


Abstract
Receptor targeting with radiolabeled peptides has become an important topic, particularly in nuclear oncology. Strong research efforts are under way in radiopharmaceutical science laboratories and in nuclear medicine departments in Europe. The target receptors belong to the large family of G-protein–coupled receptors. The prototypes of these radiopeptides are based on analogs of somatostatin targeting somatostatin receptor–positive tumors, particularly well-differentiated neuroendocrine tumors. These radiopeptides have an important impact not only on diagnosis but also on targeted radionuclide therapy of these tumors. Besides the registered radiopeptide 111In-pentetreotide, efficient SPECT tracers labeled with 99mTc and PET agents based on generator-produced 68Ga have been developed and used in the clinic. In parallel to the development of diagnostic agents, radiopeptides labeled with the β− emitters 90Y and 177Lu are also widely used. Because the same chelators and therefore the same conjugates can be used in diagnosis and therapy, they constitute ideal theranostic pairs. This progress is driven not only by scientists and clinicians but also by patient interest groups. New radiopeptides targeting other G-protein–coupled receptors are entering the clinic, among them glucagon-like peptide 1 receptor–targeting molecules. This receptor is overexpressed on literally all benign insulinomas. 111In-labeled derivatives of the insulinotropic 39-mer peptide exendin-4 were beneficial in the pre- and perioperative localization of these benign lesions. In contrast, lack of localization may indicate malignant insulinoma. The bombesin- and gastrin-releasing peptide receptor family is potentially important because these receptors are overexpressed on major human tumors such as prostate tumors, breast tumors, gastrointestinal stromal tumors, and vessels of ovarian cancer. 99mTc-labeled peptides for SPECT and 68Ga-, as well as 64Cu-labeled agonists or antagonists, have been studied in breast tumors, prostate tumors, gastrointestinal stromal tumors, and gliomas with considerable success. A phase I therapeutic study with a 177Lu-labeled agonist has been completed. There are not enough clinical data available to reveal the significance of these new modalities in patient care, but several phase I studies are under way in larger patient cohorts using PET agents. Another G-protein–coupled receptor with high overexpression on human tumors is the gastrin/cholecystokinin-2 receptor. It is overexpressed in more than 90% of cases of medullary thyroid cancer, in small cell lung cancer, and in a subgroup of neuroendocrine tumors. Correlating with in vitro receptor localization using autoradiography of 27 patients with metastasized medullary thyroid cancer, SPECT or planar imaging of these patients resulted in a 95% sensitivity of tumor localization. Finally, another G-protein–coupled receptor is found in brain tumors and peritumoral vessels. Literally all cases of glioblastoma multiforme overexpress the neurokinin type 1 receptor; the natural ligand is substance P, which was metabolically stabilized, labeled with 90Y and 213Bi, and injected into resection cavities or directly into tumors, which were critically located via a catheter system. The major advantage of this approach appeared to be the facilitated resectability of tumors, particularly in those patients who had been treated up front with the locoregional approach. It appears that neoadjuvant treatment before resection is a valid concept. Finally, another peptide family, the arginine-glycine-aspartate–based radiotracers, has made it to the clinic labeled with a variety of radioisotopes for monitoring the integrins αvβ3 overexpressed during tumor angiogenesis.
Peptides are important regulators of growth, cellular function, and intercellular communication; they act as neurotransmitters, regulating immune response and information transduction. Key classes of peptide ligands are neurotransmitters, hormones, chemokines, cytokines, and growth factors. They are oligomers of amino acids linked together into peptide bonds. There are 21 DNA-encoded natural α-amino acids and countless synthetic amino acids, allowing for a multitude of peptide modifications.
Peptides have several advantages over proteins such as monoclonal antibodies and derivatives of them as probes for tumor targeting. For example, the advantages include solid-phase peptide synthesis via easy good manufacturing practices, quick diffusion and clearance due to small size, lack of immunogenicity, and well-established bioconjugation and radiolabeling strategies. Peptides often are metabolically unstable, but synthetic strategies to improve metabolic stability and pharmacokinetics are well developed. Table 1 shows the radiopeptide probes for imaging and targeted radionuclide therapy studied routinely and in experimental trials in hospitals in Europe.
Peptide Receptors, Disease Indications, and Radiopeptide Probes in Clinical Trials in Europe
RADIOLABELED SOMATOSTATIN ANALOGS
Despite intensive research in the field of radiolabeled peptides over the last 2 decades, only radiopeptides targeting the somatostatin receptor family have a real impact on patient care, diagnostically and therapeutically. A variety of human tumors express somatostatin receptors. There are 5 human somatostatin receptor subtypes (sst1–sst5); all are expressed on tumors to some extent, but sst2 is by far the most abundant, whereas sst4 is seldomly found. The most important conjugates used clinically are based on the octapeptide octreotide and modifications thereof. They are conjugated to chelators and labeled with different radiometals (Fig. 1).
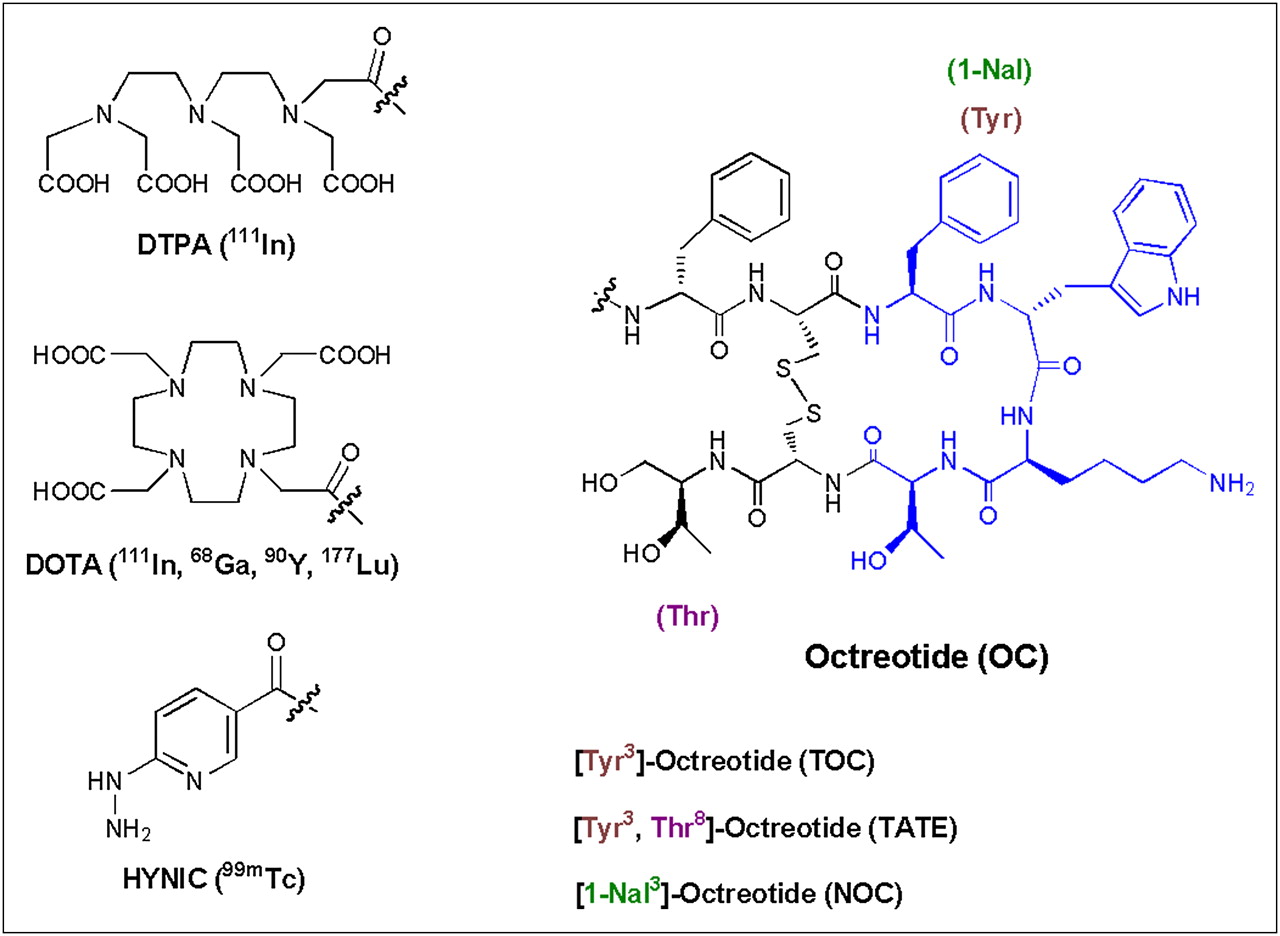
Octapeptides, chelators, and radiometals for imaging and targeted radionuclide therapy of neuroendocrine tumors in patients.
