


Abstract
Brain serotonin 7 (5-hydroxytryptamine 7, or 5-HT7) is the most recently identified serotonin receptor. It is involved in mood disorders and is studied as a target for antidepressants. Because no radioligand has yet been successfully used to study this receptor by PET neuroimaging, the objective of the present study was to develop a 5-HT7 18F-labeled radiotracer. Methods: Four structural analogs of SB269970, a specific 5-HT7 receptor antagonist, were synthesized. The nitro precursors of these analogs were radiolabeled by 18F nucleophilic substitution. Analog antagonist effects were investigated by cellular functional assay. The cerebral distribution of radiolabeled molecules was studied by in vitro autoradiography in rats, and respective selectivity was determined by competition with the 5-HT7 receptor antagonist SB269970 at different concentrations. Ex vivo small-animal PET studies in rats and in vivo PET studies in cats focused on the 1-(2-{(2R)-1-[(fluorophenyl)sulfonyl]pyrrolidin-2-yl}ethyl)-4-methylpiperidine (FP3) series. Results: Four analogs were synthesized from the SB269970 pharmacophore and divided into an FP3 (18F-4FP3 and 18F-2FP3) and an 1-(2-{(2R)-1-[(fluorophenyl)sulfonyl]pyrrolidin-2-yl}ethyl)-4-(2-methoxyphenyl)piperazine (FPMP) (18F-2FPMP and 18F-4FPMP) series. The chemical and radiochemical purities of the 4 radiolabeled molecules were greater than 98%. All presented suitable affinity for 5-HT7 (apparent dissociation constant [KD] between 1.6 and 14 nM), although the FPMP series showed moderate agonist activity for 5-HT1A receptors. Lipophilicity values were predictive of good radiotracer blood–brain barrier penetration (logD from 1.4 to 3.9). In vitro competition with a 5-HT7 antagonist, SB269970, revealed that only radioligands from the FP3 series were displaced by the 5-HT7–specific antagonist: subsequent in vivo study, therefore, focused on this series. Ex vivo 18F-4FP3 and 18F-2FP3 autoradiography was in accordance with the 5-HT7 brain distribution, with few brain radioactive metabolites. PET scans in cats showed that pretreatment with a 5-HT7 antagonist significantly reduced 18F-2FP3 but not 18F-4FP3 binding. Conclusion: The 4 PET radiotracers had suitable characteristics for 5-HT7 receptor probing in vitro, although the FP3 series seemed to be more specific for in vivo imaging of 5-HT7 receptors. In particular, on the basis of the in vivo results, 18F-2FP3 appears to be the first PET radiotracer to enable in vivo imaging of 5-HT7 receptors in animal models, possibly leading to neuroimaging studies in humans.
Serotonin (5-hydroxytryptamine, or 5-HT) is a central neurotransmitter involved in many physiologic functions and in neurologic and psychiatric disorders. Pharmacologic studies identified numerous serotoninergic receptor families and subtypes, classified by structural, functional, and pharmacologic criteria into 7 distinct receptor classes (5-HT1–7) (1).
The 5-HT7 subtype is the most recently cloned serotonin receptor (2). 5-HT7 receptors are coupled to G proteins (3) and are found in rodents, pigs, primates, and humans, with relatively high concentrations in the hippocampus, thalamus, and hypothalamus and lower levels in the cortex and amygdala (4–6). The 5-HT7 receptor exhibits a high degree of interspecies homology (∼95%) but low sequence homology with other 5-HT receptors (<40%) (3,6). Three 5-HT7 receptor isoforms, which differ only in their carboxyl terminal tails and have the same pharmacologic profile, are expressed in rats and humans (7).
The recent availability of selective 5-HT7 receptor antagonists (7–9) and of 5-HT7 receptor knockout mice (10) has considerably advanced understanding of the physiologic function of this receptor (11). 5-HT7 has been identified as important in circadian rhythms and sleep (12). It also binds several antidepressants (e.g., mianserin and maprotiline) and antipsychotics (e.g., clozapine and risperidone) with high affinity and may be a therapeutic target for schizophrenia and mood disorders. Selective 5-HT7 antagonists were indeed recommended in schizophrenia and depression (13–15). Several reports, based on receptor distribution and preliminary pharmacologic analyses, suggested 5-HT7 receptors as a new serotoninergic target for memory enhancement, epilepsy, and pain (16–19).
Therefore, serotonin 5-HT7 receptor imaging in the human brain in vivo is needed to assess directly the involvement of the receptor in neuropsychiatric diseases and possible therapies. The development of PET as a molecular imaging method provides an opportunity for in vivo observation in both animal models and humans (20). This method requires a PET radiotracer that labels 5-HT7 receptors with a high affinity and has a high selectivity, high signal-to-noise ratio, lipophilicity sufficient to penetrate the blood–brain barrier, relatively slow clearance, and low level of labeled metabolites in the brain (20).
