


Abstract
The aim of this microdosing phase 0 clinical study was to obtain initial information about pharmacokinetics, biodistribution, and specific tumor targeting of the antitenascin-C mini antibody F16SIP. Methods: Two milligrams of F16SIP, labeled with 74 MBq of 124I, were intravenously administered to patients with head and neck cancer (n = 4) scheduled for surgery 5–7 d later. Immuno-PET scans were acquired at 30 min and 24 h after injection. For pharmacokinetic analysis, blood samples were taken at different time points after infusion. Tissue uptake was extracted from whole-body PET scans. In addition, ex vivo radioactivity measurements of blood and of biopsies from the surgical specimens were performed. Results: 124I-F16SIP was well tolerated. Uptake was visible mainly in the liver, spleen, kidneys, and bone marrow and diminished over time. Tumor uptake increased over time, with all 4 tumors visible on 24-h PET images. The tumor-to-blood ratio was 7.7 ± 1.7 at the time of surgery. Pharmacokinetic analysis revealed good bioavailability of 124I-F16SIP. Conclusion: Performing a microdosing immuno-PET study appeared feasible and demonstrated adequate bioavailability and selective tumor targeting of 124I-F16SIP.The results of this study justify further clinical exploration of 124I-F16SIP-based therapies.
Head and neck squamous cell carcinoma comprises malignancies in the oral cavity, oropharynx, larynx, and hypopharynx and is the sixth leading cancer by incidence worldwide. Despite advances in treatment, overall survival of patients with head and neck squamous cell carcinoma did not improve during last few decades (1). Therefore, there is still a need for novel therapeutic approaches.
Angiogenesis is one of the distinguishing features of malignancy (2), and therefore considerable efforts have been invested in the discovery of agents that block this process. One approach is the targeted destruction of established tumor vasculature, such as by using monoclonal antibodies (mAbs) directed against angiogenesis markers (3,4). The extradomain B of fibronectin represents one of the most promising neovascular markers and is strongly expressed in the vasculature of aggressive tumors. The mini antibody L19SIP (80 kDa) has been generated to specifically target the extradomain B of fibronectin (5). This antibody format appeared superior for selective tumor targeting in comparison with intact IgG and dimeric single-chain variable fragments, has been used as a carrier of anticancer agents such as immunocytokines or 131I for radioimmunotherapy, and has shown promising preliminary clinical results (5–8).
A particularly interesting candidate for targeting angiogenesis is the F16SIP antibody, which is directed against the extradomain A1 of tenascin-C. Compared with extradomain B, tenascin-C is expressed in more tumor types and often to a higher degree. Because of the high and selective expression of tenascin-C in several tumor types, including head and neck cancer (9), and the high tumor–to–normal-organ ratios observed in preclinical biodistribution studies, F16SIP is considered even more promising for antibody-based therapeutic strategies than L19SIP (3).
During the last few years, the concept of microdosing has been introduced using radiolabeled drugs in combination with PET, to obtain initial information about biodistribution, pharmacokinetics, tumor targeting, cross-reactivity with normal tissues, and interpatient variability at an early stage of drug development with a limited number of patients (10). First-in-human microdosing PET studies are allowed because of the very low dosages (1/100 of the therapeutic dose, with a maximum of 100 μg) applied, requiring limited toxicity studies and preclinical studies before human application. For protein products, such as mAbs, a maximum of 30 nmol is allowed in microdosing studies, which corresponds to a maximum dose of 4.5 mg when an intact IgG mAb (150 kDa) is used (11). Information from microdosing (phase 0) PET studies may contribute to more clever drug selection in early clinical development, with the intention of improving the success rate once a new drug is entering clinical trials (12,13). Microdosing offers potential advantages from a drug development perspective as well as from a patient perspective. On the basis of the knowledge obtained from microdosing studies, fewer patients will probably be exposed to ineffective drug administrations in clinical phase I trials, and the risk of serious unexpected adverse events occurring in phase I clinical trials may be reduced.
Although several PET microdosing studies have been performed with small-molecule drugs (14), phase 0 studies using intact mAbs or mAb fragments have to the best of our knowledge not been described before. This is remarkable, since antibody-based products will probably contribute to most approvals in the coming years (15).
The aim of this phase 0 clinical study was to evaluate the biodistribution, pharmacokinetics, and tumor-targeting performance of the mini antibody F16SIP labeled with 124I, in patients with head and neck squamous cell carcinoma.
