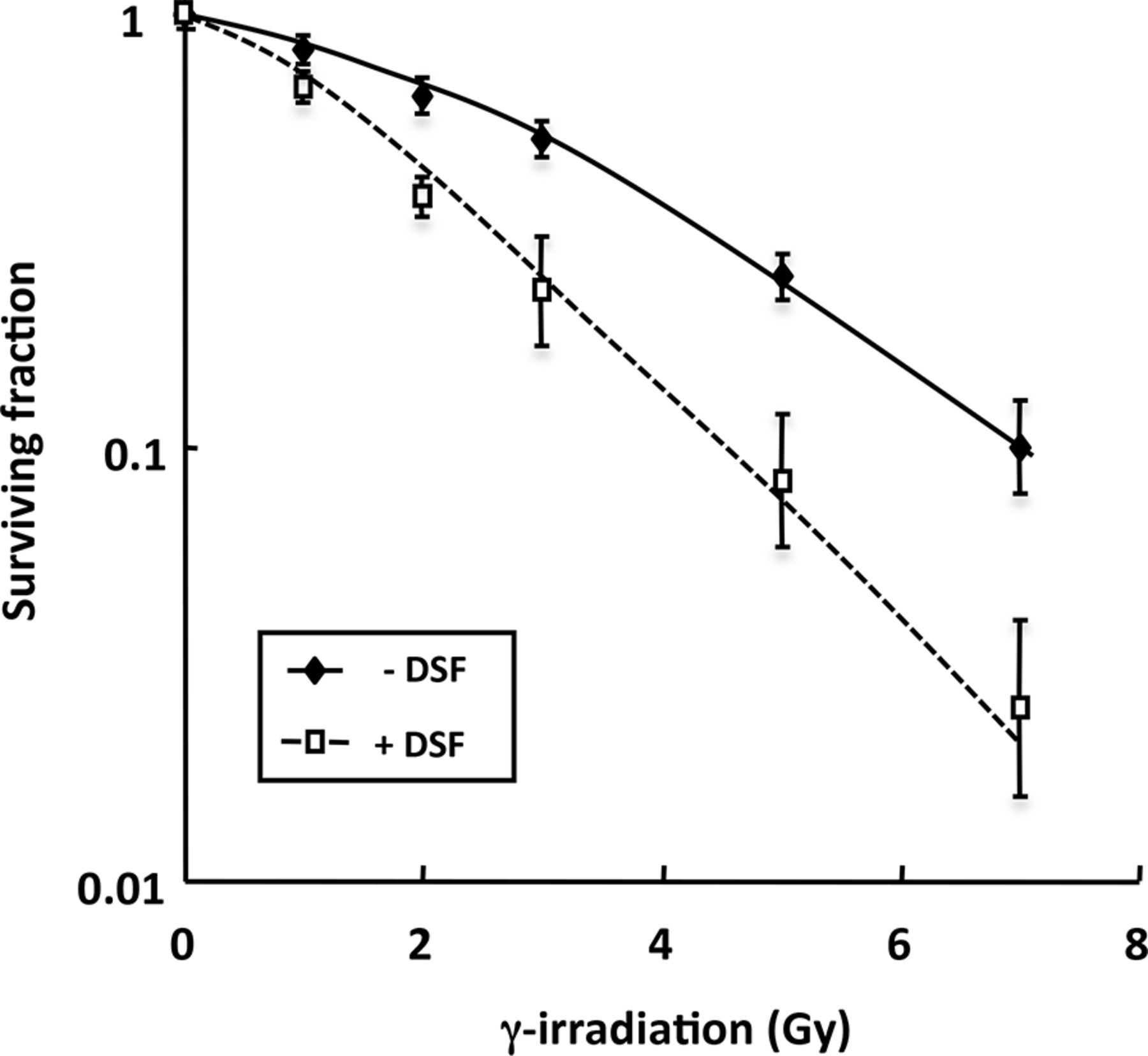
Effect of disulfiram on radiosensitivity of SK-N-BE(2c) cells. Clonogenic survival was determined after exposure of cells to γ-radiation alone or simultaneously with 0.34 mM disulfiram (n = 4) after correcting for plating efficiency and disulfiram cytotoxicity alone. Data are mean ± SEM. DSF = disulfiram.
