


Abstract
Use of 18F-FDG uptake as a surrogate marker of therapeutic response requires the recognition of biologic factors that influence cancer cell glucose metabolism. Estrogen is a potent stimulator of breast cancer proliferation, a process that requires sufficient energy, which is likely met by increased glycolysis. We thus explored the effect of estrogen on 18F-FDG uptake in responsive breast cancer cells and investigated the mediating molecular mechanisms. Methods: T47D breast cancer cells were stimulated with 17β-estradiol (E2) or bovine serum albumin (BSA)–E2 and measured for 18F-FDG uptake, lactate release, and mitochondrial hexokinase activity. The effects of antiestrogens, cycloheximide, and major protein kinase inhibitors were investigated. Immunoblots were performed for membrane glucose transporter type 1, phosphorylated phosphatidylinositol 3-kinase (PI3K), and Akt. Results: E2 augmented T47D cell 18F-FDG uptake in a dose- and time-dependent manner that preceded and surpassed its proliferative effect. With exposure to 10 nM E2, protein content–corrected 18F-FDG uptake reached 172.7% ± 6.6% and 294.4% ± 9.5% of controls by 24 and 48 h, respectively. Lactate release reached 110.9% ± 7.3% and 145.2% ± 10.5% of controls at 24 and 48 h, and mitochondrial hexokinase activity increased to 187.1% ± 31.6% at 24 h. Membrane glucose transporter type 1 expression was unaltered. The effect was absent in estrogen receptor (ER)–negative breast cancer cells and was abrogated by ICI182780, indicating ER dependence. The E2 effect was not blocked by tamoxifen and was mimicked by membrane-impermeable BSA-E2, consistent with nongenomic membrane-initiated E2 action. Inhibition by cycloheximide demonstrated the requirement of a new protein synthesis. Immunoblots displayed rapid phosphorylation of PI3K and Akt within minutes of E2 treatment, and the specific PI3K inhibitors wortmannin and LY294002 abolished the ability of E2 to elevate 18F-FDG uptake. Conclusion: Estrogen augments breast cancer cell 18F-FDG uptake by stimulating glycolysis and hexokinase activity via membrane-initiated E2 action that activates the PI3K–Akt pathway. These findings yield important insight into our understanding of the biology of breast cancer metabolism and may have potential implications for 18F-FDG uptake as a surrogate marker of therapeutic response.
Avid cancer cell uptake of the glucose analog 18F-FDG is widely exploited for the detection and staging of malignant diseases with PET. In systemic cancer therapy, the magnitude of 18F-FDG uptake can also serve as a surrogate biomarker for monitoring early therapeutic response, which may potentially allow early treatment adjustments (1–4). In breast cancer, an increasing number of patients are receiving systemic chemotherapy or hormone therapy as adjuvant, neoadjuvant, or palliative procedures. Serial PET studies have shown a correlation between treatment efficacy and reduction of breast tumor 18F-FDG uptake after primary chemotherapy (5,6) and a transient increase followed by a decrease in uptake with tamoxifen hormone therapy (7–9). Given the potential clinical value of 18F-FDG uptake as an indicator of therapeutic response, biologic factors that influence breast cancer cell glucose metabolism are of significant interest.
An exceptionally important player involved in the development and progression, treatment response, and prognosis of breast cancers is estrogen (10,11). Estrogen is a potent stimulator of proliferation in responsive cells, a process that needs to be fueled by large amounts of energy, replenished predominantly in cancer cells by stimulating glycolysis. In accordance, previous studies have observed increased glucose consumption rates in breast cancer cells exposed to estrogen (12–14). The molecular mechanisms that mediate this estrogen action, however, have not been clearly revealed. Breast cancer cells with high glucose metabolism have been associated with greater invasiveness and proliferative activity (15,16), and high breast tumor 18F-FDG uptake is considered a poor prognostic sign (17). Hence, unveiling the mechanism of estrogen-enhanced glucose metabolism could yield useful insights into tumor biology and potential therapeutic targets and may help advance the application of 18F-FDG PET for monitoring treatment response in breast cancer. In this study, we therefore investigated the effect of estradiol on 18F-FDG uptake in estrogen-responsive T47D breast cancer cells and elucidated the molecular mechanisms that mediate this metabolic effect.