Although several selective antagonists exist (8,21,22), few have been radiolabeled for use in radioligand binding and autoradiography studies (23–26). For example, 3H-SB269970 is a high-affinity selective ligand (apparent dissociation constant [KD], 1.2 nM) (27) that has been used to radiolabel the 5-HT7 receptor in vitro in rodent, pig, marmoset, and human brains (5,28).
Notably, only 1 team developed a 5-HT7 radiotracer for PET. DR4446 is a tetrahydrobenzindole derivative that possesses a higher selectivity for 5-HT7 than the other 5-HT receptors, although with only moderate affinity (apparent inhibition constant [Ki], 9.7 nM), and 11C-DR4446 was therefore synthesized and tested in monkeys (26). This preliminary PET study indicated that 11C-DR4446 had good cerebral uptake, had a specific binding component, and was metabolically stable. However, to our knowledge, no further reports have been published with this or other 5-HT7 PET radiotracers. We therefore sought to develop the first 18F-labeled agonist PET probe to image functional 5-HT7 receptors.
The present article describes the radiosynthesis of 4 original 5-HT7 antagonists belonging to the 1-(2-{(2R)-1-[(fluorophenyl)sulfonyl]pyrrolidin-2-yl}ethyl)-4-methylpiperidine (FP3) and 1-(2-{(2R)-1-[(fluorophenyl)sulfonyl]pyrrolidin-2-yl}ethyl)-4-(2-methoxyphenyl)piperazine (FPMP) series, with initial in vitro assessment. After favorable screening tests, the study focused on the 2FP3 series: ex vivo studies were performed in rats, and in vivo PET studies were performed in cats.
MATERIALS AND METHODS
Precursor Synthesis
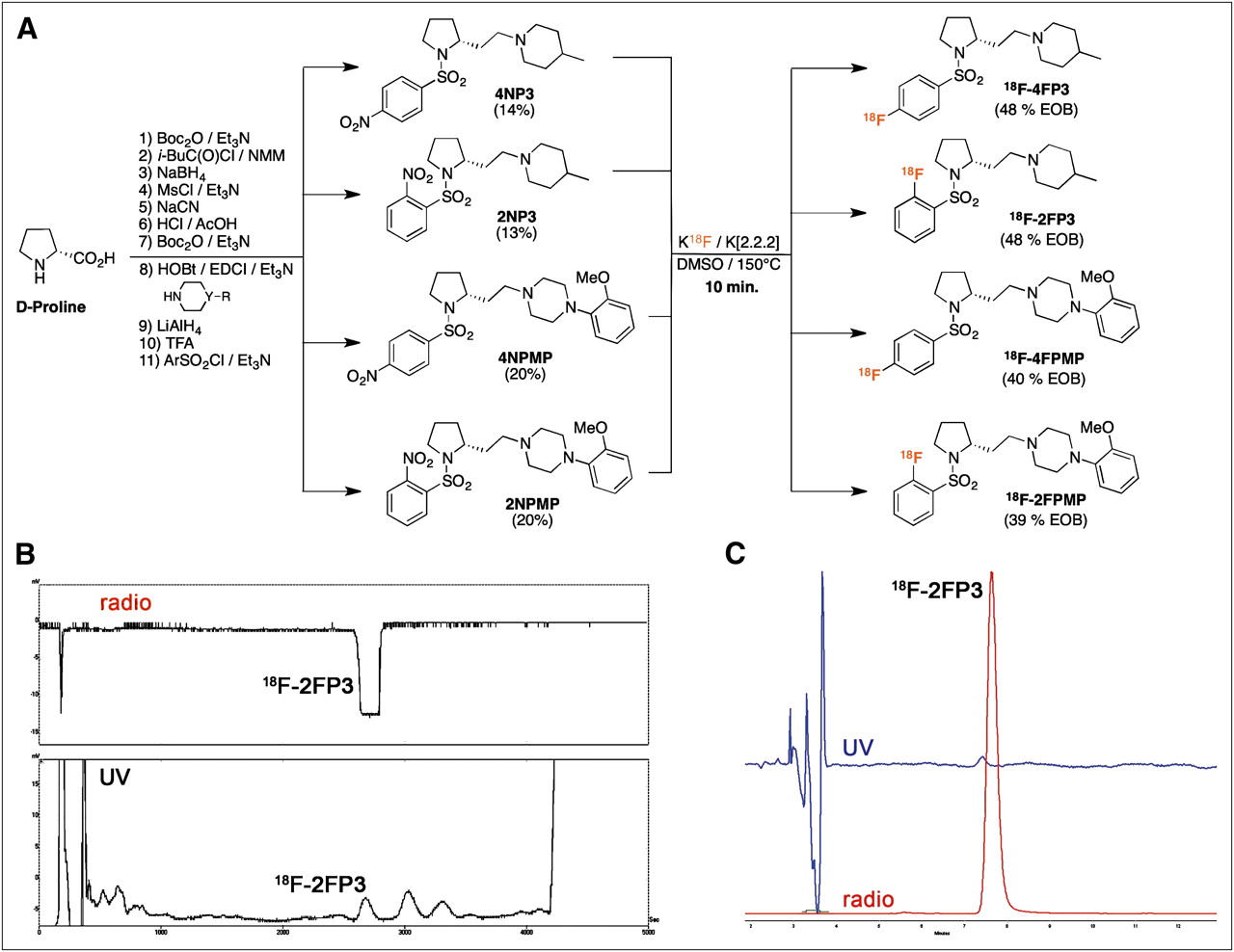
We recently synthesized 4-methylpiperidine and 4-(2-methoxyphenyl) piperazine series as possible 5-HT7 PET radioligands (29). Briefly, (d)-proline was homologated in N-Boc-(d)-homoproline by a classic multistep homologation procedure, with an overall yield of 44%. The N-Boc-(d)-homoproline was then coupled with the corresponding nitrogen heterocycle, depending on the expected series (4-methylpiperidine for the FP3 or 4-(2-methoxyphenyl) piperazine for the FPMP series), and the resulting amides were reduced in 2 steps. After removal of the Boc protection, the nitrophenylsulfonyl group (ortho- or para-nitro) was introduced, using the corresponding arylsulfonyl chloride, leading to the expected nitro precursors 1-(2-{(2R)-1-[(nitrophenyl)sulfonyl]pyrrolidin-2-yl}ethyl)-4-methylpiperidine (NP3) or 1-(2-{(2R)-1-[(nitrophenyl)sulfonyl]pyrrolidin-2-yl}ethyl)-4-(2-methoxyphenyl)piperazine (NPMP) (Fig. 1).

