


Abstract
We investigated the feasibility of using combination gene therapy and noninvasive nuclear imaging after expression of the human sodium iodide symporter (hNIS) and inhibition of the multidrug resistance (MDR1) gene in colon cancer cells. Methods: HCT-15 cells were stably transfected with a dual expression vector, in which the hNIS gene, driven by a constitutive cytomegalovirus promoter, has been coupled to an MDR1 short hairpin RNA (shRNA) cassette. Cell lines stably expressing the hNIS gene and MDR1 shRNA (designated MN-61 and MN-62) were produced, and the expression of the NIS gene and MDR1 shRNA was examined by Western blotting, reverse transcription–polymerase chain reaction, and immunostaining. The functional activities of MDR1 shRNA were determined by paclitaxel uptake and sensitivity to doxorubicin. Functional NIS expression was confirmed by the uptake and efflux of 125I and the cytotoxicity of 131I. The effect of the combination of 131I and doxorubicin was determined by an in vitro clonogenic assay. In vivo NIS expression was examined by small-animal PET with 124I. Results: The shMDR-NIS–expressing cells showed a significant decrease in the expression of MDR1 messenger RNA and its translated product, P-glycoprotein. The inhibition of P-glycoprotein expression by shRNA enhanced the intracellular accumulation of paclitaxel, the cellular retention of which is mediated by P-glycoprotein, thereby increasing sensitivity to the anticancer drug. The shMDR-NIS–expressing cells showed a significant increase of 125I uptake, which was completely inhibited by KClO4. Although the iodide efflux rate was rapid, the cell survival rate was markedly reduced by 131I treatment. Interestingly, the combination of doxorubicin and a radioiodide (131I) displayed synergistic cytotoxicity that correlated with MDR1 inhibition and NIS expression in shMDR-NIS–expressing cells. Furthermore, in mice with shMDR-NIS–expressing tumor xenografts, small-animal PET with 124I clearly visualized shMDR1-NIS–expressing tumors. Conclusion: We developed a dual expression vector with the NIS gene and MDR1 shRNA. This study represents a promising first step in investigations of the potential use of a combination of the NIS gene and MDR1 shRNA as a new therapeutic strategy allowing RNA interference–based gene therapy, NIS-based radioiodine therapy, and in vivo monitoring based on NIS imaging.
The sodium iodide symporter (NIS) is a membrane glycoprotein that mediates the active uptake of 1 iodide ion along with 2 sodium ions across the basolateral membrane of thyroid follicular cells (1,2). The cloning and characterization of the NIS gene could lead to a novel gene strategy for radioiodine therapy in thyroid cancer. Recent advances in gene therapy have led to several studies aimed at introducing NIS expression in other tumor types to provide for radioiodide therapy (3,4). These studies have indicated that the transfer of the NIS gene confers iodide uptake capacity in a wide range of tumor cells, allowing the possibility of NIS-based radioiodide therapy for many different tumors. Moreover, an NIS transgene can be used as a reporter gene for noninvasive monitoring of the therapeutic effect after radioiodide therapy by γ-scintigraphy or PET.
One of the key drug resistance mechanisms in cancer therapy is the overproduction of P-glycoprotein, which is encoded by the multidrug resistance (MDR1) gene. P-glycoprotein is a 170-kDa transmembrane protein that belongs to the adenosine triphosphate–binding cassette (ABC) transporter superfamily (5). P-glycoprotein confers cross-resistance to even unrelated drugs that differ significantly in terms of molecular structure and target specificity, such as paclitaxel, vincristine, and doxorubicin. Various attempts, such as inhibition of the activity or expression of P-glycoprotein, have been undertaken to circumvent the drug resistance of cancer cells. Neutralizing antibodies, antisense oligonucleotides, and ribozymes have been tested (6,7). However, these methods have had limited applicability and success. Recently, it was reported that double-stranded RNA interference (RNAi) had emerged as a powerful reverse genetic tool to silence gene expression in multiple organisms. Several studies have shown that small interfering RNA (siRNA) effectively inhibits the expression of MDR1 messenger RNA (mRNA) and P-glycoprotein and restores sensitivity to the anticancer drug in many multidrug-resistant cells (8–10).