Imaging of Neuroendocrine Tumors (NETs) with Somatostatin-Based Tracers
NETs have presented a diagnostic challenge for the clinician because their slow metabolic rate, small size, and variable anatomic location have limited their detection using various imaging procedures. The traditional routine diagnostic work-up of NETs has included conventional morphologic imaging procedures such as CT, ultrasound, and MRI (1,2). The successful tumor-binding studies by Reubi and Landolt (3) using [3-125I-Tyr3]octreotide brought about the idea of developing a tracer based on the γ-emitter [3-123I-Tyr3]octreotide for somatostatin receptor scintigraphy (SRS).
At the end of the 1980s, the first imaging data were reported by the Rotterdam group using [3-123I-Tyr3]octreotide; planar images were reported for a variety of tumors (4). Because of the somewhat tedious labeling procedure and high lipophilicity of the radiopeptide, it was soon replaced by 111In-diethylenetriaminepentaacetic acid (DTPA)-octreotide (OctreoScan; Mallinckrodt Inc.), which was the first registered radiopeptide, has been used for almost 20 y in the diagnosis and staging of somatostatin receptor–positive tumors, and became the gold standard for the detection of somatostatin receptor–positive tumors (5). In addition, this radiopeptide was shown to be an independent prognostic factor of survival in well-differentiated malignant endocrine tumors (6). 111In-DTPA-octreotide SPECT and SPECT/CT were shown to outperform anatomic imaging (7) but have drawbacks as well: the sst2 affinity of 111In-DTPA-octreotide is low (inhibitory concentration of 50%, 22 ± 3.6 nM), and affinity to the other somatostatin receptor subtypes is at least 10-fold lower (>200 nM). In addition, 111In is not the ideal radionuclide, because of its unfavorable nuclear physical properties, limited availability, and high price.
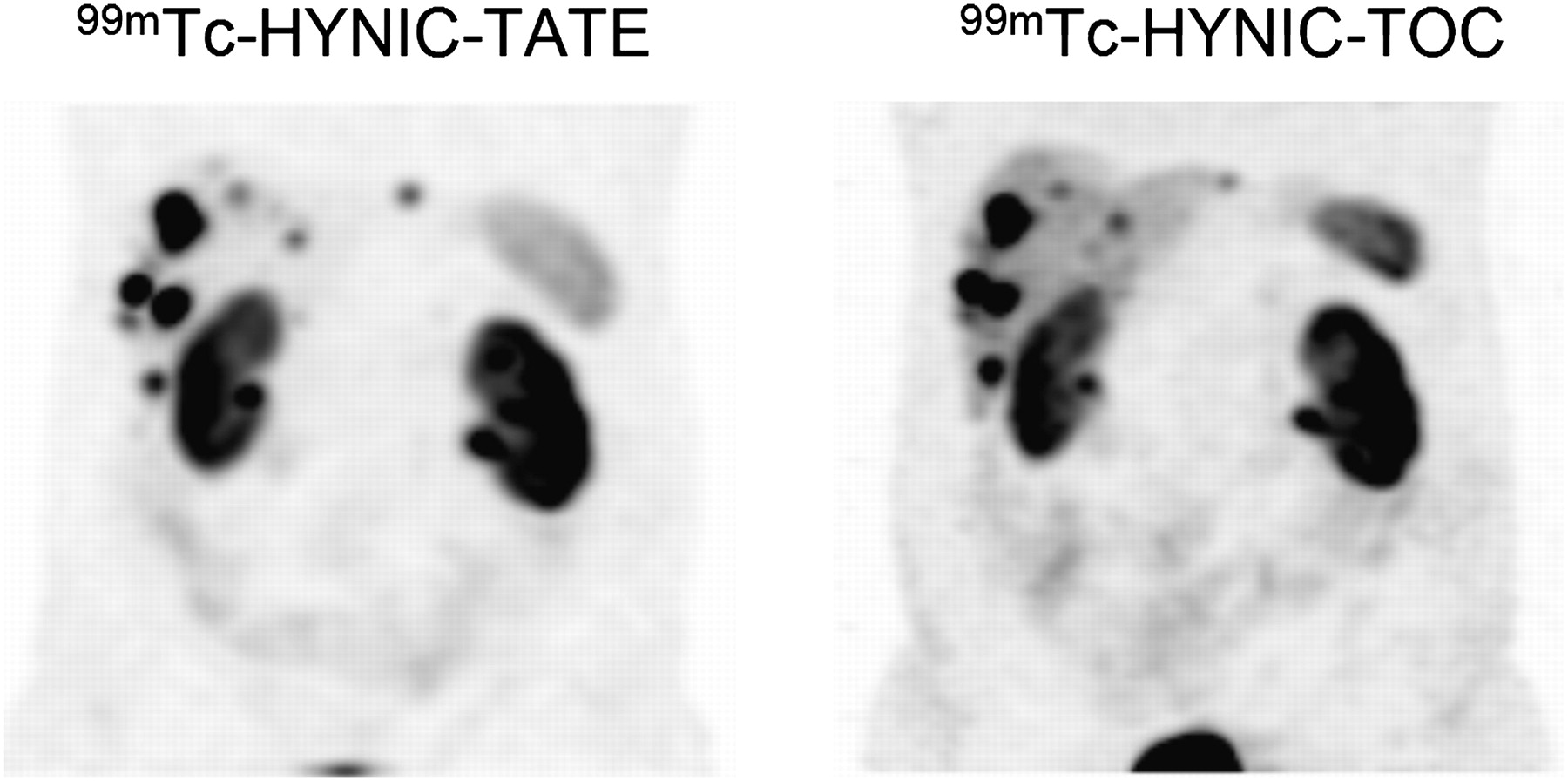
Early on, conjugates were developed for 99mTc labeling. One of the first compounds published was a bifunctional analog of hexamethylpropyleneamine oxime (8). Later, conjugates of 6-hydrazinonicotinamide (HYNIC) (9,10) and of the tetraamine derivative 1,4,8,11-tetraazaundecanecarboxy functionalized at the 6-position were developed and studied preclinically and in patients. Both families of compounds made it to the clinic and showed exquisite pharmacokinetics (11,12). The 99mTc-labeled depreotide (99mTc-NeoTect; Diatide, Inc.) is also registered in Europe; it has obtained registration for solitary pulmonary nodule imaging and showed good diagnostic accuracy for this indication but failed in comparisons with 111In-DTPA-octreotide in NETs because of its relative lipophilicity. Lebtahi et al. (13) detected more tumoral sites with 111In-DTPA-octreotide than with 99mTc-depreotide, particularly in the liver. They concluded that the detection rate of 99mTc-depreotide is much lower than that of 111In-DTPA-octreotide. Furthermore, [99mTc-ethylenediamine-N,N′-diacetic acid (EDDA)/HYNIC,Tyr3]octreotide (99mTc-HYNIC-TOC) is now available in different European countries and is registered in Poland (Tektrotyd; Polatom). The corresponding 99mTc-EDDA/HYNIC-TATE (99mTc-HYNIC-TATE) analog is also widely used. Hubalewska-Dydejczyk et al. (14) demonstrated the superiority of 99mTc-HYNIC-TATE over 111In-DTPA-octreotide in 75 patients. They found high image quality for 99mTc-HYNIC-TATE and detected distinctly more lesions than with 111In-DTPA-octreotide. The superiority was confirmed by other groups as well. The same group (15) also showed the usefulness of 99mTc-HYNIC-TATE in radioguided surgery. Cwikla et al. (16) compared the 2 tracers 99mTc-HYNIC-TOC and 99mTc-HYNIC-TATE site by site in the same 12 patients. They concluded that both tracers give excellent high-quality images at low cost and have ideal availability and a low patient dose. The 2 tracers depicted a similar number of metastatic sites. 99mTc-HYNIC-TATE seems to have some advantage in lymph node and liver metastases because of its somewhat higher hydrophilicity (Fig. 2). In summary, 99mTc-labeled somatostatin analogs have major advantages over 111In-DTPA-octreotide such as better image quality, availability, price, and patient compliance; higher sensitivity; and lower mean effective dose.