MATERIALS AND METHODS
Cells and Reagents
Estrogen receptor (ER)–positive human breast carcinoma T47D cells and ER-negative MDA-MB-231 and MDA-MB-468 cells were from the American Type Culture Collection. Culture medium, fetal bovine serum, and antibiotics were from Gibco BRL. Charcoal-stripped serum was from HyClone Laboratories. Rabbit antihuman glucose transporter type 1 (GLUT-1) antibody was purchased from Dako; goat antihuman phosphorylated phosphatidylinositol 3-kinase (p-PI3K) antibody (p85a, Tyr 508) and rabbit polyclonal anti–β-actin antibody from Santa Cruz Biotechnology; and rabbit antihuman phosphorylated Akt (p-Akt) antibody (Ser473) from Cell Signaling Technology. All other reagents, including bovine serum albumin (BSA)–E2, were from Sigma Chemicals unless otherwise specified. E2, BSA-E2, tamoxifen, and ICI182780 were prepared as stocks in absolute ethanol, and wortmannin, LY294002, and cycloheximide were prepared as stocks in dimethyl sulfoxide. Ethanol and dimethyl sulfoxide vehicles were used as controls as indicated.
Cell Culture and Estrogen Stimulation
Cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum and penicillin–streptomycin in a 5% CO2 incubator at 37°C. Cells were split 3 d before experiments and seeded with phenol red–free RPMI-1640 medium containing 5% charcoal-stripped serum. Experiments were performed when cell confluence reached approximately 80%.
The addition of E2 or cell membrane–impermeable BSA-E2 to culture medium and incubation at 37°C, 5% CO2, for indicated durations was used to stimulate the cells. Inhibition experiments were done by adding the following agents to the culture medium at 30 min before the addition of E2: the partial antiestrogen tamoxifen (0.5 μM), pure antiestrogen ICI182780 (1 μM), protein synthesis inhibitor cycloheximide (100 nM), specific PI3K inhibitors wortmannin (200 nM) and LY294002 (10 μM), specific mitogen-activated protein kinase (MAPK) inhibitor PD98059, and protein kinase C (PKC) inhibitor staurosporine (1 nM). PKC was also inhibited by prolonged (6 h) exposure to 100 nM phorbol 12-myristate 13-acetate.
Measurements of Cell Proliferation and 18F-FDG Uptake
Cell proliferation was assessed by Bradford protein assays and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays.
For uptake measurements, cells were incubated with 185 kBq (5 μCi) of 18F-FDG added to the culture medium at 37°C for 40 min. After rapid washing twice with cold phosphate-buffered saline, cells were lysed with 0.1N NaOH and measured for radioactivity on a high-energy γ-counter (Wallac). Each sample was then measured for protein content by the Bradford method, and final uptake results were expressed as protein content–corrected counts relative to those of control cells.
Measurement of Lactate Release
For lactate assays, spectrophotometric absorbance from the culture medium was minimized by changing to phenol red–free α-minimum essential medium. Cell culture medium was transferred in duplicate to a flat-bottomed 96-well microplate, and lactate concentration was assayed using a commercial kit (Eton Biosciences). The assay is based on conversion of lactate and nicotinamide adenine dinucleotide (NAD+) by lactate dehydrogenase into pyruvate and nicotinamide adenine dinucleotide hydrogen (NADH) and reduction of the tetrazolium salt INT (2-p-iodophenyl-3-p-nitrophenyl-5-phenyl tetrazolium chloride) in an NADH-coupled enzyme reaction of formazan. Briefly, after adding 50 μL of assay solution to 20 μL of culture medium, the plate was sealed and vigorously agitated for 60 min at room temperature. The reaction was quenched with 50 μL of 0.5 M acetic acid, and each sample was measured for absorbance at 490 nm on a microplate reader. Lactate concentrations were obtained by plotting a standard curve between absorbance and concentration using serially diluted standards. Final results were expressed as the percentage of lactate concentration relative to that of control cells.