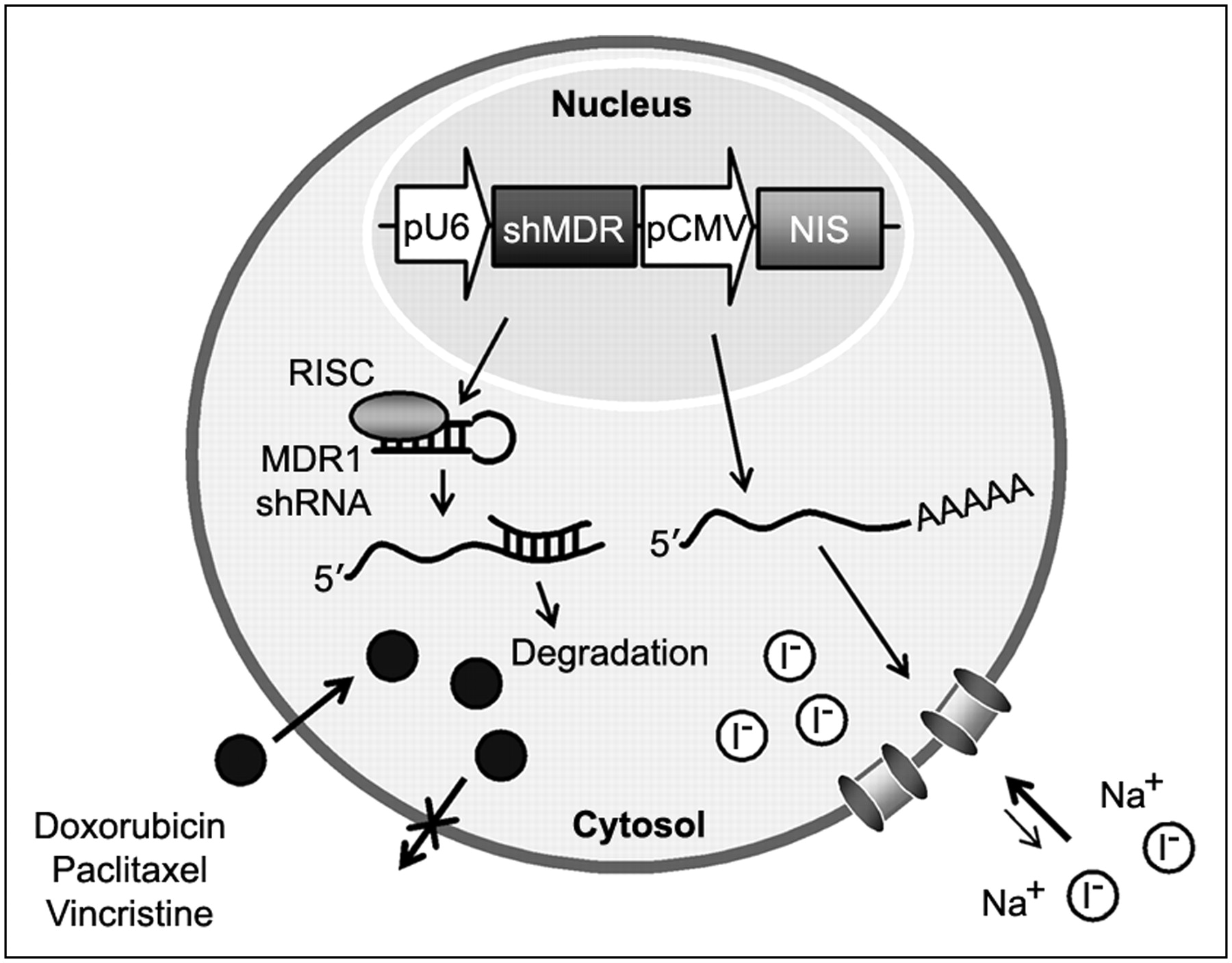
Recently, many studies reported strategies involving the coexpression of 2 therapeutic genes or linking of reporter gene activity with therapeutic gene expression in one vector system to enhance or monitor therapeutic effects (11,12). The coexpression of 2 genes is generally achieved by using dual promoters, by inserting an internal ribosomal entry site, or by fusing the 2 genes into a single translational cassette (13). In the present study, to improve NIS-based radioiodide therapy, we constructed a dual expression vector in which the NIS gene and therapeutic short hairpin RNA (shRNA) were driven by the cytomegalovirus (CMV) and U6 promoters, respectively (Fig. 1). For therapeutic shRNA, we selected MDR1, which is the major cause of the failure of effective chemotherapeutic treatment of disseminated neoplasms. We investigated the feasibility of a strategy of combining NIS-based radioiodide therapy and MDR1 shRNA–based chemotherapy in a novel dual expression vector in vitro. In addition, we also examined radioiodide accumulation in shMDR-NIS–expressing tumor xenografts in vivo to elucidate the imaging characteristics.