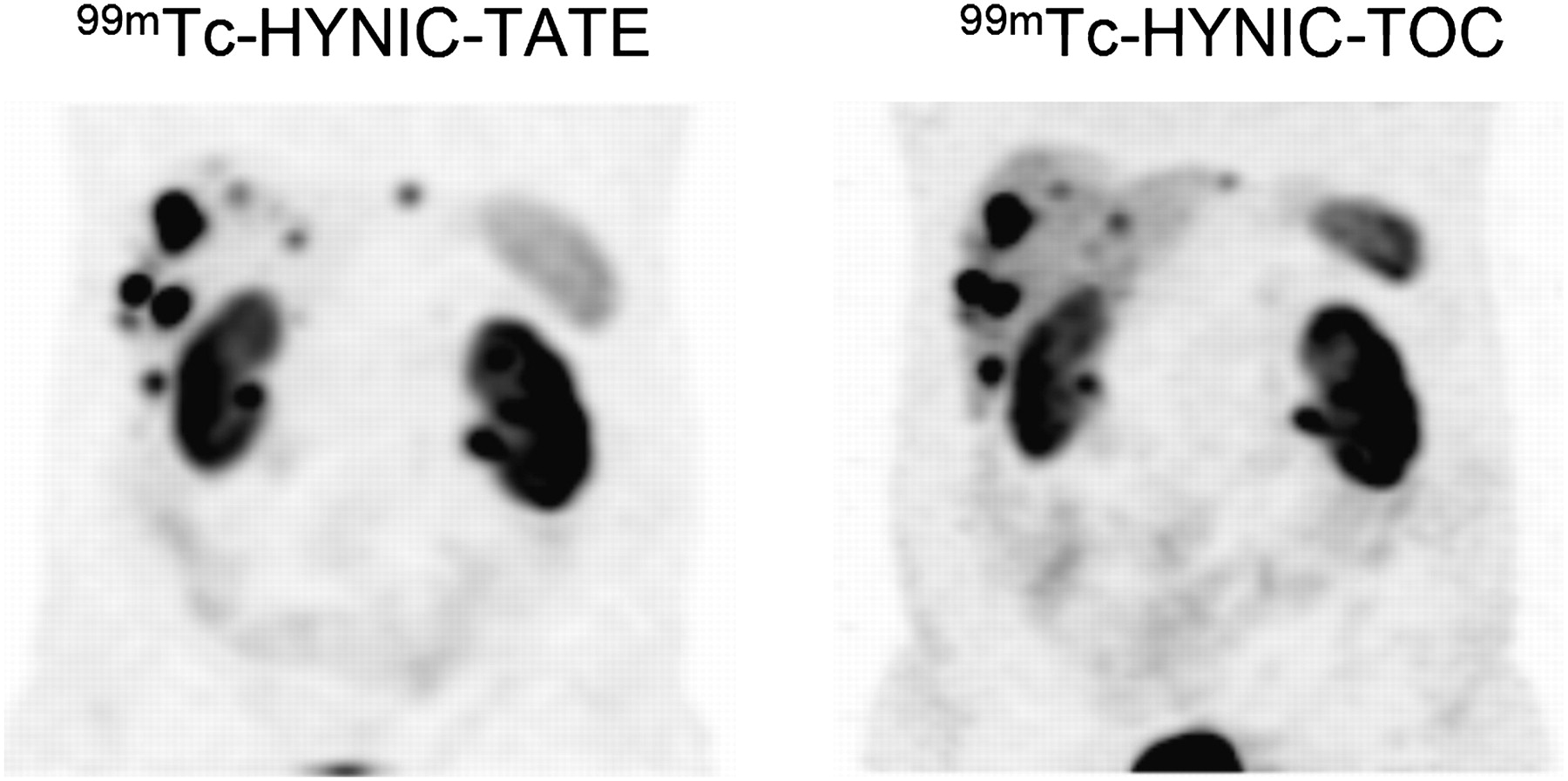
Comparative study (4 h after injection) of 99mTc-HYNIC-TOC vs. 99mTc-HYNIC-TATE in same patient showing high image quality of both tracers and lower liver but higher kidney uptake of 99mTc-HYNIC-TATE. (Courtesy of Alicja Hubalewska-Dydejczyk, Krakow University Hospital.)
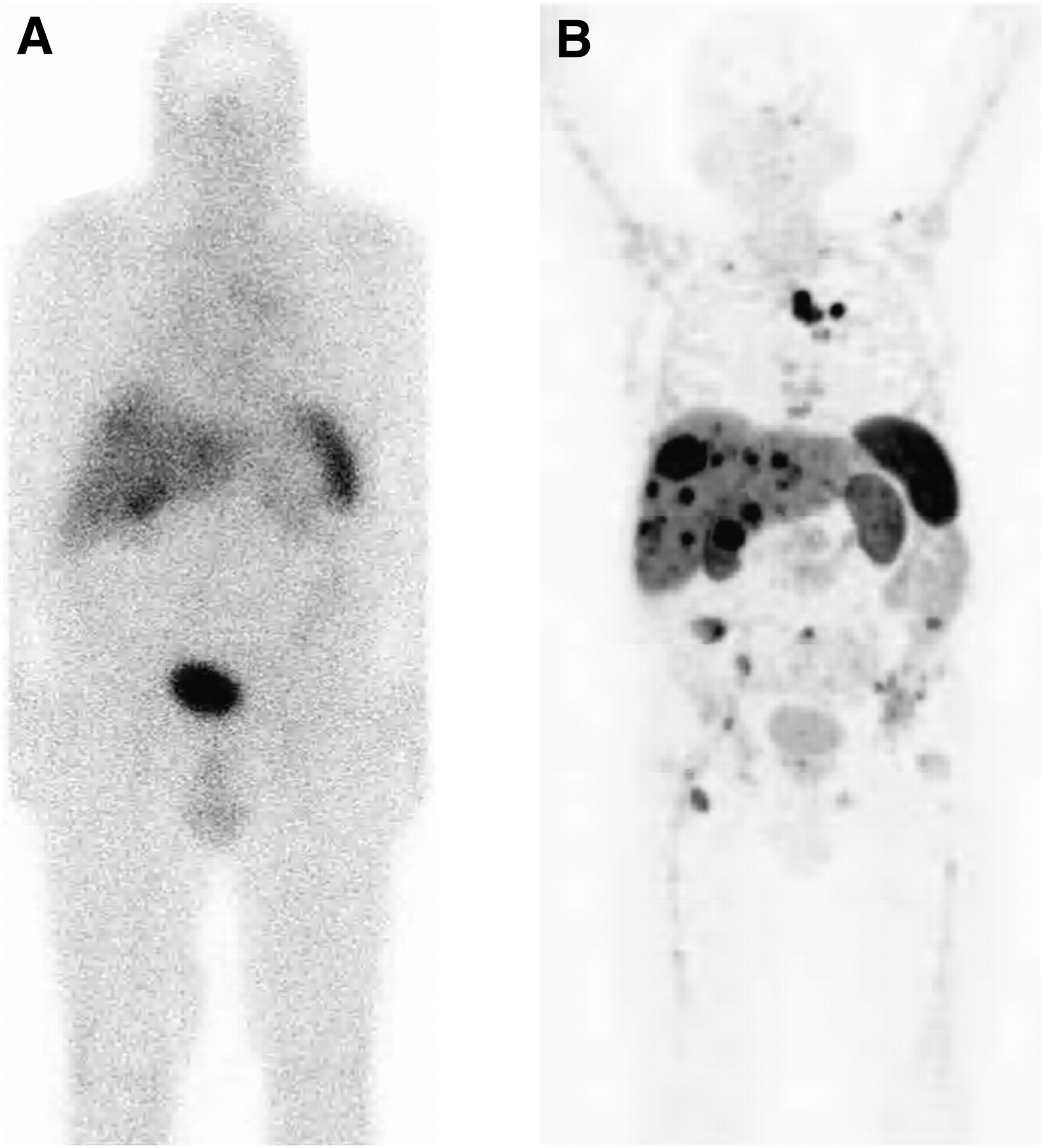
A further important step ahead in the diagnostic work-up of NETs was the introduction of novel somatostatin-based PET tracers labeled with the generator-produced radiometal 68Ga. So far, only DOTA-conjugated analogs (Fig. 1) have been studied in the clinic. There are many practical and economic advantages to using these tracers. Several 68Ge/68Ga generators are now commercially available; the long half-life of the mother (270.8 d) allows the use of the generator for approximately 9–12 mo, making its use cost-effective, and these compounds can also be synthesized relatively easily in centers without an on-site cyclotron. These factors resulted in an increasing use of this technique for the assessment of NETs over the past 5–8 y, and several clinical studies reported that in NET lesions, this technique was more accurate than either morphologic imaging or SRS. An example of the superior image quality is shown in Figure 3. Moreover, receptor-binding tracers offer the advantage of noninvasively providing data on receptor expression on NET cells, with direct therapeutic implications.

In patient with metastasized NET, planar scintigraphy image 4 h after injection of 111In-DTPA-octreotide (A) and PET image 1 h after injection of 68Ga-DOTATATE (B). Images were obtained 3 wk apart. (Courtesy of Damian Wild, University Hospital Freiburg.)
68Ga-labeled somatostatin analogs used in the clinic are based on the 3 octapeptides [Tyr3]octreotide (TOC), [Tyr3,Thr8]octreotide (TATE), and [1-Nal3]octreotide (NOC) conjugated to DOTA. Interestingly, gallium complexation to these DOTA peptides results in an improved sst2-binding affinity and an increased internalization rate, compared with other metal and radiometal (e.g., 177Lu, 90Y, and 111In)-labeled congeners (17). This factor has to be considered if dosimetric calculation and prediction of targeted therapy outcome with 90Y- or 177Lu-labeled DOTA peptides are intended. The 3 PET radiotracers differ in receptor subtype–binding profile. Whereas 68Ga-DOTATATE is sst2-selective, with the highest binding affinity of any sst2-binding peptide, 68Ga-DOTATOC binds to sst2 with high affinity and to sst5 with reasonable affinity and 68Ga-DOTANOC has high affinity to sst2, sst3, and sst5.