The Effect of Disulfiram and γ-Radiation on Combination Indices
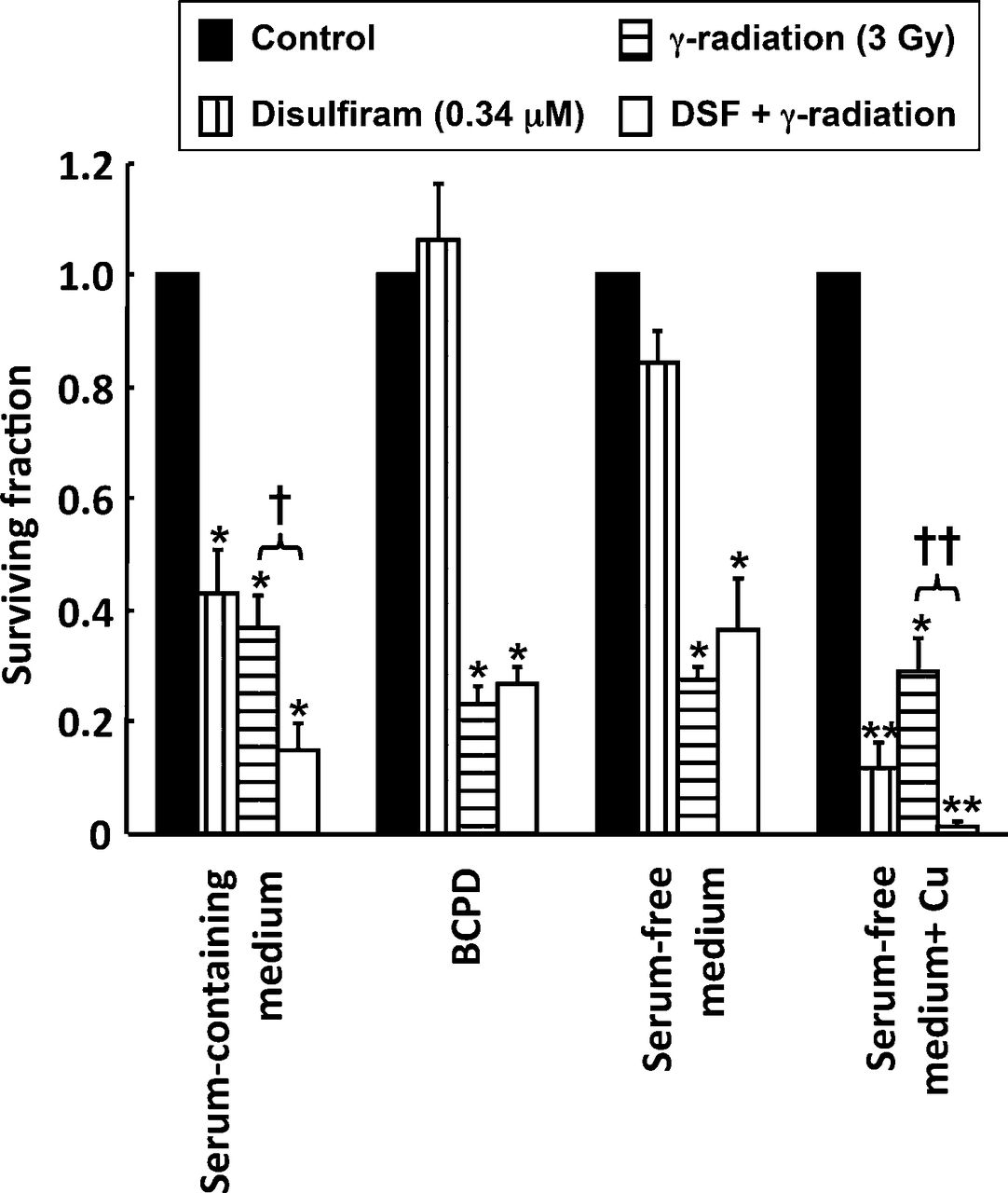
The involvement of copper in the radiosensitizing action of disulfiram was investigated by clonogenic assay of cells treated with combinations of disulfiram and γ-radiation in the presence or absence of serum, BCPD, or copper chloride. The clonogenic survival of γ-irradiated SK-N-BE(2c) cells was not significantly affected by the removal of serum or incubation with BCPD or copper chloride. Clonogenic cell kill resulting from treatment with 0.34 μM disulfiram was prevented by the removal of serum (P < 0.05) or inclusion of BCPD (P < 0.05) and enhanced by the addition of copper chloride (P < 0.05) (Fig. 5). Furthermore, the enhanced clonogenic kill induced by combined disulfiram and radiation treatment relative to single-agent treatment was prevented by serum removal (P < 0.05) or addition of BCPD (P < 0.05). In contrast, incubation with copper resulted in potentiation of the cell kill achieved by the disulfiram and radiation combination (P < 0.05).