(A) Schema for synthesis of precursors 4NP3, 2NP3, 4NPMP, and 2NPMP, and radiosynthesis of 18F-4FP3, 18F-2FP3, 18F-4FPMP, and 18F-2FPMP. (B) Chromatogram of HPLC purification of 18F-2FP3 (ultraviolet absorbance at 254 nm and radioactivity). (C) Quality control chromatogram of final 18F-2FP3 (ultraviolet absorbance at 254 nm and radioactivity) shows radiochemical purity greater than 99%. DMSO = dimethyl sulfoxide; EOB = end of bombardment; radio = radioactivity detection; UV = ultraviolet detection.
Tracer Radiolabeling and Quality Control
18F was obtained by 18O(p,n)18F reaction (Cyclone 18/9 cyclotron; IBA). Nitro–fluoro exchange was performed on a standard coincidence synthesizer (TracerLab Mx; GE Healthcare) after the automation sequence was reprogrammed. After initial fluoride preparation (collection, drying, and Kryptofix [Sigma-Aldrich] activation), 4 mg of nitro precursor of 1 of the 4 molecules was introduced, and the reaction mixture was heated at 150°C for 10 min (29). After dilution with 15 mL of water, the reaction mixture was passed through an activated C18 cartridge for prepurification, and the crude product was eluted from the cartridge with 2 mL of ethanol. Pure fluorinated radiotracer was obtained after separation on a preparative high-performance liquid chromatography (HPLC) column (SymmetryPrep C18, 7 μm, 7.8 × 300 mm; Waters) eluted with 20 mM H3PO4/tetrahydrofuran/0.1% trifluoroacetic acid at 3 mL/min, with a retention time of 35 min for 18F-4FP3, 45 min for 18F-2FP3, 50 min for 18F-4FPMP, and 70 min for 18F-2FPMP. For in vivo use, the product was diluted with 40 mL of sterile water loaded onto a C18 cartridge (SEP-Pak Light; Waters), eluted with 1 mL of ethanol, diluted with isotonic saline to an ethanol concentration of 5%, and sterilized by membrane filtration (Millex-GS, 0.22 μm; Millipore).
Quality control consisted of determining radiochemical purity and specific activity by analytic HPLC assay of an aliquot of the radiolabeled product, with comparison to the calibration curve generated from solutions of a known concentration (ultraviolet set at 254 nm and radioactive detection; C18 Nucleodur, 7.8 × 300 mm column [Macherey-Nagel]; elution with 20 mM H3PO4/0.9 mL of tetrahydrofuran/minute for FP3 and 0.8 mL/min for FPMP, with retention times of 9.5 min for 18F-4FP3, 7.6 min for 18F-2FP3, 8.8 min for 18F-4FPMP, and 6.9 min for 18F-2FPMP).