Most studies on these tracers have used 68Ga-DOTATOC: this was, in fact, the first tracer to be used in NET imaging. Many studies found a high tumor-to-nontumor contrast and a higher sensitivity for 68Ga-DOTATOC PET/CT than for SRS (18,19). The study with the largest patient population (84 patients with NET) reported a superior sensitivity (97%) for DOTATOC, compared with CT (61%) and SRS (52%), for the detection of NET lesions, especially for small tumors at the node or bone level (20). Moreover, the accuracy of 68Ga-DOTATOC for the detection of bone lesions was further investigated in detail in a recent study that compared PET data with conventional imaging findings. In 51 patients with well-differentiated NET, PET with 68Ga-DOTATOC performed better than CT or SRS for the early detection of secondary bone NET lesions (sensitivity of 97% and specificity of 92%) (21).
The special feature of 68Ga-DOTATATE is the selective and high affinity for sst2 (17,22). In a recent study, 51 patients with either negative (35/51) or equivocal (16/51) SRS findings were examined using 68Ga-DOTATATE. PET identified significantly more lesions than SRS and changed the management of most patients (70.6%), who were subsequently deemed suitable for peptide receptor radionuclide therapy (PRRT) (23).
68Ga-DOTANOC is also increasingly used in several centers (Fig. 4), and it was reported to have a more favorable dosimetry (24) and to be more accurate for NET lesion detection than was either conventional imaging or SRS. 68Ga-DOTANOC showed good sensitivity for the visualization of small lesions, particularly at the node and bone levels (25) and in cases with an unusual anatomic localization (26), and it provided valuable data for the identification of the primary tumor site in patients with confirmed secondary NET lesions (27). The clinical impact of 68Ga-DOTANOC PET/CT was evaluated in a population of 90 NET patients (28) and was found to affect either modification of therapy or stage in half the patients (e.g., initiation or continuation of hot or cold somatostatin analogs [36 patients] and surgical indication or exclusion [6/6 patients]). Considering the specific uptake of 68Ga-DOTANOC and the observation that well-differentiated NETs show a slower growth rate, higher somatostatin receptor expression, higher response rate to somatostatin analogs, and better outcome than NETs that are less well differentiated, the issue has been raised of whether it is possible to define PET/CT parameters that can predict patient outcome. Recently, in a population of 44 NET patients, 68Ga-DOTANOC PET/CT was reported to provide prognostic information (29). In fact, the maximum standardized uptake value was significantly higher in patients with stable disease (SD) or partial response (PR) at follow-up of PRRT, and a maximum standardized uptake value cutoff allowed differentiation of patients with SD or PR at follow-up from those with progressive disease (PD).
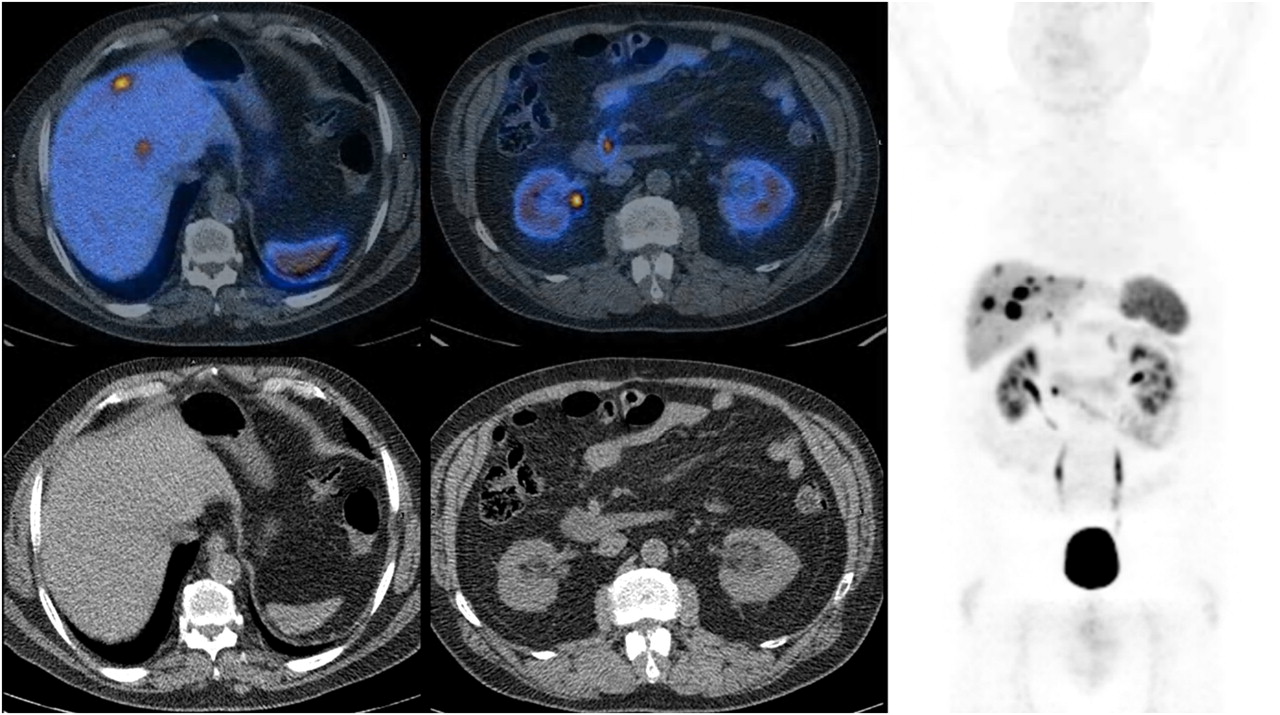
68Ga-DOTANOC PET/CT images of patient with multiple secondary NET liver lesions (maximum standardized uptake value, 36) and focal uptake in mesenteric area near duodenum (maximum standardized uptake value, 17). All shown lesions express somatostatin receptor.
The observed differences in receptor-binding affinities of 68Ga-DOTATOC, -TATE and -NOC (17) have not yet found a direct clinical correlate, but there is some indication that such differences are associated with advantages in clinical use. Wild et al. (30) studied 21 consecutive patients within 2 wk in a crossover design with 68Ga-DOTATATE and 68Ga-DOTANOC and found a distinct advantage for 68Ga-DOTANOC, which localized more lesions in, especially, the liver and pancreas. The authors concluded that the higher sensitivity may be due to the broader affinity profile of 68Ga-DOTANOC. More clinical studies that include in vitro receptor autoradiography are needed to confirm this conclusion. Currently, the use of the three 68Ga-labeled somatostatin analogs is limited to specialized centers as part of clinical trials, and each center selects its preferred peptide. One can estimate that at least 10,000 scans are being performed yearly in Europe at about 100 centers.
Although 68Ga-labeled somatostatin analogs have only recently been introduced into the clinic, the number of studies reporting better results for these analogs than for other imaging procedures has increased exponentially, as have comparative studies with other PET tracers (18F-FDG and 18F-3,4-dihydroxyphenylalanine). In a direct comparison between 68Ga-DOTANOC and 18F-3,4-dihydroxyphenylalanine in a limited patient population (13 patients), 68Ga-DOTANOC showed a higher number of lesions and in more cases identified the site of the occult primary tumor (25). A comparison of 68Ga-DOTATATE with 18F-3,4-dihydroxyphenylalanine in 25 patients with well-differentiated NET (31) showed higher sensitivity for 68Ga-DOTATATE (96% vs. 56%). It is interesting to note that since metabolic and receptor-binding PET tracers can investigate different tumor features, their use in the same patient may offer more detailed information on the biologic aspects of the tumor. Different patterns (Fig. 5) of tracer uptake (68Ga-somatostatin analogs vs. 18F-FDG) have been described within areas of the same tumor lesion or in different lesions within the same patient (32). This finding is particularly relevant because the identification of areas with high 18F-FDG uptake identifies the presence of less differentiated tumor areas that are associated with a worse prognosis and require systemic chemotherapy. In fact (33), a different pattern of tracer uptake was also documented in high- and low-grade NET: although higher 68Ga-DOTATATE uptake was reported in low-grade than in high-grade lesions, 18F-FDG uptake was significantly higher in high-grade than in low-grade lesions. 68Ga-DOTATOC was also reported to provide more accurate information than 18F-FDG in well-differentiated NET lesions (34).

68Ga-DOTANOC PET/CT images (left) and 18F-FDG PET/CT images (right) of patient with pancreatic NET. 68Ga-DOTANOC PET/CT shows increased pancreatic uptake at body and tail level (maximum standardized uptake value, 48), consistent with focal area overexpressing somatostatin receptor. 18F-FDG PET/CT images, in contrast, are negative for uptake.