Enhancement of radiation cell kill by disulfiram requires copper. Clonogenic survival after exposure of SK-N-BE(2c) cells to 0.34 μM disulfiram, 3 Gy γ-radiation, or a combination of both treatments in serum-containing medium, serum-containing medium with 300 μM BCPD, serum-free medium, or serum-free medium containing 1 μM CuCl2; n = 3. Data are mean ± SEM. Significance of differences: *P < 0.05. **P < 0.01 from untreated control. †P < 0.05. ††P < 0.01 from γ-irradiation alone. DSF = disulfiram.
Effect of Disulfiram and 131I-MIBG on Spheroid Growth Delay
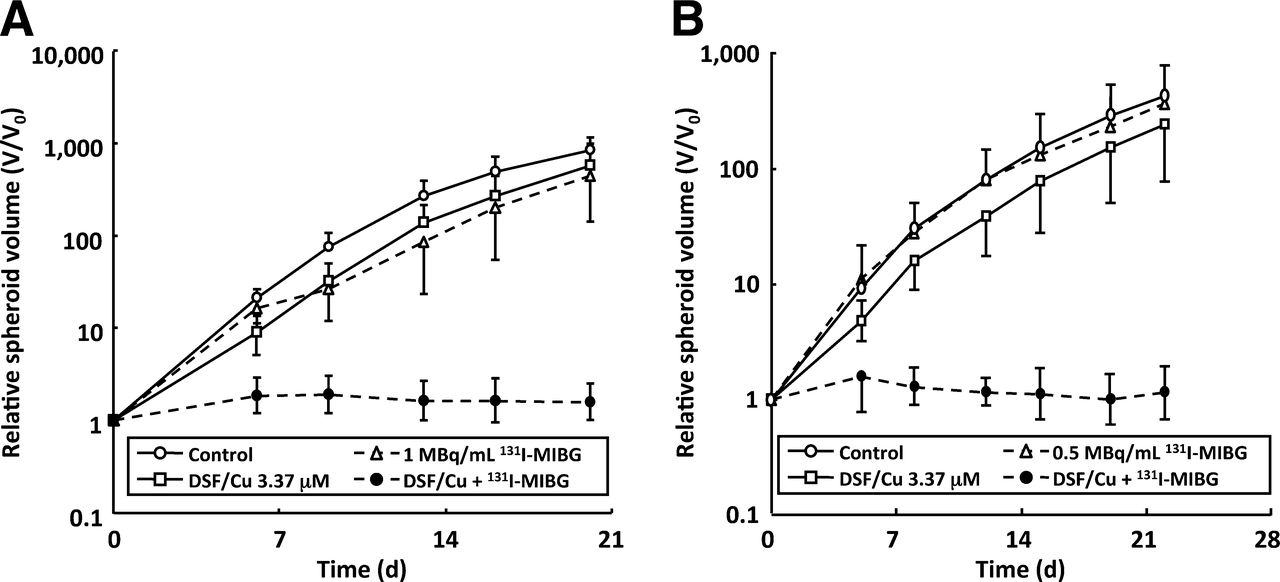
Copper chloride alone had no effect on the growth rate of spheroids composed of SK-N-BE(2c) or UVW/NAT cells, whereas disulfiram alone (at 33.7 μM) decreased the growth rate of SK-N-BE(2c) and UVW/NAT spheroids. The evaluation of a range of equimolar doses of disulfiram and copper indicated that treatment of spheroids with 3.37 μM induced growth delay but not sterilization (Supplemental Fig. 1; supplemental materials are available online only at http://jnm.snmjournals.org). Therefore, 3.37 μM (equimolar doses of disulfiram and copper) was the concentration used in subsequent evaluation of enhancement of cell kill by 131I-MIBG targeted radiation.