Animals
Adult male Sprague–Dawley rats (250–300 g of body weight; Charles River Laboratories) and European male cats (3.5–5 kg of body weight; Charles River Laboratories) were used. All animals were kept in the animal facility (room temperature, 22°C, with a 12-h day–night cycle). All experiments were performed in accordance with European guidelines for care of laboratory animals (86/609 EEC) and were approved by the animal use ethics committee of the University of Lyon (University of Lyon 1).
In Vitro Pharmacology
Respective affinity toward 5-HT7 receptors (KD) was determined by Cerep, an in vitro pharmacology company (30). Briefly, cellular functional assays were performed to search for possible antagonist and agonist effects in the 4 compounds. Competition binding experiments were performed on expressed human recombinant receptors in CHO cells. The EC50 value (concentration eliciting a half-maximal specific response) and IC50 value (concentration eliciting half-maximal inhibition of the control specific agonist response) were determined by nonlinear regression analysis of the concentration–response curves generated with mean replicate values using Hill equation curve fitting. Analysis used the commercial SigmaPlot 11.0 software (Systat Software Inc.). The KD was calculated using the modified Cheng Prusoff equation.
Lipophilicity (log P) was measured in our laboratory to evaluate the lipid solubility of radiolabeled molecules by partitioning between n-octanol and 50 mM Tris-HCl (pH 7.4) buffer. Log P was calculated as the logarithm of the concentration of radiotracers in n-octanol over the concentration in buffer.
In Vitro Studies of 18F-FPMP and 18F-FP3 Series
In vitro autoradiography was performed in rats. After euthanasia by inhaled isoflurane overdose, rat brains were carefully removed and immediately frozen in 2-methylbutane cooled with dry ice (−29°C). Briefly, coronal sections (30 μm thick) across the hippocampus and cerebellum were cut using a −20°C cryostat (SM1850; Leica), thaw-mounted on glass slides, and allowed to air-dry before storage at −80°C until use. On the day of radiotracer synthesis, the slides were allowed to reach room temperature and then incubated for 20 min in Tris phosphate-buffered saline (138 mM NaCl, 2.7 mM KCl, pH adjusted to 7.6) containing 37 kBq/mL (1 μCi/mL) of 18F-4FP3, 18F-2FP3, 18F-4FPMP, or 18F-2FPMP. For competition experiments, the slides were placed in the same buffer supplemented with SB269970, a selective 5-HT7 antagonist (10 nM, 100 nM, or 1 μM), or with serotonin (1 μM). After incubation, slides were dipped in cold buffer (4°C) for 90 s and in distilled cold water (4°C) for 90 s, and then dried and juxtaposed to a phosphor imaging plate for 60 min (BAS-1800 II; Fujifilm). Regions of interest (cingulate cortex and hippocampus) were drawn manually using Multigauge software (Fujifilm). Results were expressed in phosphostimulated luminescence per millimeter squared and converted into percentage of control.
Ex Vivo Studies of 18F-FP3 Series
The ex vivo and in vivo small-animal PET images were acquired on a ClearPET LYSO/LuYAP Phoswich scanner (Raytest) in 3-dimensional mode (31). In the first part of the experiment, rats were anesthetized with urethane (1.25 g/kg intraperitoneally), and an intravenous catheter was inserted in the caudal vein. 18F-4FP3 or 18F-2FP3 (55.5 MBq intravenously) was injected, and 20 min after radiotracer injection, the anesthetized rat was killed by decapitation. The brain was carefully and rapidly removed and centered in the field of view of the small-animal PET camera. Radioactivity was measured in a single 45-min frame. In the second part of the experiment, rats were similarly anesthetized and catheterized. SB269970, the selective 5-HT7 antagonist, was administered (5 mg/kg intravenously) 30 min before injection of 18F-4FP3 or 18F-2FP3 (55.5 MBq intravenously). In another experiment, the corresponding cold fluorinated radiotracer was administered (5 mg/kg intravenously) 30 min before injection of 18F-4FP3 or 18F-2FP3 (55.5 MBq intravenously). Twenty minutes after radiotracer injection, the brain was carefully removed and placed in the small-animal PET camera for a 45-min acquisition. The images obtained were analyzed with AMIDE software (A Medical Imaging Data Examiner; free license).