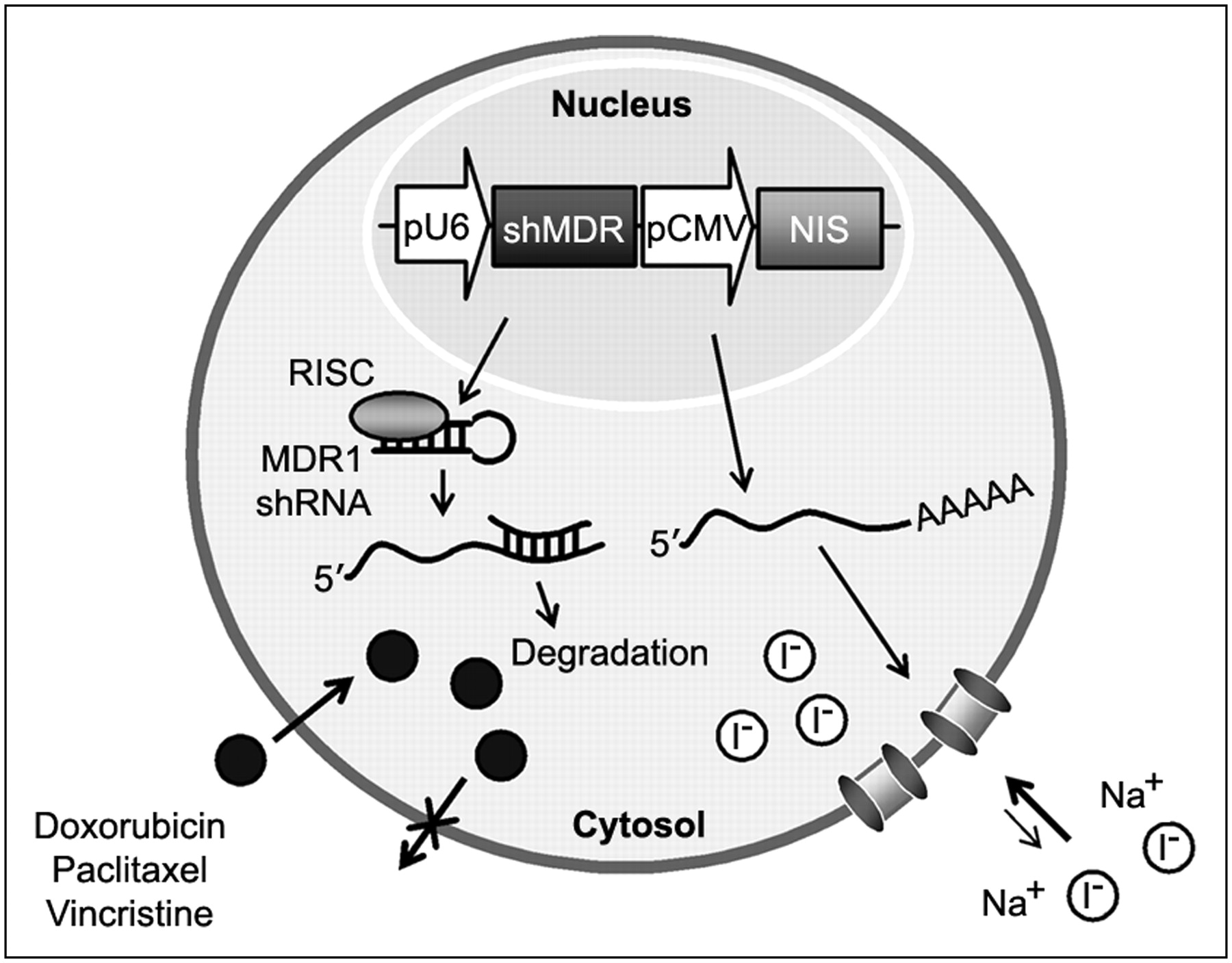
Scheme of combination gene therapy and imaging with NIS gene and MDR1 shRNA, which are simultaneously expressed from one vector system (shMDR-NIS). In the cells transfected with shMDR-NIS construct, MDR1 shRNA is processed to siRNA against MDR1 gene and then represses of P-glycoprotein expression that enhances outward transport of anticancer drug. NIS gene is transcribed to NIS mRNA and then translated to membrane transporter NIS, which mediates uptake of radioiodide across cell membrane. pCMV = CMV promoter; pU6 = U6 promoter; RISC = RNA-induced silencing complex.