Using relative spheroid volume on the last day of measurement as an index of efficacy, single-agent 131I-MIBG treatment reduced spheroid growth to 54% and 87% of control for SK-N-BE(2c) and UVW/NAT spheroids, respectively. Similarly disulfiram and copper reduced growth to 69% and 58% of control volume for SK-N-BE(2c) and UVW/NAT spheroids, respectively. In contrast, the combined treatment with 131I-MIBG and disulfiram–copper sterilized all the SK-N-BE(2c) and UVW/NAT spheroids; this sterilization result manifested as a failure to increase in size throughout the duration (20 d for SK-N-BE(2c) or 22 d for UVW/NAT spheroids) of the experiments (Fig. 6).

Disulfiram enhances delay of spheroid growth induced by 131I-MIBG. Multicellular spheroids derived from SK-N-BE(2c) (A) or UVW/NAT cells (B) were treated with disulfiram plus CuCl2 (3.37 μM), 131I-MIBG (1 MBq/mL for SK-N-BE(2c) and 0.5 MBq/mL for UVW/NAT), or a combination of both. Data are expressed as mean spheroid volume at every time point divided by original volume (V/V0) ± SD of 13–23 spheroids per treatment. DSF = disulfiram.
Antitumor Activity of Disulfiram and Radiation
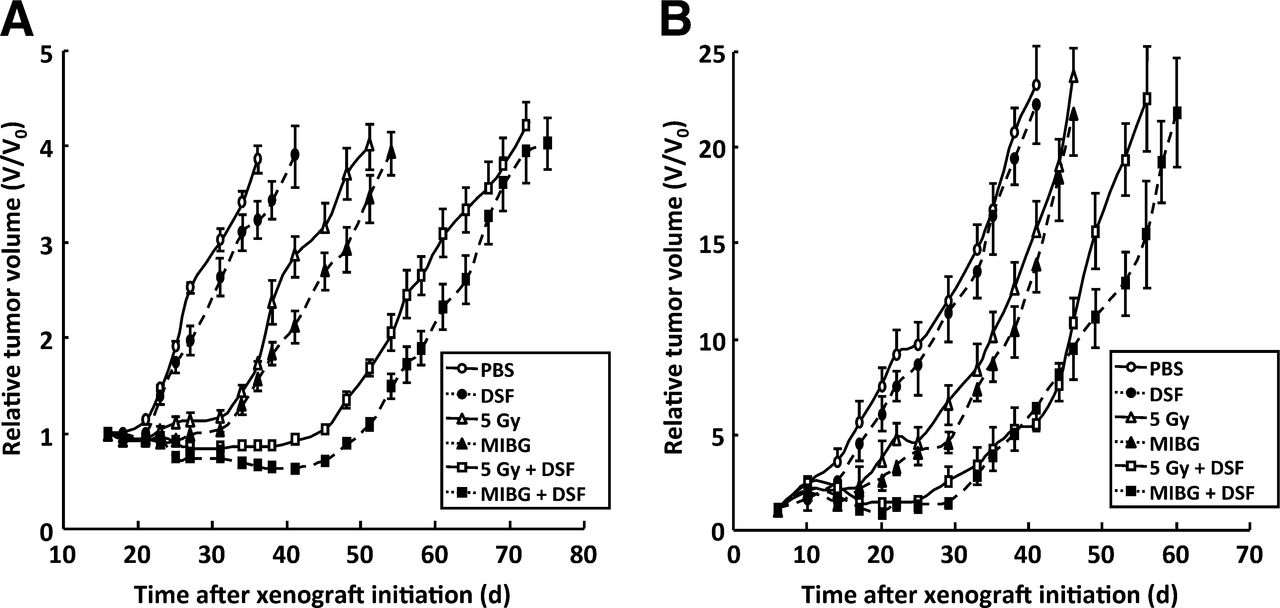
The antitumor effect of disulfiram alone or in combination with radiation treatment was studied in vivo using subcutaneous tumor xenografts grown in athymic mice. None of the animals in this study showed signs of distress. Alternative radiation modalities were examined: external-beam γ-rays from a 60Co-cobalt source and the NAT-seeking radiopharmaceutical 131I-MIBG administered by intraperitoneal injection.