Measurement of Mitochondria-Associated Hexokinase Activity
Mitochondria-enriched fractions of cells were isolated by a previously reported method (18). Briefly, cells suspended in an isolation buffer containing 0.1 M Tris-(4-morpholinepropanesulfonic acid), 0.1 M ethylene glycol tetraacetic acid/Tris, and 1 M sucrose (pH 7.4) were homogenized by repeated stroking in a glass potter. After cell debris was eliminated by 600g centrifugation at 4°C for 10 min, the homogenate was centrifuged in a glass tube at 7,000g at 4°C for 10 min. The pellet was washed with cold isolation buffer and centrifuged at 7,000g at 4°C for 10 min to obtain the mitochondria pellet, which was resuspended in a minimal volume of isolation buffer.
A buffer, comprising the homogenization buffer containing 0.5 mM glucose, 5 mM adenosine triphosphate, and 0.25 mM reduced nicotinamide adenine dinucleotide phosphate and 6 units glucose-6-phosphate dehydrogenase, was prepared and preincubated at 20°C for 15 min. An aliquot of the mitochondrial sample was added to the buffer, and the mixture was measured for absorbance. Hexokinase activity was determined from a standard curve, with 1 unit defined as enzyme activity that phosphorylates 1 μmol of glucose per minute at 20°C, and final results were expressed as protein-corrected enzyme activity relative to that of control cells.
Immunoblots for Membrane GLUT-1, p-PI3K, and p-Akt
Plasma membrane GLUT-1 protein was prepared as previously described (19). Briefly, cells were treated with 10 strokes on a dounce homogenizer in HES buffer (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.4, 1 mM ethylenediaminetetraacetic acid, and 250 mM sucrose) containing 5 mM benzamidine, 1 μM aprotinin, 1 μM leupeptin, 1 μM pepstatin, and 1 mM phenylmethylsulfonyl fluoride. After cell debris was eliminated by 10 min of centrifugation at 2,000g, the sample was centrifuged at 9,000g for 20 min to obtain the first pellet. The supernatant was transferred to a high-speed microtube and centrifuged at 180,000g for 90 min. The pellet was suspended with 500 μL of phosphate-buffered saline with protease inhibitor, loaded on a sucrose gradient, and centrifuged in a Beckman SW 40.1 rotor at 48,000 rpm for 55 min to obtain the second pellet. Protein from pellets 1 and 2 was separated on a 10% polyacrylamide gel, electroblotted to a hydrobond ECL nitrocellulose membrane (Amersham), and reacted with a polyclonal antibody against human GLUT-1 (1:1,000 dilution). Immune reactive protein was visualized by incubation with horseradish peroxidase–conjugated secondary antirabbit IgG antibody (1:3,000 dilution; 0.13 μg/mL), followed by 1 min of soaking in Amersham ECL Western Blotting Detection Reagent (GE Healthcare) and exposure to a high-performance chemiluminescence film. Protein-band intensities were measured using a calibrated densitometer and Quantity One software (Bio-Rad Laboratories).
For immunoblotting of p-PI3K, cells were lysed in a buffer containing 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 5 mM ethylenediaminetetraacetic acid, 1% NP-40, 1 μM aprotinin, 1 μM leupeptin, 1 μM pepstatin, and 1 mM phenylmethylsulfonyl fluoride on a rocker at room temperature for 30 min. Protein (20 μg) obtained from the supernatant after 16,000g centrifugation for 15 min was separated on a 15% polyacrylamide gel, membrane-transferred, and incubated with a polyclonal antibody against human p-PI3K (p85a, Tyr 508; 1:1,000 dilution; 0.2 μg/mL). Immune-reactive protein was visualized as above using a secondary antigoat IgG antibody (1:1,000 dilution; 0.4 μg/mL). The membrane was subsequently stripped with a buffer containing 62.5 mM Tris (pH 6.8), 2% sodium dodecyl sulfate (SDS), and 100 mM 2-mercaptoethanol at 50°C for 30 min and immunoblotted with a rabbit polyclonal antibody against β-actin (1:5,000 dilution; 0.04 μg/mL) as a loading control.