In Vivo Studies of 18F-2FP3
Cat PET Scan Acquisition
PET scans were acquired for European male cats anesthetized with isoflurane (Baxter) administered at 5% for 5 min and then lowered to 2.5%. The cat was placed in an acrylic stereotactic apparatus with ear bars, and a catheter was inserted into the forearm branch of the brachiocephalic vein, continuously perfused with NaCl 0.9%. PET scans were acquired on an Exact ECAT HR+ (CTI-Siemens) in 3-dimensional mode. The first 10 min of acquisition was the transmission scan, performed with 3 rotating 68Ge and 68Ga sources, followed by a 74-MBq (2-mCi) injection of 18F-2FP3. Radioactivity was measured in a series of 31 sequential frames of increasing duration from 30 s to 10 min; total scan time was 90 min. Images were reconstructed in the frontal plane. Regions of interest (cingulate cortex, thalamus, hippocampus, and cerebellum) were manually drawn using CAPP software (CTI-Siemens) according to the stereotactic atlas of the cat brain (32,33) and the MR images. The radioactivity of different regions attributed to the fixation of 18F-2FP3 was expressed in becquerels per centimeter cubed.
Cat MR Image Acquisition
The same cats were anesthetized with isoflurane administered at 5% for 5 min and then lowered to 2.5%. The head of the animal was immobilized in a stereotactic acrylic frame with ear bars and orbital and hard palate parts. MR image acquisition (1.5-T Magnetom scanner; Siemens AG) comprised a 40-min 3-dimensinal anatomic T1-weighted sequence. The anatomic volume covered the whole brain with 0.7 mm3 voxels.
Determination of Unchanged 18F-2FP3 in Rat Brain
Three series of 2 rats were anesthetized by intraperitoneal injection of urethane (1.7 g/kg), and a catheter was inserted into the caudal vein. The rats were killed by decapitation at 10, 20, 30, and 40 min after a bolus injection of 18F-2FP3 (55.5 MBq [1.5 mCi]). The brains were rapidly removed; the cerebral hemisphere was homogenized by an ultrasonic homogenizer in 400 μL of perchloric acid (0.4 mol/L), and the mixture was neutralized by 120 μL of 4 M potassium acetate. After centrifugation at 5,000g for 10 min, the supernatant was filtered (0.45 μm) before HPLC. The HPLC system consisted of a C-18 reversed-phase column (Nucleodur, 7.8 × 300 mm; Macherey-Nagel) (elution with H3PO4 [20 nM]/tetrahydrofuran: 85/15 at a flow rate of 0.9 mL/min). During elution, 2-min fractions were collected and counted for radioactivity with an automated γ-counter (Cobra II; Packard).
RESULTS
Nitro Precursor Synthesis and 18F-4FP3, 18F-2FP3, 18F-4FPMP, or 18F-2FPMP Radiosynthesis
The synthetic routes for the preparation of the FP3 and FPMP series and their radiolabeling are summarized in Figure 1A. Precursors were synthesized in 9 steps from (d)-proline, with an overall yield in the range of 13%–20%.
The radiolabeling of the 4 radiotracers was obtained from their nitro precursors at 150°C, with a radiochemical yield between 39% and 48% corrected for decay and 100–120 min radiosynthesis time (including HPLC purification and formulation), using a reprogrammed automated fluorination module. No radioactive by-products were observed (Fig. 1B), and the selected HPLC conditions ensured good separation of 18F-4FP3, 18F-2FP3, 18F-4FPMP, or 18F-2FPMP from their respective nitro precursors, as confirmed on quality control (Fig. 1C). Radiochemical purity was better than 98%, and the specific activity of the 4 molecules was between 40 and 130 GBq/μmol, corrected at the end of synthesis.
Partition Coefficient Measurement
The lipophilicity of the fluorinated molecules, determined in vitro by the octanol–water partition coefficient (log P), was found to be between 1.2 and 1.4 for the FP3 series and between 3.4 and 3.9 for the FPMP series (average of 3 experiments). These values were confirmed with the calculated log D (ACD/ChemSketch software, version 7.0; ACD/Labs).
In Vitro Pharmacology
The measured KD demonstrated good affinity of the fluorinated molecules for the 5-HT7 receptor, in particular for the FPMP series, with a value below 2 nM and antagonist properties (18F-4FP3 KB, 1.92; 18F-2FP3 KB, 1.43; 18F-4FPMP KB, 3.89; and 18F-2FPMP KB, 3.38). In terms of 5-HT1A affinity, the FPMP series showed a 5-HT1A agonist contribution (EC50, 1.4 × 10−7 for 18F-4FPMP and 5.8 × 10−8 for 18F-2FPMP), unlike the FP3 series, which was without 5-HT1A contribution (EC50 >> 10−5). No radiotracers showed 5-HT6 affinity (EC50 >> 10−5).