MATERIALS AND METHODS
Cell Culture
The human colon cancer cell line HCT-15 was obtained from the Korean Cell Line Bank (KCLB number 10225) and grown in RPMI 1640 medium supplemented with 20% heat-inactivated fetal bovine serum (FBS) and the appropriate antibiotics. HCT-15 transfectants were grown in RPMI 1640 medium supplemented with 20% FBS and G418 at 500 μg/mL. The cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Plasmid Constructs
The siRNA sequence targeting MDR1 corresponded to the coding region at positions 3491–3509: 5′-GGC CTA ATG CCG AAC ACA T-3′ (GenBank accession number NM_000927). Oligonucleotides with a sequence predicted to induce efficient RNAi of MDR1 (containing sense and antisense sequences linked by a hairpin loop: CGAA) were synthesized (forward: 5′-gatccc GGCCTAATGCCGAACACAT CGAA ATGTGTTCGGCATTAGGCC ttttttccaac-3′; reverse: 5′-tcgagttggaaaaaa GGCCTAATGCCGAACACAT TTCG ATGTGTTCGGCATTAGGCC gg-3′). These oligonucleotides were annealed in STE buffer (10 mM Tris [pH 8.0], 50 mM NaCl, and 1 mM ethylenediaminetetraacetic acid) at 94°C for 5 min and cooled gradually. The double-stranded product was cloned downstream to the human U6 promoter of the pRNAT/U6 vector (Genscripts) and designated as pRNAT-shMDR.
For the construction of a dual expression vector, a green fluorescent protein complementary DNA (cDNA) sequence coupled to the CMV promoter was removed from the pRNAT-shMDR vector, and an NheI–SalI restriction site was introduced into the pRNAT-shMDR vector by the polymerase chain reaction (PCR). Next, human NIS (hNIS) cDNA (kindly provided by Dr. Je-Yeol Cho, Kyungpook National University, Daegu, Republic of Korea) was amplified by PCR with the sense primer, which contains an NheI restriction site, and the antisense primer, which contains an SalI restriction site. After restriction digestion with NheI and SalI of pRNAT-shMDR, NIS cDNA was cloned into the pRNAT-shMDR vector. The resulting plasmid construct containing the MDR1 shRNA sequence and NIS cDNA coupled to the U6 and CMV promoters, respectively, was designated as shMDR-NIS and confirmed by DNA sequencing.
Transfection
The human colon cancer cell line HCT-15 was transfected in OptiMEM I medium with Lipofectamine (Invitrogen) in accordance with the manufacturer's instructions. For stable transfection, selection was performed with neomycin (500 μg/mL; Invitrogen) in RPMI 1640 medium containing 20% FBS for approximately 3 wk from the day after transfection. The expression of MDR1 shRNA and the expression of the NIS gene were analyzed by reverse transcription (RT)–PCR and Western blotting, respectively. Surviving clones were isolated and subjected to screening for iodide and paclitaxel uptake activities. Two stable cell lines (designated MN-61 and MN-62), which showed the highest levels of expression of hNIS and MDR1 shRNA among approximately 70 colonies screened, were obtained. Negative control clones (HCT/Mock) were randomly selected from the transfection with the empty vector.