p-Akt was immunoblotted with immunoprecipitated protein. Cell lysate obtained by a 15-min treatment in 4°C with radioimmunoprecipitation assay buffer (10 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM ethylenediaminetetraacetic acid; 0.1% SDS; 1% Triton X-100 [Sigma Chemicals]; and 0.1% sodium–deoxycholic acid) was centrifuged at 14,000g for 15 min. The supernatant was precleaned with 100 μL of protein-A Sepharose bead slurry (50%) at 4°C for 10 min on a rocker, followed by 14,000g centrifugation at 4°C for 10 min. Samples were then incubated with a monoclonal antibody against p-Akt (ser473) for 2 h at 4°C, followed by the addition of 50 μL of protein-A Sepharose bead slurry (50%) and overnight incubation at 4°C. After the cold radioimmunoprecipitation assay buffer was washed 3 times, protein was eluted in 30 μL of Laemmli 2× buffer (30% glycerol, 4% SDS [pH 6.8], 12 mM Tris-HCl, and bromophenol blue) [Sigma Chemicals], heated at 95°C for 5 min, and separated on a 10% SDS–polyacrylamide gel. After the protein was transferred to a polyvinylidene difluoride membrane, immunoreactive protein was detected and quantified as above using the above p-Akt primary antibody (1:1,000 dilution) and antirabbit secondary antibody (1:2,000 dilution; 0.2 μg/mL).
Statistical Analysis
Data were analyzed by nonpaired t tests and presented as mean ± SD. Differences were considered significant when the P value was less than 0.05.
RESULTS
E2 Augments T47D Cell 18F-FDG Uptake, Preceding and Surpassing Proliferative Effects
The exposure of ER-positive T47D cells to 10 nM E2 caused a substantial augmentation of 18F-FDG uptake of 208.7% ± 18.2% of control cells at 24 h (P < 0.001; Fig. 1A). In comparison, cell content as measured by protein concentration and MTT assays increased only 31.6% ± 6.1% and 31.1% ± 4.4% at 24 h (Fig. 1A) and 41.7% ± 5.5% and 68.6 ± 5.3 at 48 h (data not shown), respectively. Hence, enhancement of 18F-FDG uptake by E2 clearly preceded and surpassed its proliferative effect.

Effects of E2 on T47D cell 18F-FDG uptake and proliferation. (A) 18F-FDG uptake, protein content, and MTT assays in cells exposed to 10 nM E2 for 24 h. (B) Time course of increase in protein content–corrected 18F-FDG uptake, compared with protein content, after exposure to 10 nM E2. (C) Concentration-dependent effect of 24-h E2 stimulation on 18F-FDG uptake. E2 or vehicle was added to cells in phenol red–free culture medium containing 5% charcoal-stripped serum. All results are mean ± SD of percentage control levels of triplicate samples obtained from representative experiment of 2 independent experiments. *P < 0.05. †P < 0.005 vs. controls. ‡P < 0.001 vs. controls.
The time course of E2 effect on protein content–corrected 18F-FDG uptake displayed a mild increase at 8 h (120.6% ± 2.3% of controls) that progressively increased with longer exposure, reaching 172.7% ± 6.6% by 24 h and 294.4% ± 9.5% by 48 h (both P < 0.001; Fig. 1B). Dose-dependent experiments showed that increased 18F-FDG uptake began when E2 concentrations were 0.1 nM and peaked at 1–10 nM (Fig.1C). These concentrations can be considered biologically relevant, because plasma levels of estradiol in women of menstrual phase or under replacement therapy are known to range around 0.1–1 nM.
18F-FDG Effect Is Accompanied by Enhanced Glycolysis and Hexokinase Activity
The enhancement of 18F-FDG uptake by E2 was accompanied by an elevated lactate release into culture medium to 110.9% ± 7.3% and 145.2% ± 10.5% (P < 0.001) of controls by 24 and 48 h, respectively (Fig. 2, left). Mitochondrial hexokinase activity was substantially augmented to 187.1% ± 31.6% of control levels by 24 h (P < 0.001; Fig. 2, middle). In contrast, plasma membrane–expressed GLUT-1 levels were uninfluenced by E2 (Fig. 2, right). These findings demonstrate significant contributions from stimulated glycolysis and hexokinase activity on the metabolic effect of E2 on T47D cells.