In Vitro Distribution Studies
The distributions of 18F-4FP3, 18F-2FP3, 18F-4FPMP, and 18F-2FPMP were assessed by semiquantitative autoradiography in rat brains (Fig. 2A). Autoradiograms obtained after incubation with a constant in vitro radiotracer concentration demonstrated the presence of structures able to concentrate radioactivity—that is, cortex, hippocampus, thalamus, and cerebellum. These binding areas corresponded to those described as rich in 5-HT7 receptors (6). The binding level in the FPMP series (18F-4FPMP and 18F-2FPMP) was higher in the hippocampus than in other regions, whereas the level of hippocampus binding in the FP3 series (18F-4FP3 and 18F-2FP3) was similar to that in other regions (experiments performed in triplicate for each radiotracer).

(A) In vitro autoradiograms of rat brain sections incubated with 18F-4FP3, 18F-2FP3, 18F-4FPMP, or 18F-2FPMP show locations and density of 5-HT7 receptors (proportional to gray level) in hippocampus (Hip), cortex (Cx), and thalamus (Thal). (B) Histograms obtained by in vitro autoradiography in rats and competition between 18F-4FP3, 18F-2FP3, 18F-4FPMP, 18F-2FPMP, and SB269970, a 5-HT7 receptor antagonist. Radioactivity in cortex is represented at different concentrations and expressed as percentage of control. Mean values (±SEM) for each group are significantly different (*P < 0.05) from controls on unpaired Student t test. DMSO = dimethyl sulfoxide; EOB = end of bombardment; radio = radioactivity detection; UV = ultraviolet detection.
In Vitro Competition Studies
18F-4FP3 and 18F-2FP3 radioactivity measured in displacement experiments in the hippocampus and cingulate cortex was reduced markedly and dose-dependently after the addition of SB269970, a 5-HT7 receptor antagonist (performed in triplicate). Diminution in 18F-4FP3 binding in cingulate cortex was −40% and −70% with 100 nM and 1 μM SB269970, respectively, and in the hippocampus −20%, −40%, and −60% with 10 nM, 100 nM, and 1 μM, respectively. Diminution in 18F-2FP3 binding in the hippocampus was −40%, −60%, and −70% with 10 nM, 100 nM, and 1 μM SB269970, respectively, and in the cingulate cortex −40%, −60%, and −70% with 10 nM, 100 nM, and 1 μM, respectively (Fig. 2B). At the same concentrations of SB269970 and in the same brain areas, 18F-4FPMP and 18F-2FPMP binding remained unchanged (Fig. 2B).
In other experiments, incubation with 1 μM serotonin reduced 18F-2FP3 binding by 40% in the cingulate cortex and hippocampus but failed to modify 18F-4FP3, 18F-4FPMP, or 18F-2FPMP binding (performed in triplicate, results not shown).
Ex Vivo 18F-4FP3 and 18F-2FP3 Binding in Rat Brain
The small-animal PET images obtained in isolated rat brain showed 18F-4FP3 and 18F-2FP3 binding in a 5-HT7–rich region (cortex and cerebellum). Preinjection of unlabeled 4FP3 or 2FP3 significantly decreased 18F-4FP3 and 18F-2FP3 binding in all brain areas (experiments performed in duplicate, Fig. 3). Preinjection of SB269970 (5 mg/kg) decreased 18F-4FP3 and 18F-2FP3 binding in all brain areas (experiments performed in duplicate, Fig. 3).

Ex vivo coronal small-animal PET images of 18F-4FP3 or 18F-2FP3 binding in isolated rat brain (45-min acquisition), 20 min after radioligand injection.
In Vivo 18F-4FP3 and 18F-2FP3 Binding in Cat Brain
In vivo PET images in cats revealed that after intravenous injection of 18F-4FP3 and 18F-2FP3, there was rapid accumulation of the radioligand (Fig. 4). Time–activity curves showed high initial uptake in the hippocampus, cingulate cortex, thalamus, and cerebellum. Binding ratios were obtained with time curves integrated from 5 to 45 min.