PET/CT with 68Ga-DOTA-somatostatin analogs is an accurate imaging modality for evaluating patients with well-differentiated NET lesions. All described compounds have agonistic properties, which were considered mandatory because of the ability of these compounds to induce internalization of the peptide–receptor complex. Recently, 111In-labeled somatostatin-based antagonists with affinity to sst2 and sst3, which internalize poorly, were shown to provide potentially even better tumor visualization than do agonists (35). One of the first articles describing 68Ga- and 64Cu-labeled sst2 antagonists indicated the high potential of these radiopeptides in PET/CT (36). This new family of tracers may even present a better imaging option.
PET/CT with 68Ga-DOTA peptides not only accurately visualizes NET lesions but also noninvasively provides valuable information on somatostatin receptor expression patterns on tumor cells, with direct therapeutic implications. In fact, patients with somatostatin receptor–expressing lesions are candidates for targeted therapy with either hot or cold somatostatin analogs.
PRRT of NETs with Somatostatin-Based Analogs
PRRT is a new treatment modality and is well established in Europe for patients with unresectable somatostatin receptor–positive NETs; thousands of patients have been treated successfully with this approach. However, there is a variety of treatment protocols using different somatostatin analogs and radionuclides. To our knowledge, no controlled, randomized multicenter study has yet been published. Nevertheless, the results are convincing. Roughly 25% of all treated patients achieve an objective tumor response. Serious side effects have been rare.
The first radiopharmaceutical used in PRRT was 111In-DTPA-octreotide, which was developed for diagnostic purposes with activities between 10 and 160 GBq (37–40). The rationale for its use was represented by the fact that, in addition to the γ-radiation, 111In emits Auger electrons (low-energy electrons with a short tissue penetration range of 0.02–10 μm). They exert their cytotoxic potential within the nucleus at close proximity to the DNA after receptor internalization. A major breakthrough was achieved by the conjugation of somatostatin analogs with the chelator DOTA, which stably binds all M3+ radiometals, such as 90Y (high-energy β+ emitter, 2.28 MeV; half-life of 64 h) and 177Lu (low-energy β− emitter, 0.5 MeV; half-life of 6.7 d) as well as some positron (β+) emitters (e.g., 68Ga), and permits the use of such nuclides for therapy and imaging purposes (41,42). Several studies using 90Y-DOTATOC, 90Y-DOTA-lanreotide, and 90Y-DOTATATE, as well as 177Lu-DOTATATE and 177Lu-DOTATOC, alone or in combination have been published.
Studies Using 90Y-DOTATOC, 90Y-DOTA-Lanreotide, and 90Y-DOTATATE
The first report on PRRT using 90Y-DOTATOC was published in 1997 by a group at Basel University (43). Biodistribution and clearance were better for 90Y-DOTATOC than for 111In-DTPA-octreotide. The kidney-to-tumor ratio was 1.9 times lower for 90Y/111In-DOTATOC than for 111In-DTPA-octreotide (43). One year later, the same group reported results for 10 patients. In this small cohort, 50% of patients experienced PR and 50% experienced SD (44). In the following studies, patients were treated with a cumulative dose of 6.0 or 7.4 GBq of 90Y-DOTATOC per square meter in 4 cycles. The overall response (OR) was about 25% (45,46). Similar results were found in a more extensive study of 116 patients, who were treated with the same dose (an OR of 27% was found) (47).
A research group from the European Cancer Institute in Milan used a higher range of cumulative dose (5.9–11.0 GBq in 2 cycles) in 21 patients with NETs, achieving an OR of 29% (48). In a more recent study (49) of 141 patients with various somatostatin receptor–positive tumors, an OR of 26% and an SD rate of 56% were found. The favorable response rates were higher in patients who presented with SD before therapy than in patients whose disease was progressive before therapy.
Long-term follow-up and survival data for 90Y-DOTATOC were published by Valkema et al. (50). In this study, 58 patients were treated in a dose-escalating study with 1.7–32.8 GBq of 90Y-DOTATOC. The response rates were comparable to those of other studies using 90Y-labeled somatostatin analogs, but in addition to the encouraging response rates a significant longer overall survival (36.7 mo) was found, compared with a group treated with 111In-DTPA-octreotide (median survival, 12.0 mo) (39). Overall survival was significantly better in patients who had SD at baseline than in patients who had PD at baseline, in patients without liver metastases than in patients with liver metastases, and in patients with a high Karnofsky performance score than in patients with a low Karnofsky performance score (50).
In a multicenter phase I study (40,51), the goal was to determine the maximum tolerated injected activity in 1 or 4 cycles. Escalating doses of 90Y-DOTATOC up to 9.3 GBq/m2 as a single injection and up to 14.8 GBq/m2 in 4 cycles were administered to 60 patients, of whom 54 could be treated with their maximum allowed activity, achieving an OR of 7%. The median time to progression was not reached until 26 mo after the last treatment cycle. However, the maximum tolerated injected activity could not be determined, because on the basis of 86Y-DOTATOC dosimetry, the dose to the red marrow would be too high.
Recently, the Basel group reported retrospectively on the response, survival, and safety profile for 90Y-DOTATOC in 1,109 patients with NETs (52). Patients were recruited from 29 countries and more than 100 centers between October 1997 and February 2010. Biochemical, morphologic, and clinical response rate were determined: 34.1% of the patients had a morphologic response, 15.5% had a biochemical response, and 29.7% had a clinical response. Survival benefit correlated with morphologic and clinical response. Adverse events such as severe transient grade 3 or 4 hematologic toxicity were reported in 12.8% of patients. One patient developed myelodysplastic syndrome, and 1 developed acute myelotic leukemia. One hundred two patients (9.2%) developed severe renal toxicity (grade 4 or 5). The authors concluded that their study examined the outcome of 90Y-DOTATOC therapy in a large cohort of patients with a wide spectrum of NETs, that the high tumor uptake in the pretherapeutic diagnostic scans with 111In-DTPA-octreotide was significantly associated and predictive of lengthened survival, and that high kidney uptake in the diagnostic scan predicted a risk of severe kidney toxicity.
The analog 90Y-lanreotide was investigated in a European multicenter trial that included 154 patients who received cumulative treatment activities ranging from 1.9 to 8.6 GBq of 90Y-DOTA-lanreotide. The therapy entry criterion was PD at the time of planned therapy. Preliminary treatment results for 154 patients indicated SD in 41% and PR in 14% (53).
Clinical data on 90Y-DOTATATE have been published by Baum et al., who reported an objective response rate (PR) of 39% (54,55). Cwikla et al. reported on the effect of 90Y-DOTATATE treatment in 60 patients with histologically proven gastroenteropancreatic NETs, treated with a cumulative activity of 15.2 GBq (56). At 6 mo after the final treatment, a radiologic PR was observed in 13 patients (22%), and the remaining patients had SD. Median progression-free survival was 17 mo, and median overall survival was 22 mo. In patients with early PD, progression-free survival was 4.5 mo and overall survival 9.5 mo, whereas in those with SD or PR, progression-free survival and overall survival were 19.5 and 23.5 mo, respectively (56).
In summary, OR rates in patients treated with 90Y-DOTATOC, 90Y-DOTATATE, and 90Y-DOTA-lanreotide ranged from 6% to 39% despite differences in the protocols used. These results and the prolonged overall survival represent an improvement in therapeutic effectiveness, compared with studies of 111In-DTPA-octreotide.
Studies Using 177Lu-DOTATATE and 177Lu-DOTATOC
In 2003, the first study using 177Lu-DOTATATE was published (57). In this study, 35 patients with gastroenteropancreatic NETs were treated with escalating injected activities up to a final cumulative activity of 22.2–29.6 GBq. An OR of 38% was found. The effects of the therapy on tumor size could be assessed in 34 patients. Three months after the last administration, a complete response was found in 1 patient (3%), PR in 12 (35%), SD in 14 (41%), and PD in 7 (21%), including 3 patients who died during the treatment period. Tumor response correlated positively with high uptake on pretherapeutic octreotide scintigraphy, a limited hepatic tumor mass, and a high Karnofsky performance score. No serious side effects were reported (57).
In a more recent evaluation, 310 patients were treated with up to a cumulative activity of 27.8–29.6 GBq, usually in 4 treatment cycles, with treatment intervals of 6–10 wk. Serious adverse events that were likely attributable to the treatment included myelodysplastic syndrome in 3 patients and temporary, nonfatal, liver toxicity in 2 patients. In 2 patients, serious kidney toxicity was observed but was judged to be unrelated to the treatment. Complete and partial tumor remissions occurred in, respectively, 2% and 28% of the patients. A minor tumor response occurred in 16% of patients. Thus, OR occurred in 46% of patients. Median time to progression was 40 mo; median overall survival from the start of treatment was 46 mo, and median overall survival from the time of diagnosis was 128 mo. The authors concluded that this therapy has few serious adverse effects and can be regarded as safe. Compared with historic controls, there was a survival benefit of 40–72 mo from diagnosis (58).