Effects of E2 on lactate release (A), mitochondrial hexokinase activity (B), and membrane GLUT-1 expression (C). Cells were treated with 10 nM E2 or vehicle for 24 or 48 h, as indicated. P1 and P2 are first and second pellet fractions of centrifuged membrane samples, respectively. Results are mean ± SD obtained from 2 independent experiments (n = 6) for lactate and hexokinase assays and from single representative experiment (n = 3) for GLUT-1. **P < 0.01 vs. controls. ‡P < 0.001 vs. controls.
Increased 18F-FDG Uptake Occurs Through Membrane-Initiated E2 Action
Unlike T47D cells, 18F-FDG uptake in ER-negative MDA-MB-231 and MDA-MB-468 breast cancer cells was totally unresponsive to E2, indicating ER dependence of the metabolic effect (Fig. 3A). In T47D cells, pretreatment with the pure antiestrogen ICI182780 abrogated the ability of E2 to augment 18F-FDG uptake. However, the partial antiestrogen tamoxifen was incapable of blocking the E2 effect (Fig. 3B). When cell membrane–impermeable BSA-E2 was used to examine the role of membrane ER, 18F-FDG uptake was enhanced in a dose-dependent fashion, closely resembling that of E2. Hence, 18F-FDG uptake was increased 63.1% ± 1.8% and 91.5% ± 7.6% over controls with 10 and 100 nM of BSA-E2, respectively (both P < 0.001; Fig. 3C).

Evidence for membrane ER–initiated E2 action. (A) Effects of E2 on 18F-FDG uptake in ER-positive T47D cells and ER-negative MDA-MB-231 and MDA-MB-468 cells. (B) Effects of partial antiestrogen tamoxifen (0.5 μM) and pure antiestrogen ICI182780 (1 μM) on 18F-FDG uptake of T47D cells. (C) Dose-dependent effect of membrane-impermeable BSA-E2 on 18F-FDG uptake of T47D cells. Results are mean ± SD of triplicate samples from single representative experiment (A and C) or 3 independent experiments (n = 9; B). E2(+) = cells treated with 10 nM E2 for 24 h; ICI = ICI294002; Tmx = tamoxifen. †P < 0.005 vs. controls. ‡P < 0.001 vs. controls.
Rapid PI3K–Akt Activation Is Present and Is Necessary for E2-Augmented 18F-FDG Uptake
Western blot analysis of phosphorylated forms of PI3K and Akt protein revealed rapid activation of both kinases, beginning within minutes and persisting for at least 2 h after initiation of E2 treatment (Fig. 4).

Rapid activation of PI3K–Akt pathway in E2-stimulated T47D cells. (A) Time course of immunoblot findings and protein-band intensities of p-PI3K and p-Akt after exposure to 10 nM E2. (B) Western blots of p-PI3K and β-actin after 10 nM E2 stimulation. Bars are mean ± SD of protein-band intensities of p-PI3K relative to that of β-actin. (C) Western blots and protein-band intensities of p-Akt from immunoprecipitated samples after 10 nM E2 stimulation. Bars are mean ± SD of percentage control levels. *P < 0.05 vs. controls. †P < 0.005 vs. controls.
Inhibition experiments with cycloheximide showed that the E2 effect required new protein biosynthesis (Fig. 5A). The specific PI3K inhibitors wortmannin and LY294002 reduced the E2 effect to levels even lower than those in untreated controls (Fig. 5B), indicating dependence of the effect on the PI3K signaling. In contrast, inhibition of the MAPK pathway with PD98059 or the PKC pathway with staurosporine or prolonged phorbol 12-myristate 13-acetate exposure (known to inhibit PKC) had no influence on E2-stimulated 18F-FDG uptake (Figs. 5C and 5D).