MATERIALS AND METHODS
Patients
Four patients with previously untreated head and neck squamous cell carcinoma and scheduled for surgery 5–7 d later were included (Table 1). A World Health Organization performance status of 0–1 was required. The study was approved by the Medical Ethics Committee of the VU University Medical Center and the National Competent Authority. All patients signed an informed consent form before inclusion. Tenascin-C expression was confirmed by immunohistochemical staining in all tumors.
Patient and Tumor Characteristics
Safety
Before and up to 1 wk (until surgery) after administration of 124I-labeled F16SIP, routine laboratory analyses were performed. Vital signs including heart rate, blood pressure, body temperature, and respiratory rate were recorded before and up to 6 h after injection. For thyroid blocking, potassium perchlorate (900 mg/d) was administered orally from day −2 until 3 d after administration of 124I-F16SIP.
Synthesis of 124I-F16SIP
Clinical-grade F16SIP was provided by Philogen. Since F16SIP binds to a noninternalizing extracellular matrix target, we selected 124I as the positron-emitting isotope. 124I (870 MBq/mL; Cyclotron BV) was coupled to F16SIP via the so-called IODOGEN (Pierce)-coated mAb method (16). For a description of the procedure, including quality control results, see the supplemental data (available online at http://jnm.snmjournals.org). Two milligrams (25 nmol) of F16SIP labeled with 74 MBq of 124I were intravenously injected as a bolus.
Pharmacokinetics
Serial venous blood samples were taken to determine activity in the blood at 15 min, 2 h, 6 h, 20–24 h, and 120–168 h after injection. The 120- to 168-h samples were taken under general anesthesia just before surgery. After centrifugation, radioactivity in blood and plasma was measured in a well counter (Wallac 1480 Wizard; PerkinElmer Life Sciences). Uptake was expressed as the percentage of the injected dose per kilogram (%ID/kg).
PET Acquisition and Quantification
To minimize patient discomfort, imaging procedures were performed in a 2-d period. PET/CT scans were obtained at an early time point (30 min after injection of 124I-F16SIP) and at a later time point (24 h after injection). Whole-body PET was performed on a Gemini TF64 scanner (Philips). Manually defined regions of interest were drawn in the freely available software AMIDE 0.9.2 (17). The following regions of interest were defined: liver, spleen, kidneys, thoracic vertebra to represent bone marrow uptake, descending aorta (blood-pool activity), and, if visible, tumor. For all organs the decay-corrected mean uptake (in Bq/mL) was converted into %ID/kg.
Tissue Biopsies
In all 4 patients, biopsy samples of the primary tumor and several other accessible tissues (i.e., muscle, skin, mucosa, fat, and submandibular gland) were isolated from the surgical specimens. Lymph node sampling was not performed to avoid disturbance of routine histopathologic diagnosis. All biopsy samples were weighed, the amount of 124I was measured in the well counter and corrected for radioactivity decay, and uptake was expressed as %ID/kg. A tumor-to-blood ratio was calculated using the blood sample obtained just before surgery.
RESULTS
Patients received 71.0 ± 1.5 MBq (mean ± SD) of 124I-F16SIP (2 mg) intravenously. All patients tolerated the administration of 124I-F16SIP well, without side effects.
Figures 1 and 2 show the biodistribution of the conjugate over time. The initial PET/CT scan (performed 0.6 ± 0.3 h after injection) showed blood-pool activity and activity in liver, spleen, bone marrow, and kidneys. In one patient (patient 1), remarkably high uptake in the spleen was visible (>2-fold higher than in the other patients) (Fig. 2). At these early scans, we observed 124I-F16SIP uptake in the primary tumor in only 1 patient (1.4 %ID/kg, patient 4). The second PET/CT scan (acquired 23.5 ± 2.5 h after injection) revealed a decrease (%ID/kg) in all measured compartments (liver, 58% ± 5%; spleen, 62% ± 9%; kidneys, 34% ± 21%; blood pool, 68% ± 6%; and bone marrow, 52% ± 6%), with a concomitant increase of radioactivity at all 4 primary tumor sites. Uptake of 124I in the thyroid and stomach, representing dehalogenization of 124I-F16SIP, was negligible. Since liver as well as kidneys are visible on the scans, the main route of clearance did not become clear. Dose escalation studies might provide further insight on this aspect.