Although disulfiram alone had no significant effect on tumor growth, the growth-inhibitory effect of γ-radiation or 131I-MIBG was enhanced by combination treatment with disulfiram in both tumor types (Fig. 7). The times after treatment for 4-fold (SK-N-BE(2c)) or 10-fold (UVW/NAT) increase in tumor volume are shown in Table 2. These results indicate that combination treatment with disulfiram and radiation produced a greater tumor growth delay than either agent alone, and this delay was apparent in 2 different xenograft models and using 2 different sources of radiation.
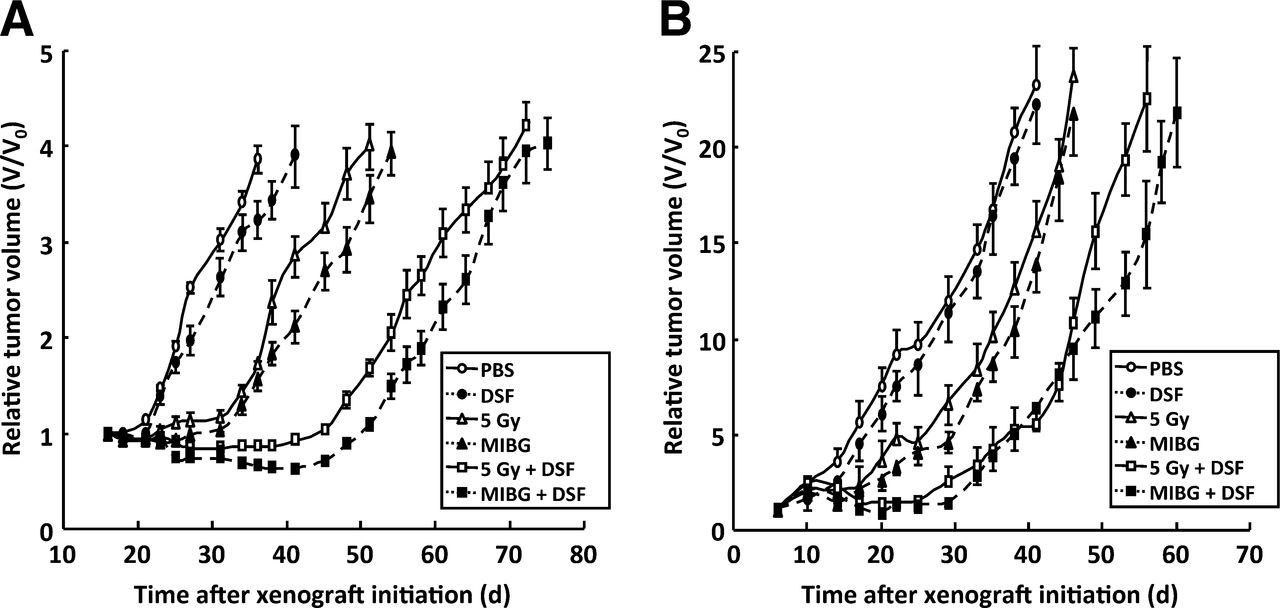
Disulfiram enhances radiation-induced growth delay of experimental tumors. Growth of human tumor xenografts in athymic mice exposed to phosphate-buffered saline, disulfiram alone (200 mg/kg), γ-radiation (5 Gy), 131I-MIBG (10 MBq), or combinations of disulfiram with γ-radiation or 131I-MIBG. Xenografts were derived from SK-N-BE(2c) cells (A) or UVW/NAT cells (B). Data are expressed as mean tumor volume at every time point divided by original volume (V/V0) ± SD. DSF = disulfiram; PBS = phosphate-buffered saline.