Effects of cycloheximide and specific protein kinase inhibitors. (A) Effect of protein biosynthesis inhibition with 100 nM cycloheximide on E2-stimulated 18F-FDG uptake. Effects of specific PI3K inhibitors wortmannin (200 nM) and LY294002 (10 μM) (B), specific MAPK inhibitor PD98059 (50 μM) (C), and PKC inhibitor staurosporine (1 nM) and prolonged phorbol 12-myristate 13-acetate (100 nM) (D) exposure on E2-stimulated 18F-FDG uptake. Results are mean ± SD of percentage control level obtained from 3 (n = 9; A–C) or 2 independent experiments (n = 6; D). cyclohex = cylcoheximide; PMA = phorbol 12-myristate 13-acetate; stau = staurosporine; wortman = wortmannin. ‡P < 0.001 vs. E2-stimulated uptake in absence of inhibitors.
DISCUSSION
In this study, we demonstrate that E2 induces a dose- and time-dependent augmentation of 18F-FDG uptake in T47D breast cancer cells, in a manner that significantly precedes and surpasses any proliferative effect. The effect occurred through enhancement of glycolytic flux and hexokinase activity but not membrane GLUT-1 expression. Evidence that indicated the ER dependence of this metabolic response included the absence of an effect in ER-negative breast cancer cells and the complete abolishment of the effect by the pure antiestrogen drug ICI182780.
Increased release of lactate from E2-treated cells in our experiments indicates that a significant portion of the excess glucose taken up was used for glycolysis. The first step of the glycolytic pathway involves hexokinase as the rate-limiting enzyme that catalyses the conversion of glucose to glucose-6-phosphate. In our study, E2 induced a 1.9-fold increase in mitochondrial hexokinase activity in T47D cells. Mitochondrial hexokinase is the driving force for high glycolytic activity in cancer cells, because association to the outer mitochondrial membrane confers direct access to mitochondria-generated adenosine triphosphate and renders the enzyme less sensitive to glucose-6-phosphate inhibition (20). Energy requirements of rapidly proliferating cells are substantially different from those of quiescent cells. Hence, there is a need for biologic stimulators of proliferation to also propagate signals that reorganize the cell's metabolic activity. Malignant cells are heavily dependent on glucose as their energy fuel, and the association between tumor growth and increased glucose consumption is well established. The elevation of hexokinase activity by proliferative stimuli may, thus, represent an adaptive response to meet the increased energy demand of replicating cells (21) or, as has been suggested, may be a modification able per se to drive mitotic activity (22). Unlike hexokinase activity, we did not see any change in expression of membrane GLUT-1, a facilitative hexose transporter widely overexpressed in breast cancers and mainly responsible for 18F-FDG uptake in T47D cells (23,24). GLUT-1 levels have been reported to be relatively low in T47D cells (24), and they appear to have a limited role in augmenting glucose accumulation by E2 in these cells.
In our results, unlike ICI182780, the partial antiestrogenic agent tamoxifen showed no inhibitory effect on E2-stimulated 18F-FDG uptake. Estrogen action is now recognized to occur either through the classic nuclear ER or the more recently identified plasma membrane–localized ER (25,26). Tamoxifen causes conformational changes on ER that precludes its binding to response elements on the DNA (27). Therefore, the antiestrogenic effect of tamoxifen is restricted to nuclear ER, with little influence on membrane ER. On the other hand, ICI182780 is generally known to block the actions of both nuclear and membrane ER (28). Therefore, our observation that E2-induced 18F-FDG uptake is abrogated by ICI182780 but unaffected by tamoxifen is consistent with membrane-initiated estrogen action. Interestingly, when tamoxifen was tested in the absence of estrogen influence, 18F-FDG uptake slightly decreased, compared with control cells. This finding was rather unexpected given the partial estrogen agonist property of the drug and previous observations that ER-positive breast tumors increase their 18F-FDG uptake in response to tamoxifen therapy (7–9). This latter phenomenon has been explained by an initial agonistic effect of tamoxifen before its antagonist effect supervenes (7–9). Our findings that tamoxifen does not directly stimulate the glucose metabolism of breast cancer cells, however, indicate that other contributing factors may need to be identified to explain the metabolic flare response of breast tumors seen on 18F-FDG PET.
Further evidence that E2-enhanced 18F-FDG uptake is mediated by membrane ER–initiated action was provided by a cell-impermeable plasma protein–conjugated form of estrogen (BSA-E2) that can act only on membrane-localized ER (29). As a result, exposure to BSA-E2 closely simulated the effect of E2, causing significant increases of 18F-FDG uptake in a dose-dependent manner. Taken together, these results indicate that nongenomic membrane–initiated action is responsible for the metabolic effect of estrogen on breast cancer cells.