(A and B) Coronal whole-body PET images acquired at 30 min (A) and 24 h (B) after injection of 124I-F16SIP. (C) Transversal PET images obtained 24 h after injection demonstrating tumor targeting (arrows) in all 4 patients. 1 = liver; 2 = spleen; 3 = kidney; 4 = descending aorta. *Artifact due to patient motion between CT and PET examinations (arms) resulting in incorrect attenuation and scatter corrections.

Uptake of 124I-F16SIP in organs as derived from PET/CT images acquired at 30 min (A) and 24 h (B). *No tumor uptake was visible on PET/CT in patients 1–3.
Figure 1C illustrates evident tumor targeting of 124I-F16SIP after 24 h. Tumor-involved lymph nodes (1 micrometastasis and 1 macrometastasis with a diameter of 2.5 cm) were not detectable. There were no other unexpected sites of increased uptake of 124I-F16SIP throughout the body.
Blood clearance of 124I-F16SIP is illustrated in Figure 3. Whole-blood analysis revealed a mean %ID/kg of 10.4 ± 3.0 at 15 min and 3.1 ± 0.8 at 24 h after injection. For plasma, these numbers were 20.8 ± 6.8 and 5.6 ± 1.6 %ID/kg, respectively.
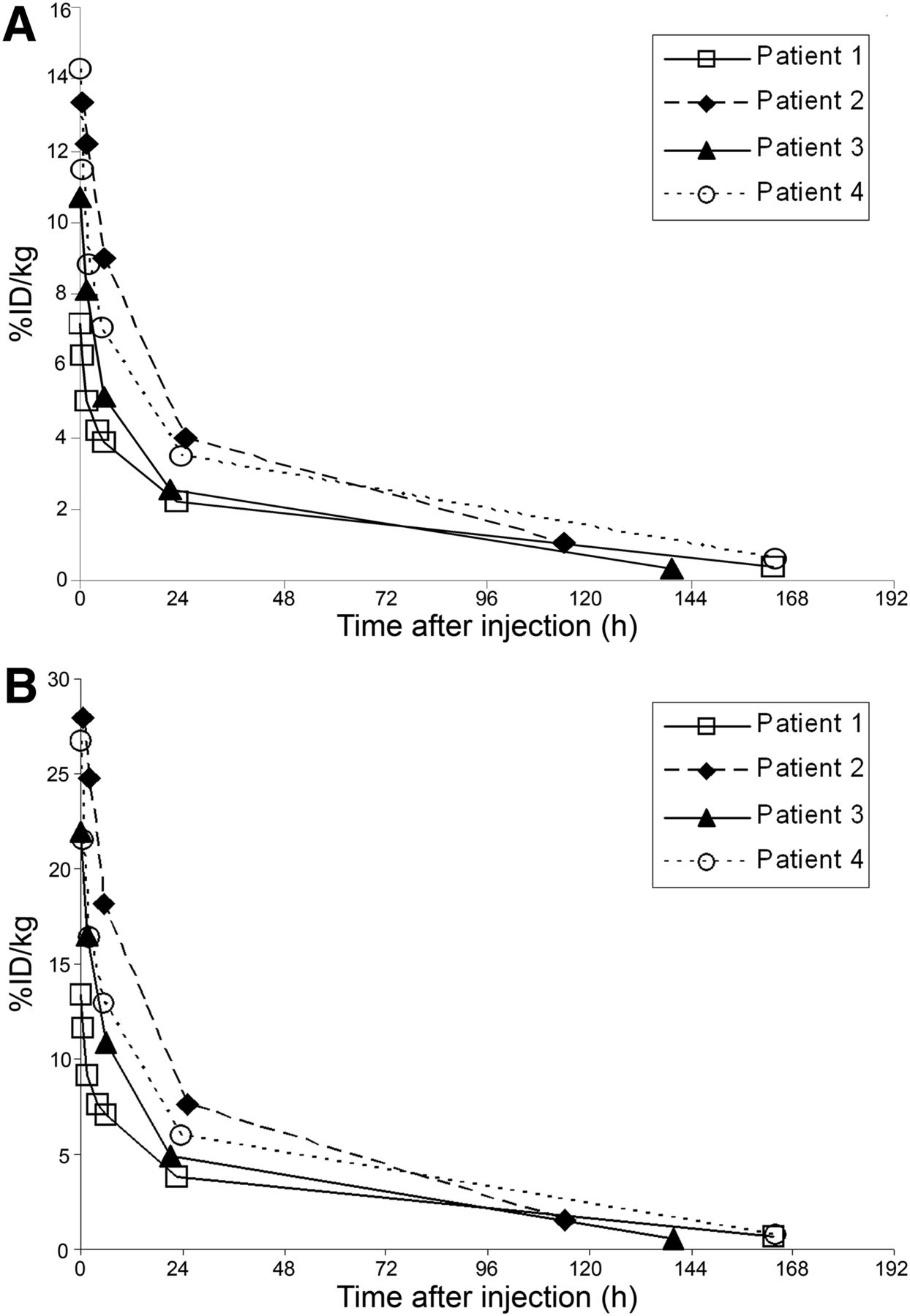
Full blood (A) and plasma (B) pharmacokinetics of 124I-F16SIP.