The first results on 177Lu-DOTATOC were reported in 2005 for 27 patients after relapse from 90Y-DOTATOC therapy (59). The inclusion criterion was that the patients achieved at least SD after 90Y-DOTATOC treatment and thereafter experienced PD again. After restaging, the study found PR in 2 patients, minor response in 5 patients, SD in 12 patients, and PD in 8 patients, and no serious side effects were seen. The authors concluded that 177Lu-DOTATOC therapy in patients who experienced relapse after 90Y-DOTATOC treatment is feasible, safe, and efficacious. Frilling et al. used 177Lu-DOTATOC treatment in 20 patients with metastatic nonresectable NETs refractory to 90Y treatment (60). In 8 patients, the treatment was repeated more than once. No serious adverse events were documented. After restaging, a PR was found in 5 patients, SD in 11 patients, and PD in 4 patients.
A recent preliminary study from the National Cancer Institute in Milano reported the effect of 177Lu-DOTATOC in patients with NETs refractory to conventional therapies. Patients were treated with an original plan including 4 therapeutic cycles alternating 177Lu-DOTATATE (5.55 GBq) and 90Y-DOTATATE (2.6 GBq) (61). Treatment was well tolerated, with no adverse event found during a median follow-up time of 7 mo. A PR was found in 66% of patients evaluated 3 mo after the fourth treatment, and SD was found in 27% of the patients.
A similar protocol was chosen by Kunikowska et al. in a prospective study of 50 patients with disseminated NETs (62). One group (group A) was treated with 90Y-DOTATATE, and a second group (group B) received 1:1 90Y:177Lu-DOTATATE. The administered activity was 3.7 GBq/m2 of body surface in 3–5 cycles. The patients were matched with respect to age; sex; origin, number, and location of tumors; and size of metastases. The median survival time was 26.2 mo in group A and was not reached during the observation period (54 mo) in group B. Median event-free survival was also superior in group B (29.4 mo) versus group A (21.4 mo). The safety of both protocols was comparable; side effects were mild and rare.
Overall, these studies show that PRRT combining 90Y- and 177Lu-labeled somatostatin analogs is a promising approach to maximize the therapeutic effects and minimize the toxicity of PRRT.
Generally, PRRT can be regarded as a relatively safe treatment, and severe side effects are rare, especially when compared with the side effects of chemotherapy. The side effects of PRRT can be categorized as acute effects or as more delayed effects caused by radiation toxicity. The acute effects occurring up to a few days after injection of the therapy include nausea, vomiting, and increased pain at tumor sites. These side effects are generally mild, are minimized by a slow infusion, and can be prevented or reduced by symptomatic treatment. Severe toxicity may rarely occur as a result of the radiation absorbed dose in healthy organs. The organs at risk are mainly the kidneys, the bone marrow, and to a lower extent the liver.
RADIOLABELED GLUCAGON-LIKE PEPTIDE-1 ANALOGS
The targeting of another receptor entity belonging to the large family of G-protein–coupled receptors, the glucagonlike peptide 1 (GLP-1) receptor, may change the diagnostic work-up and therefore current medical practice with regard to the localization and imaging of insulinoma. The GLP-1 receptor is highly overexpressed on essentially all benign insulinomas but also on gastrinomas (63). Benign insulinomas also may present a diagnostic challenge; they arise from the β-cells of the pancreas and present as small nodules (usually <2 cm). They secrete insulin, leading to potentially life-threatening hypoglycemia. Complete tumor resection is the only way to cure these patients. Preoperative localization may cause problems with normal radiologic techniques, and the sensitivity of ultrasound, MRI, and CT is low, with a detection rate of less than 40% of the lesions. The use of endoscopic ultrasound has higher sensitivity, ranging from 57% to 94%. Intraarterial selective Ca2+ stimulation and venous sampling improved the sensitivity even further to up to 96% (64,65). But this method is invasive and somewhat risky, and the exact location of the tumor cannot be determined. There is a clear need for a safe, sensitive, and easy-to-perform imaging method. Preclinical studies using 111In-, 99mTc-, and 68Ga-labeled peptides based on the 39-mer peptide exendin-4 looked promising (66) and allowed the first clinical studies. Preliminary studies using the peptide Lys40(Ahx-DOTA-111In)NH2-exendin-4 in 6 patients provided proof of the concept that this new tracer allowed sensitive preoperative localization of the tumor; localization was possible in all 6 patients (tumor size between 9 and 18 mm). One insulinoma not detectable with any of the conventional imaging methods (CT, endoscopic ultrasound, 111In-DTPA-octreotide scintigraphy, intraarterial selective Ca2+ stimulation, and venous sampling) could be localized with the GLP-1 receptor scan and turned out to be an ectopic insulinoma. In addition, precise intraoperative localization of the insulinoma using a γ-probe was feasible up to 14 d after tracer injection (67,68). After surgical removal of the insulinoma, the presence of high GLP-1 receptor expression was assessed in the tumor tissue in vitro by GLP-1 receptor autoradiography. These studies along with autoradiographic studies in 33 patients, 32 of which were positive in regard to GLP-1 receptor expression, indicate that this approach is likely to affect the algorithm of pre- and intraoperative localization of suspected insulinoma. Interestingly a recent study of 11 patients with malignant insulinoma showed that, in contrast to benign insulinoma, malignant insulinoma often lacks the GLP-1 receptor; only 4 of the 11 patients had positive findings on a 111In-DTPA-exendin-4 scan (and on in vitro autoradiography) whereas 8 patients showed positive findings on 68Ga-DOTATATE PET/CT (and autoradiographic sst2 expression). The authors concluded that negative GLP-1 receptor findings may potentially indicate the presence of a malignant insulinoma (66). Unfortunately, the high kidney uptake of these tracers precludes any radiotherapeutic application at the moment.
RADIOLABELED BOMBESIN/GASTRIN-RELEASING PEPTIDE (GRP) ANALOGS
Bombesin is a tetradecapeptide neurohormone and an amphibian homolog of mammalian GRP (a 27-mer peptide). Bombesin-like peptide receptors also belong to the family of G-protein–coupled receptors and include 4 subtypes: the neuromedin B receptor, the GRP receptor, the bombesin 3 receptor, and the bombesin 4 receptor (69). These receptors have been found in different tumor types. In particular, GRP receptors have been demonstrated in major human tumors such as breast cancer (70%) and prostate cancer (30 of 30 invasive prostatic cancers) and in high-density prostatic intraepithelial lesions, peritumoral vessels of ovarian cancer, renal cell cancer, small cell lung cancer, and gastrointestinal stromal tumors (70–75).
99mTc-Labeled Bombesin-Based Peptides
Various bombesin analogs have been developed for 99mTc labeling and SPECT. In particular, the truncated bombesin(7–14) was coupled to an N3S-chelator via a Gly-5-aminovaleric acid spacer (99mTc-RP527) and was studied preclinically and in humans with prostate and breast cancer (76,77). The tracer was excreted via the kidneys and gastrointestinal tract; high contrast was possible in the thoracic region. Imaging of abdominal tumors was somewhat problematic because of the lipophilic character of the peptide. 99mTc-RP527 showed specific uptake in 4 of 6 breast cancer lesions, including lymph nodes and bone metastases, and in 1 of 4 prostate carcinomas; the tracer showed pronounced pancreas uptake. The authors concluded that the application of 99mTc-RP527 was safe and resulted in specific tumor localization and good imaging characteristics. In this group of patients, no immunohistochemical or autoradiographic data were available to confirm the presence of GRP receptors. In a recent article (78), the authors presented imaging data on 9 tumor patients with a diagnosis suggestive of breast carcinoma and in an additional 5 breast tumor patients with tamoxifen-resistant bone metastases. The authors found 99mTc-RP527 uptake in the primary tumor of 8 of 9 patients and in involved lymph nodes. No uptake was found in the tamoxifen-resistant tumors. A strong correlation was found with the presence of GRP receptor as determined by immunohistochemistry.
Bombesin Analogs for PET of Tumors
The currently available data for potential use of bombesin analogs as PET tracers are mainly preclinical. Several bombesin analogs and bombesin antagonists have been developed (79–85) and labeled with different PET radioisotopes (68Ga, 64Cu, 18F) and using different chelators (86). Overall, good-quality images were obtained in preclinical models using bombesin-like peptides as tracers to image bombesin receptor–expressing tumors (84).