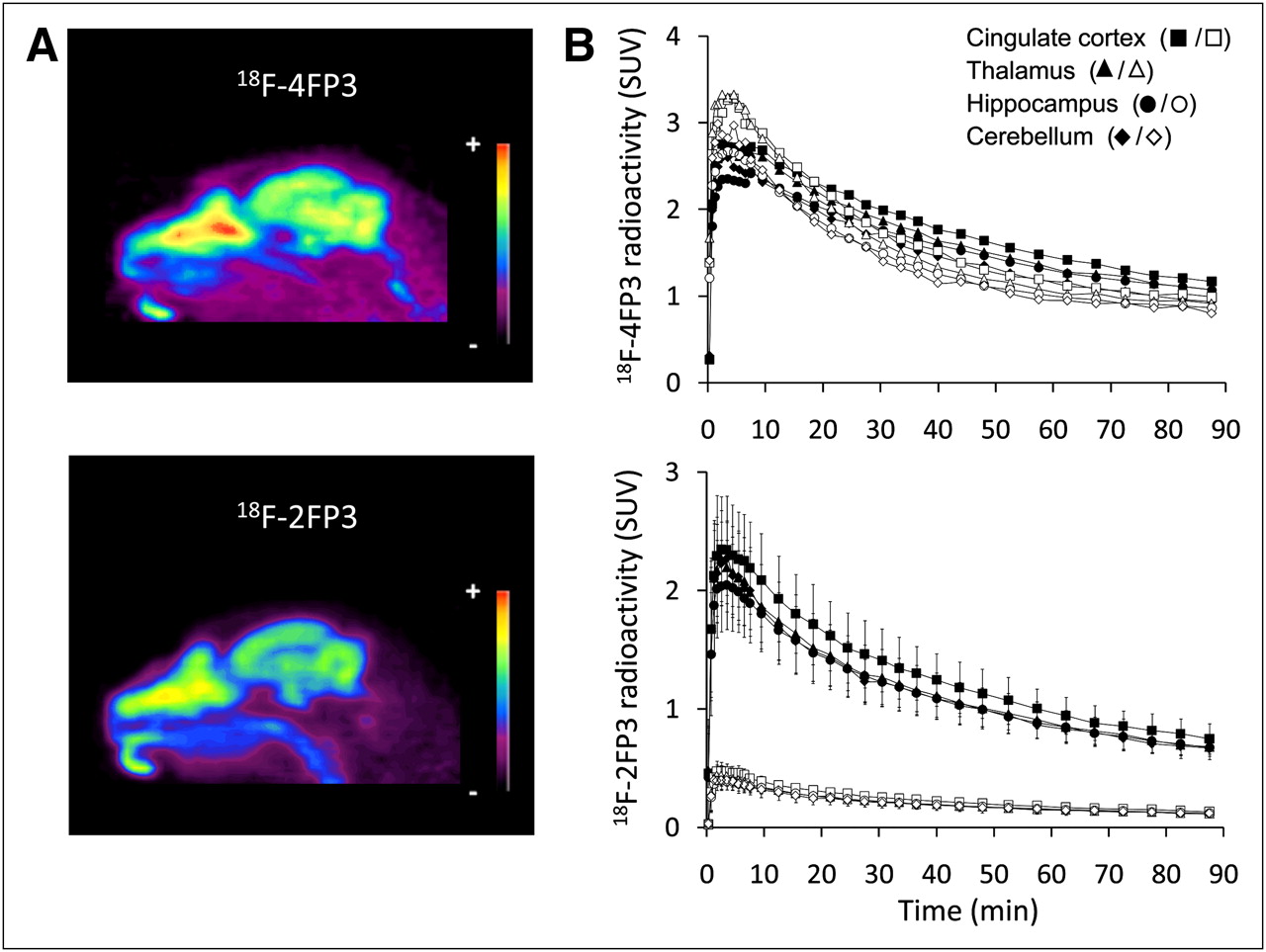
(A) Representative sagittal PET images summed for 0–90 scans, showing 18F-4FP3 and 18F-2FP3 distribution in cat brain. (B) Time–activity curves of 18F-4FP3 and 18F-2FP3 PET tracers at baseline (closed symbols) and after blockade with intravenous SB269970 (open symbols), a 5-HT7 antagonist (5 mg/kg, 30 min before radiotracer injection) (duplicated experiments).
After SB269970 preinjection, 18F-2FP3 concentrations fell to near zero in all regions, whereas 18F-4FP3 binding remained unchanged (experiments performed in duplicate for each radiotracer)
Unmetabolized 18F-2FP3 in Rat Brain
In rat brain, over the time scale considered (10, 20, 30, and 40 min after injection), the only radioactive peak detected after HPLC separation was due to the parent compound, unchanged 18F-2FP3 (95% for each time point; experiments performed in duplicate).
DISCUSSION
The aim of the study was to select and evaluate a potential PET radioligand for in vivo imaging of brain 5-HT7 receptors. The strategy was to synthesize 4 molecules and their nitro precursors and to focus in vivo study on the best molecules in terms of specificity and in vitro properties.
From a chemical standpoint, the choice of FP3 and FPMP series structures was inspired by a pharmacophore model (34) and by the structure of SB269970, the prototypical 5-HT7 antagonist (27,35). The ortho and para positions were chosen for fluorine nucleophilic substitution, and 2 hydrophobic groups were selected, leading to the synthesis of the FP3 series (4FP3 with 18F in para, and 2FP3 with 18F in ortho) and FPMP series (4FPMP with 18F in para, and 2FPMP with 18F in ortho). The labeling of the nitro precursor of the 4 tracer candidates was straightforward, with excellent radiochemical purity and a satisfactory specific activity.