The Effect of Single-Agent or Combination Treatment on Tumor Growth
DISCUSSION
In response to treatment of neuroblastoma and glioma cells with disulfiram, we observed a biphasic pattern of clonogenic cell kill. Similar dose–response patterns have been reported in disulfiram-treated myeloma cells (27), in murine leukemia cell lines incubated with the reduced form of disulfiram (diethyldithiocarbamate) (28), and in pheochromocytoma cells treated with the structurally related compound pyrrolidine dithiocarbamate (PDTC) (29).
It has been suggested that many of the effects of disulfiram are due to its ability to induce oxidative stress. For example, disulfiram depleted antioxidant levels in rat cortical astrocytes (2) and elevated ROS levels in melanoma cells (3). Moreover, the toxicity of disulfiram to melanoma cells was prevented by exogenous antioxidants (3). In the present study, the antioxidant NAC prevented the reduction in clonogenic survival induced only by high concentrations (>10 μM) of disulfiram but had no effect on disulfiram-induced toxicity at lower concentrations. Clinical studies have indicated that after a single oral dose of 500 mg, or repeated doses of 250 mg, plasma concentrations of disulfiram were less than 2 μM (30,31). Therefore, a ROS-independent mechanism is expected to predominate at clinically achievable concentrations of disulfiram.
PDTC is structurally similar to disulfiram and has been shown to cause serum-dependent apoptosis of breast tumor cells (32). We also observed that the presence of serum was necessary for disulfiram-induced toxicity at low (<10 μM) disulfiram concentrations. The toxicity of some metal-chelating compounds, including disulfiram, may be dependent on the presence of metal ions such as copper (33), and high copper concentrations have been noted in commercially prepared fetal calf serum (34). Therefore, it is possible that the presence of copper in serum was, at least partly, responsible for the serum-dependent disulfiram toxicity.
Disulfiram-induced toxicity to rat astrocytes was prevented by the cell-impermeable copper chelator BCPD (2). We also observed the inhibition of clonogenic cell kill, at disulfiram concentrations less than 10 μM, by the inclusion in the medium of BCPD, suggesting a role for copper in disulfiram-induced cytotoxicity. At concentrations of disulfiram greater than 10 μM, the ROS-dependent cytotoxic effect predominated. Similarly, PDTC-induced apoptosis of breast cancer cells was also suggested to be a result of its copper-binding activity (35).
The reduced potency of disulfiram resulting from BCPD treatment was similar to that observed in response to incubation with serum-free medium, suggesting that serum in the medium was sufficient and necessary for the disulfiram-induced toxicity. The inclusion of exogenous copper in the incubation medium was sufficient to reinstate the toxic effect of disulfiram in the absence of serum, whereas BCPD, in molar excess relative to disulfiram, prevented the toxic effect of copper, presumably by competing with disulfiram for extracellular copper binding.
Clonogenic cell kill was maximal when disulfiram and copper concentrations were equimolar. Increasing concentration of disulfiram in molar excess of copper progressively reduced the cytotoxic effect of the disulfiram–copper complex, suggesting that the concentration of copper relative to disulfiram is crucial with respect to cytotoxicity.
The increased potency of disulfiram observed in the presence of serum may also be explained by copper chelation. Serum contains approximately 15 μM copper (36). Therefore, cells cultured in 10% or 15% (v/v) serum would be exposed to 1.5 or 2.3 μM copper. In serum-containing medium, the greatest cell kill resulting from disulfiram treatment occurred at 1.7 μM. This observation is consistent with the requirement for copper chelation for optimal effectiveness of disulfiram. It also suggests that there would be sufficient copper in human and murine serum to allow the formation of effective levels of the disulfiram–copper complex.