Only a few human studies have used positron-emitting bombesin analogs. Seventeen patients with gastrointestinal stromal tumor treated with imatinib (because of an unresectable primary tumor, a recurrent gastrointestinal stromal tumor, or metastatic disease) were studied by dynamic PET using both 68Ga-BZH3 (a pan–bombesin analog binding to neuromedin B receptor, GRP receptor, and bombesin 3) (87) and 18F-FDG (88). 18F-FDG studies were superior: 14 of 17 patients were 18F-FDG–positive, whereas only 7 of 17 patients showed 68Ga-BZH3 uptake. The authors concluded that only a subgroup of gastrointestinal stromal tumor patients showed significant 68Ga-BZH3 uptake; the authors therefore did not recommend the routine use of bombesin analogs in gastrointestinal stromal tumor patients. Interestingly, the authors reported a single case of recurrent gastric gastrointestinal stromal tumor that was 68Ga-BZH3–positive but false-negative with 18F-FDG: they suggested that the low proliferative activity within the tumor might explain the differential pattern of tracer uptake. Dimitrakopoulou-Strauss et al. (89) recently described the use of 68Ga-BZH3 in a small patient population (15 patients) with recurrent gliomas. All patients were studied with 18F-FDG PET/CT for metabolic staging and with 68Ga-BZH3 PET/CT to detect bombesin receptor expression. In fact, available treatment options for gliomas are largely dependent on tumor grade; therefore, the possibility of noninvasively assessing tumor grade by PET is interesting. The authors observed that 68Ga-BZH3 was helpful for differentiation between low- and high-grade gliomas, in particular for the identification of high-grade tumors. In fact, in their small patient population, 18F-FDG correctly identified 3 of 6 patients with World Health Organization (WHO) grade II disease, 3 of 6 patients with WHO III, and none of the 3 patients with WHO IV. In contrast, 68Ga-BZH3 revealed enhanced uptake in 3 of 6 patients with WHO II, 4 of 6 patients with WHO III, and 3 of 3 patients with WHO IV.
Recently, bombesin antagonists were studied clinically in primary prostate cancer using PET. A statin-based peptide labeled with 64Cu (90) showed focal uptake in the primary tumor in 3 of 4 patients. The pharmacokinetics are promising, because fast washout from the kidneys and the pancreas was observed.
The paucity of available data in the literature on the use of bombesin peptides for PET does not allow generalization of results obtained in only small patient populations; however, the results obtained so far seem promising, and further studies are needed to better ascertain the role of these compounds in clinical practice.
Only a single report is available on the radiotherapeutic application of a 177Lu-labeled bombesin derivative. Bracco developed a bombesin(7–14) derivative coupled to DOTA via a Gly-4-aminobenzoyl spacer. Convincing data were obtained in mouse models. Seven patients with hormone-refractory metastatic prostate cancer were studied in a phase 1 study with increasing 177Lu activities (≤4.17 GBq cumulative) (91). An interim analysis found uptake in the lesions of 5 of 7 patients. Side effects included abdominal cramps, nausea, and diarrhea and were controlled by specific drugs. Strong uptake was seen in the pancreas, which was the dose-limiting organ. At 3 mo after the end of treatment, all patients showed progression. One problem with this therapeutic agent appeared to be its low plasma stability.
RADIOLABELED GASTRIN/CHOLECYSTOKININ 2 ANALOGS
The gastrin receptor may be an attractive target for radiopeptide imaging and therapy of several human tumors. Reubi et al. (92) have identified by autoradiography that the gastrin receptor is overexpressed on medullary thyroid carcinoma (92%), small cell lung carcinoma (57%), astrocytoma (65%), stromal ovarian tumors (100%), and gastroenteropancreatic tumors (22%). The first human studies were performed by Behr et al. (93). Using an 111In-DTPA-octreotide–conjugated minigastrin derivative, they visualized medullary thyroid carcinoma tumors.
The same group (94) reported a study of 60 consecutive patients (51 carcinoids, 3 gastrinomas, 2 glucagonomas, 3 paragangliomas, and 1 insulinoma) comparing gastrin receptor scintigraphy with SRS. The detection rate was 74%. Eleven patients had negative SRS findings, and 6 were gastrin receptor scintigraphy–positive. On the basis of the number of lesions, gastrin receptor scintigraphy performed better in 13 patients and SRS performed better in 24. Overall, gastrin receptor scintigraphy detected additional tumor sites in 20% of the patients. There was no Pearson correlation between the scan results and the grade of tumor differentiation, but this aspect appears to deserve more attention. In a similar comparative study of 29 patients with metastatic medullary thyroid cancer, Gotthardt et al. (95) found that tumor detection with gastrin receptor scintigraphy was 94.2%, whereas with SRS the detection rate was only 40.7%, correlating with in vitro autography data.
Radiotherapeutic studies were also reported by the same group using 90Y-DTPA-d-Glu-Glu5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2 in 8 patients with rapidly progressing, metastatic medullary thyroid carcinoma (96). Overall, 2 patients had a PR, 4 had SD, and 2 had PD. The study had to be stopped because of serious kidney and hematologic toxicity. Recently, the development of new radioligands with the macrocyclic chelator DOTA was reported (97–99). They show increased serum stability and improved tumor-to-kidney ratios. A study of medullary thyroid cancer with these highly improved radiopeptides has been initiated within a European project involving 10 hospitals.
RADIOLABELED RGD PEPTIDES
PET of αvβ3 integrin expression using radiolabeled RGD peptides has made its way from bench to bedside. The first clinical data are on the glycosylated peptide 18F-galacto-RGD in a limited number of patients with malignant melanoma, sarcoma, osseous metastasis from renal cell carcinoma, and villonodular synovitis (100). Tracer uptake was found to have good intra- and interindividual heterogeneity in the lesions, indicating good diversity in αvβ3 expression. Beer et al. investigated whether 18F-galacto-RGD uptake correlates with αvβ3 expression in patients with various solid tumors, such as musculoskeletal system tumors, melanoma, head and neck cancer, glioblastoma, and breast cancer (101). The patients were examined with 18F-galacto-RGD PET before surgical removal of the lesions. Sections from representative areas of the lesions with low uptake and representative areas with intense uptake were examined using immunohistochemistry. A significant correlation was found between standardized uptake values and tumor-to-blood ratios by means of the intensity of immunohistochemical staining and by microvessel density. Moreover, tumors negative on 18F-galacto-RGD PET showed a lack of αvβ3 expression. The biodistribution of 18F-galacto-RGD in cancer patients showed specific receptor binding and rapid, predominantly renal, excretion, resulting in low background activity in most regions of the body (102).
Side-by-side comparison of 18F-galacto-RGD and 18F-FDG uptake in patients with different primary or metastatic cancers, including non–small cell lung carcinoma, renal cell carcinoma, rectal cancer, and others, showed no substantial correlation between glucose metabolism (18F-FDG uptake) and αvβ3 expression in malignant lesions (103). Consequently, 18F-FDG and 18F-galacto-RGD provide different information. Whereas 18F-FDG PET was more sensitive for tumor staging, 18F-galacto-RGD PET has potential value for selection of patients entering clinical trials with αvβ3-targeted antiangiogenic therapies, considering the correlation of tracer uptake with αvβ3 expression.
The feasibility of using 18F-galacto-RGD PET in patient groups that differ in αvβ3 expression has been investigated in a study of squamous cell carcinoma of the head and neck (104), in which immunohistochemistry confirmed αvβ3 expression to be mainly on tumor vessels, and in a study of breast cancer (105), in which αvβ3 expression was found to be predominantly on microvessels and, to a lesser extent, on tumor cells. In glioblastoma patients (106), uptake was found to be heterogeneous and substantially lower in glioblastoma than in tumors outside the central nervous system, probably because 18F-galacto-RGD does not cross the intact blood–brain barrier. A higher accumulation was seen in the highly proliferating and infiltrating areas of the tumors, whereas normal brain tissue did not show significant uptake. Immunohistochemical staining was prominent in microvessels and glial tumor cells, and tracer uptake correlated with the αvβ3 expression of corresponding tumor samples.
The second clinically evaluated tracer was 99mTc-NC100692, introduced by GE Healthcare. In breast cancer patients, this tracer could detect 19 of 22 histopathologically confirmed malignant lesions (107). However, the immunohistochemical correlation with αvβ3 expression was not reported.
A phase I trial has been performed with 18F-AH111585 (also called 18F-fluciclatide) on a small number of patients with metastasized breast cancer, for whom all tumors detected by CT were visible on 18F-AH111585 PET (108). Both the primary tumor and metastases in the lungs, pleura, bones, and lymph nodes showed increased uptake, compared with background. However, liver metastases were indirectly identified as photopenic regions because of the high background activity in normal liver tissue. The tracer is being evaluated in a phase II study on patients with brain tumors, lung cancer, squamous cell carcinoma of the head and neck, differentiated thyroid carcinoma, sarcoma, and melanoma to correlate 18F-AH111585 PET with histologic parameters of angiogenesis (including αvβ3 expression) (109).
The tracer 18F-RGD-K5, used in parallel with 18F-FDG in a small number of patients with breast cancer, has shown high 18F-RGD-K5 uptake in most lesions identified by 18F-FDG (110). 18F-RGD-K5 uptake showed no correlation with 18F-FDG uptake, as was also the case for 18F-galacto-RGD. Although most lesions (80%) showed higher 18F-FDG uptake, in a few cases (4%) 18F-RGD-K5 uptake was higher than 18F-FDG uptake. 18F-RGD-K5 is being used in a phase II clinical study to assess its usefulness for predicting efficacy and for early response monitoring of a combination of anti-VEGF antibody bevacizumab therapy plus chemotherapy (111).