Iodide Uptake Studies
The uptake of 125I by control and transfected HCT-15 cells was determined at steady-state conditions as described previously (14). In brief, the cells were plated on 6-well plates (2 × 105 cells per mL) and grown for 48 h. Next, the growth medium was aspirated and replaced with Hank's balanced salt solution (HBSS) supplemented with 10 mM NaI, Na125I at 3.7 MBq/mL, and 10 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid (HEPES) at pH 7.3. The cells were incubated for 45 min at 37°C in the presence or absence of 10 μM KClO4. At the end of incubation, the cells were quickly washed with ice-cold phosphate-buffered saline (PBS) and lysed with 1N NaOH, and trapped iodide was measured with a γ-counter (Perkin-Elmer).
Iodide Efflux Studies
Iodide efflux studies of 125I were performed as described previously (14). In brief, MN-61 cells were plated on 6-well plates (2 × 105 cells per well) and incubated with HBSS supplemented with 10 mM NaI, Na125I at 3.7 MBq/mL, and 10 mM HEPES at pH 7.3 and 37°C for 30 min. The medium was replaced every 3 min with fresh HBSS. The content of 125I in the collected supernatant was measured with a γ-counter (Perkin-Elmer). Just after the last time point, the cells were quickly washed with ice-cold PBS and lysed with 1N NaOH, and trapped iodide was measured with a γ-counter.
Cell Proliferation Assay
Cell viability was assessed with a CellTiter 96 Aqueous One Solution cell proliferation assay (Promega) as previously described (15). In brief, 103 cells per well were seeded in 96-well plates, a doxorubicin dilution series was added in triplicate, and the cells were incubated for 4 d under standard culture conditions. At the end of incubation, a 20-μL volume of the detection solution was added to each well and incubated for 90 min before the absorbance at 490 nm was measured with a microplate reader (BioRad).
RNA Isolation and RT–PCR
Total RNA was extracted from cells with TRIzol reagent (Invitrogen) and quantified by ultraviolet light absorbance spectroscopy. The level of expression of MDR1 mRNA was examined as previously described (16). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) amplification was used as an internal control.
Western Blotting
Cells were washed twice with PBS containing 1 mM phenylmethylsulfonyl fluoride, scraped off the dishes, and centrifuged at 500g for 10 min. Cell pellets were then lysed in cold lysis buffer (20 mM Tris-HCl [pH 7.4], 200 mM NaCl, 1% Triton X-100 [Sigma], 1 mM phenylmethylsulfonyl fluoride, and 1% aprotinin) for 30 min with occasional rocking. Identical amounts (50 μg of protein) of cell lysates were resolved by 7% sodium dodecyl sulfate (SDS)–polyacrylamide gel electrophoresis (PAGE) and then transferred to nitrocellulose membranes. The membranes were incubated in blocking solution, consisting of 5% skim milk in TBS-T (10 mM Tris-HCl [pH 8.0], 150 mM NaCl, and 0.1% polysorbate 20], for 1 h at room temperature and then immunoblotted with anti–P-glycoprotein (clone C219; Calbiochem), anti-NIS (clone C-2; Labvision), or anti-actin (Sigma) antibody. Immunoreactive bands were visualized with an enhanced chemiluminescence kit (Amersham).
Immunostaining and Confocal Microscopy
HCT/Mock and MN-61 cells were seeded at a density of 4 × 104 cells per well on collagen-precoated chamber slides (Nunc) and grown for 48 h. The cells were washed twice with PBS (pH 7.4) and fixed with 4% paraformaldehyde in PBS for 10 min at room temperature. The fixed cells were then permeabilized with 0.2% Triton X-100 in PBS for 3 min, washed 3 times with PBS, blocked for 30 min with 2% bovine serum albumin (Sigma) in PBS, and stained with anti-NIS antibody (clone 44; Chemicon) diluted in 1% bovine serum albumin for 1 h at room temperature. After the cells were washed 3 times with PBS, a rhodamine-conjugated secondary antibody (Molecular Probes) was applied for 40 min at room temperature. The cells were washed with PBS and counterstained with DAPI (1 μg/mL). The slides were mounted with a solution of SlowFade (Molecular Probes), covered with glass cover slips, and examined by use of a laser confocal scanning system with Leica TCS SP2.