Similarly, it has been observed that the toxicity of disulfiram to Salmonella typhimurium was increased by the elevation of copper sulphate levels to equimolar concentration but was decreased in the presence of excess disulfiram (37). The presence of surplus disulfiram may result in competition between free disulfiram and disulfiram–copper complexes for cellular uptake or intracellular binding. This competition for targets may then explain the effect of increasing survival observed with increasing disulfiram concentration above copper concentration.
The dithiocarbamates disulfiram and PDTC have been reported to be effective inhibitors of proteasome activity (5), especially when complexed with copper (6). Because the proteasome plays a pivotal role in the control of cell cycle progression and survival after potentially toxic insult, proteasome inhibitors are regarded as promising anticancer agents. They can induce apoptosis as a consequence of the accumulation of undegraded proteins and the prevention of nuclear factor-κB activation. Disulfiram also facilitates copper uptake (33), increasing its intracellular availability. Therefore, it appears that disulfiram has potential as an anticancer agent. In particular, the absence of antitumor effect by disulfiram administered as a single agent as opposed to its enhancement of radiotoxicity suggests that the efficacy of disulfiram should be characterized by tumor specificity and minimal damage to tissues that fail to accumulate radiopharmaceutical.
It has been suggested that proteasome inhibitors may be effective radiosensitizers (13). Therefore, it is possible that disulfiram or its metabolites have radiosensitizing properties. Indeed, diethyldithiocarbamate has previously been shown to enhance radiation sensitivity in rapidly proliferating cells (14). We observed that disulfiram acted synergistically with γ-radiation to potentiate clonogenic cell kill at all levels of toxicity. The clonogenic survival of cells exposed to γ-radiation was not affected by removal of serum or the addition of BCPD or copper(II) chloride, indicating that radiation-induced clonogenic cell kill was not copper-dependent. Conversely, clonogenic cell kill by disulfiram at 0.34 μM, a concentration previously shown to inhibit proteasome activity in cell-based assays (5), was prevented by removal of serum or addition of BCPD and enhanced by copper supplementation. Moreover, the enhanced clonogenic cell kill resulting from combined disulfiram and γ-radiation treatment was prevented by serum removal or incubation with BCPD, whereas the addition of copper reinstated the toxicity of γ-radiation combined with disulfiram in serum-free conditions. This result suggests that copper was necessary for radiosensitization by disulfiram.
CONCLUSION
We have demonstrated in vitro and, to our knowledge in vivo, that disulfiram acted as a radiosensitizer. The effect of disulfiram alone was less appreciable in spheroids than in monolayers. Resistance associated with aggregates of cells is a commonly observed effect and has been attributed to intercellular linkage; interactions between cell and matrix; and limited penetration resulting in gradients of oxygen, nutrients, and drugs. Disulfiram did, however, enhance the radiation-induced delay of the growth of spheroids derived from 2 different tumor cell lines and retarded the growth of 2 human tumor xenografts in athymic mice. Disulfiram not only enhanced the antitumor efficacy of γ-radiation delivered from an external source but also potentiated the efficacy of 131I-MIBG targeted radionuclide therapy, in both spheroid and xenograft models. Significantly, the synergistic activity in vivo was achieved at a disulfiram dosage that had negligible effect on xenograft tumor growth and has previously been demonstrated to be associated with insignificant side effects (9). Such features are highly desirable in a radiosensitizing agent. The positive interaction between disulfiram and 131I-MIBG is worthy of further investigation.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This work was supported by grants from the Neuroblastoma Society, Molecular Insight Pharmaceuticals Inc, Sport Aiding Medical Research for Kids (SPARKS), Children with Cancer U.K. and Great Ormond Street Hospital Children’s Charity, Cancer Research U.K., and the Scottish Governmental Chief Scientist Office. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Apr. 24, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication August 29, 2012.
- Accepted for publication December 27, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}