Nongenomic membrane–initiated estrogen action is now understood to mediate several important biologic responses in breast cancer cells (25,26), including E2-induced proliferation (30). These responses are similar to the action mechanisms of peptide growth factors in that E2 sets off cytoplasmic signaling cascades via activation of major protein kinases within minutes of receptor interaction. Because ERs do not have intrinsic kinase activity, membrane-initiated intracellular signaling in the presence of E2 depends on the formation complexes that include ER, Src protein, and the regulatory subunit of PI3K. This formation of complexes triggers activation of the catalytic subunit of PI3K that leads to the activation of downstream Akt, a serine–threonine protein kinase that mediates many of the cellular effects of PI3K (30). The PI3K–Akt cascade thus constitutes a major signaling pathway through which estrogens mediate membrane-initiated biologic actions.
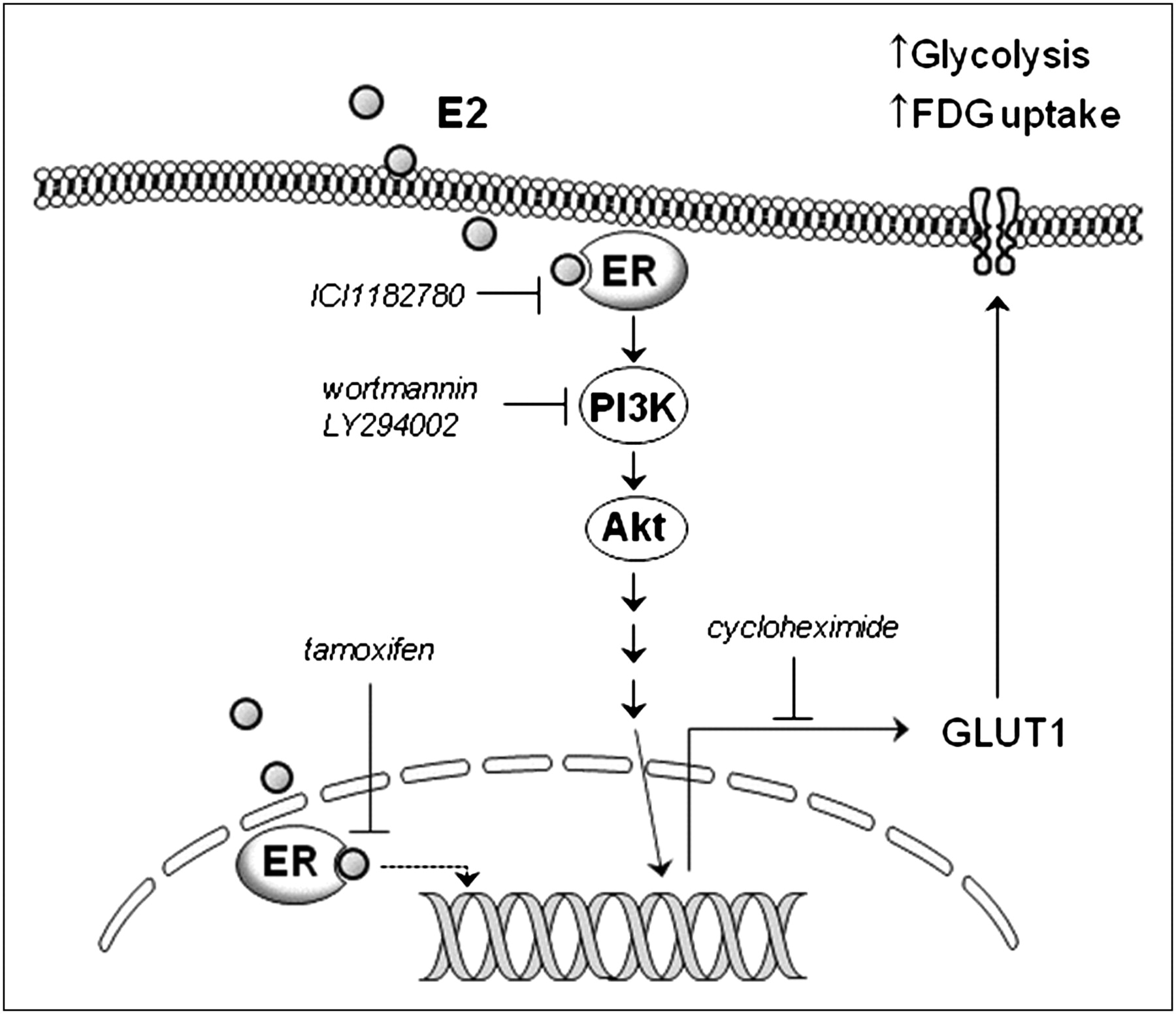
In our study, we thus tested the effects of blocking the PI3K pathway and the MAPK and PKC pathways, which are the major pathways reported to mediate membrane-initiated estrogen actions, on the metabolic effect of E2. As a result, the ability of E2 to augment 18F-FDG uptake was found to be dependent on PI3K activity but not on MAPK or PKC activity. Furthermore, both PI3K and Akt were found to be rapidly activated by E2 treatment. These findings indicate a central role, which is more likely causal than associated, for the PI3K–Akt pathway in mediating the metabolic effect of E2 in breast cancer cells. PI3K and Akt are also important in growth factor– and insulin-stimulated glucose metabolism in certain cells. Akt can also directly stimulate glycolytic activity in cancer cells in a manner correlating to tumor aggressiveness (31) and can induce many of the metabolic changes reflected in the Warburg effect (32). Our results, therefore, provide a link in breast cancer cells between E2-induced augmentation of glycolytic activity and PI3K–Akt signaling, which has critical roles in cell proliferation, survival, and metabolism. A schematic representation of the signaling pathways that lead to enhanced glycolysis and 18F-FDG uptake in E2-stimulated breast cancer cells is shown in Figure 6.

Schematic representation of intracellular signaling pathways leading to enhanced glycolysis and 18F-FDG uptake in E2-stimulated breast cancer cells.