Paclitaxel and Doxorubicin Accumulation
Steady-state paclitaxel and doxorubicin accumulation was evaluated as previously described (8). In brief, parental HCT-15 cells and transfectants were seeded in 6-well plates and grown for 48 h. The growth medium was aspirated and replaced with 1 mL of RPMI 1640 medium containing 50 nM 3H-paclitaxel (3.7 × 1011 Bq/mmol; Moravek Biochemicals). After incubation for 2 h at 37°C, the cells were cooled on ice, washed 3 times with ice-cold PBS, and solubilized with 0.2 mL of 1% SDS. The radioactivity in each sample was determined by scintillation counting. To assess the steady-state doxorubicin accumulation, HCT-15 or shMDR-NIS–expressing cells were incubated with 25 μM doxorubicin for 2 h. At the end of incubation, the cells were washed 3 times with PBS and observed under a confocal microscope with a ×400 lens (Leica).
In Vitro Clonogenic Assay
Exponentially growing cells were incubated for 7 h with Na131I at 18.5–37.0 MBq/10 mL in HBSS supplemented with 10 mM NaI and 10 mM HEPES at pH 7.3. After incubation with 131I, the cells were trypsinized, seeded at a density of 103 cells per well in 6-well plates to which a doxorubicin dilution series (25–50 nM) was added in triplicate, and incubated for 10 d under standard culture conditions. At the end of incubation, the resulting cell colonies were stained with a Diff Quick staining kit (IMEB Inc.), and colonies containing more than 10 cells were counted. Parallel experiments were performed with HBSS without Na131I or doxorubicin, and all values were adjusted for plating efficiency. Cell survival was expressed as the percentage of colonies relative to that in the untreated control.
Small-Animal PET
Small-animal PET was performed with a Concorde microPET R4 rodent-model scanner (Concorde Microsystems Inc.) (17). An animal bearing tumor xenografts was injected with 18.5 MBq of 124I via a tail vein. At 1, 3, and 15 h after injection, the animal was placed in a prone position on the bed of the small-animal PET scanner. Anesthesia was performed with 1%−2% isoflurane in 100% O2 during injection and imaging. The images were reconstructed with a 2-dimensional ordered-subsets expectation maximum algorithm. Corrections were not required for attenuation or scattering. Activity was quantified by viewing the regions of interest in the tumors and averaging the activity concentrations over the contained voxels.
Statistical Analysis
All numeric data were expressed as mean ± SD. The statistical significance of differences was assessed by analysis of variance (ANOVA). P < 0.05 was considered to be statistically significant.
RESULTS
Stable Expression of MDR1 shRNA and NIS Gene in HCT-15 Cells
As shown in Figure 1, hNIS cDNA was cloned into an expression vector under the control of the CMV promoter, whereas the MDR1 shRNA sequence was recombined under the control of the U6 promoter. For characterization of the dual expression vector, the HCT-15 colon cancer cell line, which has high levels of endogenous MDR1 expression, was stably transfected with the shMDR-NIS dual expression construct. Two stable cell lines, MN-61 and MN-62, were obtained by selection with G418, and the levels of expression of MDR1 mRNA and P-glycoprotein were determined by RT–PCR and Western blotting, respectively. As shown in Figure 2A, the levels of expression of endogenous MDR1 mRNA were markedly decreased in MN-61 and MN-62 cells relative to those in parental HCT-15 and HCT/Mock cells. In contrast, no significant differences were observed in glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA expression in any of the cell lines. The expression of P-glycoprotein was also significantly decreased in MN-61 and MN-62 cells, in agreement with the results of the RT–PCR analysis (Fig. 2B). The expression of actin was not affected by the expression of MDR1 shRNA. hNIS protein expression was further confirmed by Western blotting and immunohistochemical analysis. Western blotting of cell lysates derived from MN-61 and MN-62 cells with a monoclonal antibody that recognizes hNIS protein revealed a band with a molecular mass of approximately 90 kDa, which was not detected in parental HCT-15 or HCT/Mock cells (Fig. 3A). In addition, NIS-specific immunoreactivity in MN-61 cells was revealed by immunofluorescence staining. In contrast, HCT/Mock cells did not show NIS-specific immunoreactivity. Control slides stained with isotype-matched nonimmune immunoglobulin were consistently negative (Fig. 3B).