The results of tissue biopsies from the surgical specimen taken at 5–7 d after injection are presented in Figure 4. The tumor-to-blood ratio at this time point was 7.7 ± 1.7, and the tumor-to-muscle ratio was 15.2 ± 13.6.

Uptake of 124I-F16SIP in tissues obtained from surgical specimen at 5–7 d after injection. SMG = submandibular gland.
DISCUSSION
To the best of our knowledge, this was the first phase 0 microdosing clinical PET study using an antibody as the investigational agent. The results of this study clearly demonstrate the potential value of a phase 0 clinical study: information about biodistribution, pharmacokinetics, and proof-of-concept tumor targeting of the antibody 124I-F16SIP, obtained from a small number of patients (n = 4). Interpatient variability was limited with respect to biodistribution and pharmacokinetics, and there was evident tumor targeting in all 4 patients, with a tumor-to-blood ratio of about 8 at 5–7 d after injection.
The principal aim of a microdosing study is to obtain information about cross-reactivity, efficacy of tumor targeting, and interpatient variability under conditions that are not likely to cause toxicity. PET is a highly sensitive method to visualize and quantify these tissue-based variables (as opposed to, for example, plasma pharmacokinetics). However, there is a major concern when mAbs are evaluated in microdosing clinical trials: if the target antigen is highly expressed in a well-accessible normal organ, a large part of the infused labeled mAb may sink into that tissue, leaving just a small part of mAb available for such uses as tumor targeting. This phenomenon was demonstrated in a recent clinical immuno-PET study performed by Dijkers et al. (18). They found that a relatively high mAb dose (≥50 mg of 89Zr-trastuzumab) was required to allow reliable HER2 imaging in breast cancer patients. At a lower mAb dose of 10 mg, rapid (hepatic) clearance of 89Zr-trastuzumab was observed, most likely because of high levels of shed extracellular domain of HER2 in the plasma. After the dose had been increased, the plasma sink was saturated and the optimal biodistribution and pharmacokinetics were identified (18). This finding indicates that lack of tumor targeting in microdosing studies with mAbs is not a show-stopper per se for further clinical development, since biodistribution might improve when higher mAb dosages are used. However, if tumor targeting is observed in microdosing studies with mAbs, perspectives for clinical application are clearly better, and biodistribution and targeting might even improve when the mAb dosage is increased.
Although our study showed increased splenic uptake in 1 patient (patient 1; Fig. 1), scans also showed bone marrow uptake and there were no dominant sink organs. Furthermore, the pharmacokinetics of the 124I-F16SIP mini antibody were comparable to those of the other mini antibody, L19SIP, which was shown to be effective in clinical radioimmunotherapy studies (data not shown). Therefore, the bioavailability of 124I-F16SIP appeared to be nearly optimal, with selective tumor targeting observed in all patients despite the microdose that was used.
CONCLUSION
The promising results of the L19SIP studies (6–8), together with the information on the presented phase 0 clinical study, encourage further clinical evaluation of 124I-F16SIP–based therapeutic strategies (e.g., radioimmunotherapy) in which escalating doses will be used. Moreover, this study proved the feasibility of phase 0 clinical studies using mAbs.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. This project was financially supported by Cancer Center Amsterdam VUmc foundation and by the European Union FP7, ADAMANT. The publication reflects only the authors’ views. The European Commission is not liable for any use that may be made of the information contained. Dario Neri is cofounder and shareholder of Philogen, the company that owns the F16 antibody. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Gerard Visser (Nuclear Medicine and PET Research) for helping to develop the labeling procedure and Marijke Stigter-van Walsum (Otolaryngology/Head and Neck Surgery, VU University Medical Center) for preparing 124I-F16SIP.
Footnotes
Published online Jan. 18, 2013.
- © 2013 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication July 16, 2012.
- Accepted for publication October 10, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}