Recently, 68Ga-NOTA-RGD was used along with 18F-FDG in patients with hepatic metastases of colorectal cancer before a combination of bevacizumab therapy and oxaliplatin, 5-fluorouracil, and leucovorin therapy (112). Although all hypermetabolic liver lesions were visualized on 18F-FDG PET, half the patients showed mild 68Ga-NOTA-RGD uptake in these lesions and the other half showed no uptake. Interestingly, the patients with 68Ga-NOTA-RGD uptake in the hepatic metastases showed a PR to the antiangiogenic combination therapy, whereas the other patients had SD or PD. These findings support the hypothesis that PET of αvβ3 might help in the selection of patients for antiangiogenic therapies.
RADIOLABELED SUBSTANCE P AND CHLORATOXIN
PRRT, a locoregional therapy that is more invasive than most radionuclide therapies, has been applied to brain tumors. It requires the stereotactic implantation of a catheter system into the tumor or into the resection cavity. Given the biology of brain tumors—95% of glioblastomas manifest as a unifocal lesion that recurs within a 2-cm margin at the primary site, and most patients die from the primary tumor or local recurrence—a locoregional approach is suggested for these malignancies. To date, 3 categories of peptide-based radiotracers have been used in clinical phase I and II studies. However, despite promising results, the number of patients treated has been low and only limited data are available.
In therapy studies using 90Y-DOTATOC on brain tumors, the most promising responses have been found in low-grade gliomas (113,114). The higher expression of somatostatin receptors on low-grade gliomas than on high-grade gliomas (113) may account for this finding. In contrast to sst2, the neurokinin type 1 receptor is consistently overexpressed on glioma cells and on tumor vessels (115). This overexpression was the rationale for the use of the radiolabeled 11-amino-acid peptide substance P to target malignant gliomas.
Autoradiography disclosed overexpression of neurokinin type 1 receptor in 55 of 58 gliomas of WHO grades 2–4. Internalization of substance P conjugated to the chelator 1-(1-carboxy-3-carboxypropyl)-1,4,7,10-tetraazacyclododecane-4,7,10-triacetic acid (DOTAGA) was found to be specific, and in a first clinical study using 90Y- and 213Bi-labeled DOTATAGA-substance P, 20 patients have been enrolled (116). Patients with high-grade lesions (n = 16) were included after progression of disease or denial of conventional treatment options. The 4 low-grade glioma patients selected local targeted radiotherapy instead of an observational phase or external-beam radiotherapy. In all cases, the drug was injected via a stereotactically implanted catheter system either into the tumor or into the resection cavity. Application of the radiopharmaceutical was straightforward. The radiopharmaceutical was distributed according to tumor geometry. Only transient toxicity was seen: symptomatic radiogenic edema in 1 patient. The observation period ranged from 7 to 66 mo after completion of targeted radiopeptide therapy. Disease stabilization or improved neurologic status was observed in 13 of 20 patients. Secondary resection disclosed widespread radiation necrosis with improved demarcation of the tumor.
In addition to the preclinical and clinical evaluation of DOTATAGA-substance P, a more sophisticated dosimetry protocol was applied to this novel therapy option (117). The aim was to establish a reproducible dosimetry for intratumoral radiopeptide therapy so that the doses of this modality could be compared with external-beam radiotherapy and the effective dose range identified. In 12 patients with malignant gliomas, 2 MBq of 111In-substance P and 370–3,330 MBq of 90Y-substance P were sequentially applied, and each time serial SPECT scans were acquired over a period of 24 h. Quantitative voxelwise dose distribution maps (in Gy/GBq) were computed from these data. The correlation between pretherapeutic and posttherapeutic dosimetry was accurate. The calculated absorbed dose to the tumor was as high as 40–483 Gy/GBq (mean, 155 Gy/GBq) for the first therapeutic injection with 90Y-substance P. Given an average therapeutic activity of 30 mCi (1.11 GBq), the result was a mean absorbed radiation dose of 172 Gy within the tumor.
On the basis of these encouraging results, 2 further studies were initiated with radiolabeled substance P. The previous study (116) showed that from a surgical point of view, resectability was facilitated by improved demarcation and by the radiation-induced antiangiogenic effect in these patients who had been treated up front with locoregional radiopeptide therapy. Therefore, a study (118) analyzed whether neoadjuvant treatment before tumor resection is also a valid concept for improved therapy of malignant brain tumors. Seventeen glioblastoma patients were treated by local injection of 90Y-DOTAGA-substance P before tumor resection. The patients were treated by 4 cycles of intratumoral radiopeptide therapy with 90Y-DOTAGA-substance P at monthly intervals. The cumulative injected activity ranged from 3,700 to 12,765 MBq. Briefly, the study found that resectability was markedly improved after neoadjuvant radiopeptide therapy, comparable to the situation with recurrent gliomas. In all patients, pseudocapsulation was detected intraoperatively, and reduced bleeding due to the radiation effects facilitated surgical resection. Histopathologic examination of the resected lesion disclosed widespread radiation necrosis. Therefore, neoadjuvant therapy of glioblastoma using locally injected radiopeptides was concluded to be feasible, of low toxicity, and deserving of further assessment as a first-line option in the therapeutic cascade of glioblastoma treatments. No conclusions could yet be drawn on survival.
In the most recent study, the feasibility and effectiveness of targeted α-radionuclide therapy for brain tumors was assessed, using the α-radiation–emitting radionuclide 213Bi, with a mean tissue penetration range of 81 μm, instead of 90Y, with a mean tissue penetration range of 5 mm. This pilot study included 5 patients with critically located gliomas (119). Local injections of 213Bi-DOTA-[Thi8,Met(O2)11]-substance P were given. Application of the radiopeptide was straightforward and well tolerated by all patients. No additional neurologic deficit was observed. Repeated MRI was suggestive of radiation-induced necrosis and demarcation of the tumors, and this finding was validated by subsequent resection. Therefore, the study concluded that targeted local radiotherapy with 213Bi-DOTA-[Thi8,Met(O2)11]-substance P may represent an innovative and effective treatment strategy for critically located malignant gliomas, because primarily nonoperable gliomas may become resectable over the course of treatment. However, the study group was too small for conclusions on prognosis to be drawn.
Finally, to date only 1 study using a synthetic chlorotoxin (TM-601), a 36-amino-acid neurotoxin, has been published (120). The peptide TM-601 has been linked covalently to 131I (131I-TM-601). A phase I clinical trial has been completed in 18 patients with recurrent glioma (17 glioblastoma and 1 anaplastic astrocytoma). Two weeks after surgery, the patients received a single dose of 131I-TM-601 from 1 of 3 dosing panels (0.25, 0.50, or 1.0 mg of TM-601), each labeled with 370 MBq of 131I. This study demonstrated that a single dose of 370 MBq of 131I-TM-601 was well tolerated for 0.25- to 1.0-mg TM-601 and may have an antitumoral effect. The ligand was found to bind malignant glioma with high affinity and for long durations.
Acknowledgments
No potential conflict of interest relevant to this article was reported.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.
- 72.
- 73.
- 74.
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.
- 81.
- 82.
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.↵
- 91.
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.
- 99.↵
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.↵
- 118.↵
- 119.↵
- 120.↵
- Received for publication November 8, 2011.
- Accepted for publication November 8, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- 18F-Labeled Somatostatin Analogs as PET Tracers for the Somatostatin Receptor: Ready for Clinical Use
- 18F-Labeled Somatostatin Analogs as PET Tracers for the Somatostatin Receptor: Ready for Clinical Use
- A Half-Century of Nuclear Medicine
- Biodistribution, Pharmacokinetics, and Dosimetry of 177Lu-, 90Y-, and 111In-Labeled Somatostatin Receptor Antagonist OPS201 in Comparison to the Agonist 177Lu-DOTATATE: The Mass Effect
- Somatostatin Receptor Antagonists for Imaging and Therapy
- Bombesin Antagonist-Based Radiotherapy of Prostate Cancer Combined with WST-11 Vascular Targeted Photodynamic Therapy
- Highly Increased 125I-JR11 Antagonist Binding In Vitro Reveals Novel Indications for sst2 Targeting in Human Cancers
- Molecular Imaging and Therapy with a Purpose: A Renaissance of Nuclear Medicine
- Tumor-Specific Binding of Radiolabeled PEGylated GIRLRG Peptide: A Novel Agent for Targeting Cancers
- Decoding Intratumoral Heterogeneity of Breast Cancer by Multiparametric In Vivo Imaging: A Translational Study
- Optimization of RGD-Containing Cyclic Peptides against {alpha}v{beta}3 Integrin
- Radiopeptides for Imaging and Therapy: A Radiant Future
- Cerenkov Luminescence Imaging for Radiation Dose Calculation of a 90Y-Labeled Gastrin-Releasing Peptide Receptor Antagonist
- Improved Modeling of In Vivo Kinetics of Slowly Diffusing Radiotracers for Tumor Imaging
- Targeted Radionuclide Therapy: Proceedings of a Joint Workshop Hosted by the National Cancer Institute and the Society of Nuclear Medicine and Molecular Imaging
- A Comparative Study of Radiolabeled Bombesin Analogs for the PET Imaging of Prostate Cancer
- Peptide Receptor Radionuclide Therapy in the United States