2FP3 series lipophilicity measured in vitro was compatible with blood–brain barrier uptake. On the other hand, the relatively high lipophilicity of the FPMP series exposed it to higher nonspecific binding after radiolabeling, although this parameter is not systematically predictive for radiotracers (36). In vitro assay revealed the nanomolar affinity of the 4 molecules for the 5-HT7 receptor, as previously reported for SB269970, the prototypical 5-HT7 antagonist (28). Such affinities (in the range of 1–10 nM) are an important finding because the 5-HT7 receptor density observed in various species (28) is less than a tenth of that of other serotonin receptors (37).
Although 5-HT7 presents a low-sequence homology with other 5-HT receptors (<40% according to Plassat et al. (38), Shen et al. (39), and Tsou et al. (40)), we chose to test the affinities of the 4 molecules toward 5-HT1A and 5-HT6 receptors. This choice was justified by reports of difficulty for some 5-HT7 ligands in discriminating between 5-HT1A (21,41,42) and 5-HT6 receptors (43). We also showed that molecules of the FPMP series presented 5-HT1A agonistic affinity (EC50 > 10−5). The FP3 series was more specific, showing neither 5-HT1A nor 5-HT6 affinity.
In vitro autoradiography revealed distribution patterns of the 4 radiotracers in accordance with 5-HT7 localization. Previous brain distribution studies revealed the presence of 5-HT7 receptors in numerous brain areas (including the cerebellum), with elevation in the hippocampus, thalamus, hypothalamus, and cerebral cortex (6,22). The possible 5-HT1A contribution revealed by in vitro pharmacology for the FPMP series was confirmed by higher hippocampus uptake in this series on autoradiography.
The in vitro competition studies confirmed that the FP3 series specifically binds 5-HT7 receptors, because addition of SB269970, the specific 5-HT7 antagonist, displaced radiotracer binding dose-dependently. The lack of competition between SB269970 and either radiotracer of the FPMP series encourages us to focus future in vivo studies on the FP3 series (18F-4FP3 and 18F-2FP3).
18F-4FP3 and 18F-2FP3 uptake and distribution in rat brain after ex vivo small-animal PET showed high uptake and displaceable binding after blockade by the corresponding cold molecules. Further, the 5-HT7 selectivity of in vivo brain 18F-2FP3 binding was confirmed in the blocking study, in which 18F-2FP3 binding was decreased by a SB269970 bolus, whereas 18F-4FP3 binding was unchanged. This result confirmed that 18F-2FP3 presents the best radiopharmacologic properties of our 4 radiotracer candidates.
Final tests, to check the absence of radioactive metabolites in the brain, showed that more than 95% of brain radioactivity was due to unchanged 18F-2FP3, excluding any significant contribution of radiolabeled metabolites to specific binding in the brain. Taken together, the results indicate that 18F-2FP3 is a promising PET tracer for visualization and quantification of brain 5-HT7 receptors.
Although these first studies were preclinical, we are confident that the radiopharmacologic profile of 2FP3 will be appropriate for clinical studies, because the 5-HT7 receptor exhibits a high degree of interspecies homology, from rodents to humans (∼95% (6)). However, a quantification approach with kinetic models is needed to complete this work and prepare its translation to human studies. In this aim, arterial input measures are currently in progress. Optimally, better affinity for 5-HT7 receptors is to be preferred, because it is known that the specific binding of a radiotracer is a function of its affinity for the receptor relative to the density of the binding site. Consequently, the development of 2FP3 analogs is also being pursued, with chemical structures modified to optimize PET tracer properties, particularly in terms of affinity, which should be subnanomolar.
CONCLUSION
To our knowledge, this is the first report of a successful fluorinated PET radioligand for in vivo imaging of the serotonin 5-HT7 receptor. The 4 PET radiotracers we synthesized had characteristics suitable for in vitro 5-HT7 receptor probing; 18F-2FP3 notably showed specific and reversible in vivo binding to 5-HT7 receptors. Further evaluation of this radioligand is warranted to investigate its potential as a 5-HT7 radiotracer for human brain imaging.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Marion Alvarez for her help with radiochemistry and Sylvain Fieux for his help with animal procedures. The work was funded in part by the French National Research Agency ANR and a joint grant with the French Ministry of Research and Advanced Accelerator Applications (Saint Genis-Pouilly, France). No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Oct. 11, 2011.
- © 2011 by Society of Nuclear Medicine
REFERENCES
- Received for publication February 14, 2011.
- Accepted for publication July 19, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}