In addition to their biologic significance, the findings of this study have several implications for clinical PET. Our data support and provide the molecular basis for the proposal that circulating estrogens influence 18F-FDG uptake in responsive normal and malignant breast tissues (9,33). This finding does not imply that breast tumors with high 18F-FDG uptake likely have estrogen-stimulated ER. Rather, breast cancer is a heterogeneous disease, with complex biology, whose metabolism is governed by a myriad of factors including tumor size, grade, and stimulation by growth factors and hormone receptor status. Nonetheless, 18F-FDG uptake enhancement by estradiol challenge could be a useful biomarker of hormone responsiveness in postmenopausal women as previously proposed (9). Although tamoxifen did not directly simulate 18F-FDG uptake in our in vitro experiments, the usefulness of metabolic flare seen on 18F-FDG PET for indicating responsive tumors (7–9)—which may require tamoxifen action in an in vivo environment—is not invalidated. More recently, novel targeted agents that involve or directly target the PI3K–Akt pathway are emerging for the treatment of breast cancer (34). The success of such targeted therapies requires biomarkers of tumor response to guide individualized treatment decisions. In this regard, our findings also suggest a potential role for 18F-FDG PET in monitoring response of breast cancers to therapeutic agents that target the PI3K–Akt pathway.
CONCLUSION
Estradiol augments 18F-FDG uptake in T47D breast cancer cells via increased glycolysis and hexokinase activity, which is mediated by membrane-initiated estrogen action that leads to rapid activation of PI3K–Akt signaling. These findings not only yield insight into our understanding of the biology of breast cancer metabolism but also provide the molecular basis for the use of 18F-FDG uptake as a marker of responses to systemic therapies that target estrogen action or downstream signaling pathways.
Acknowledgments
This work was supported by a Korean Science and Engineering Foundation (KOSEF) grant funded by the Korean government (MOST) (20090066109).
Footnotes
↵* Contributed equally to this work.
- © 2010 by Society of Nuclear Medicine
REFERENCES
- Received for publication January 6, 2010.
- Accepted for publication April 28, 2010.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}