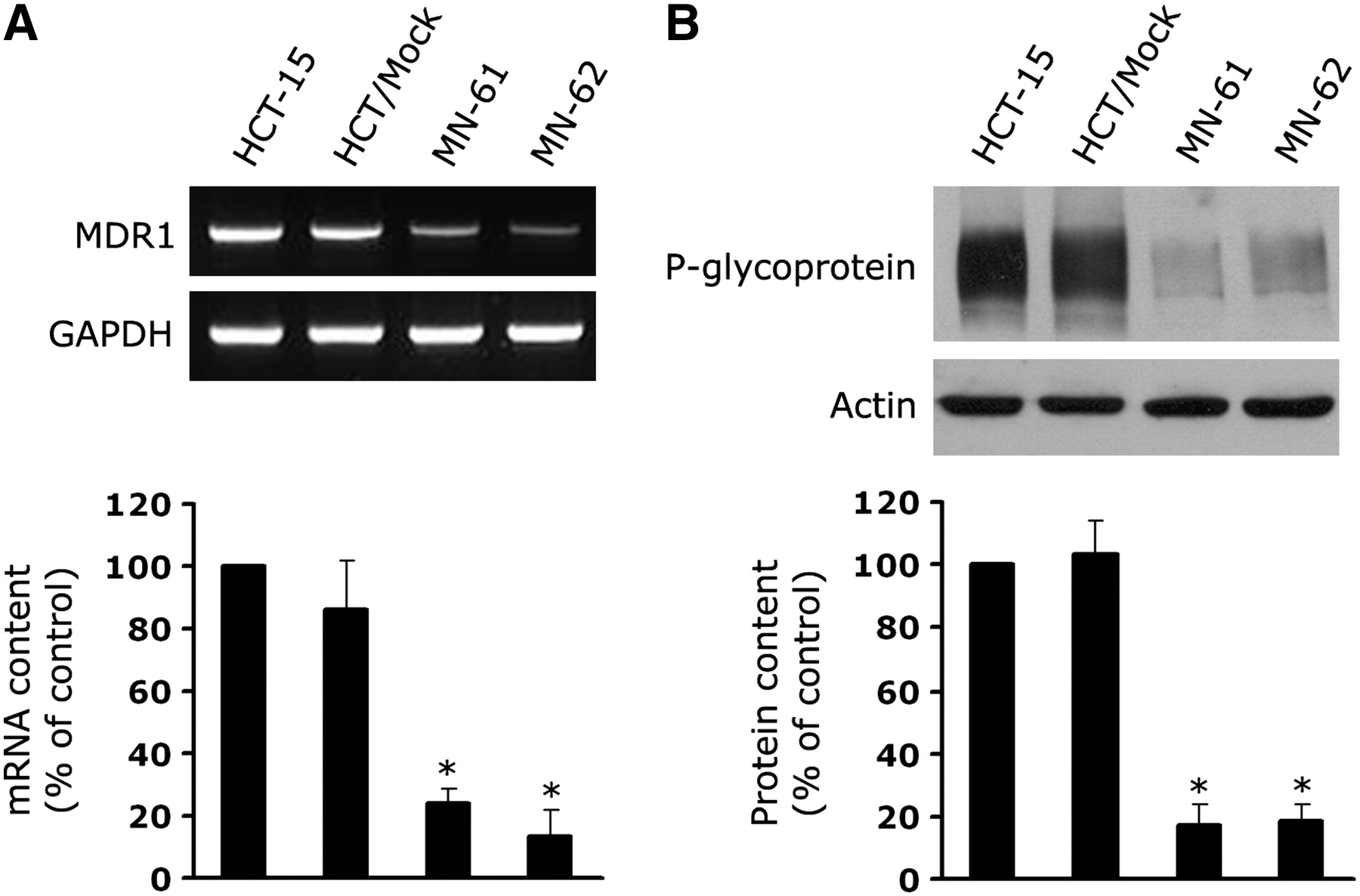
Suppression of MDR1 mRNA and P-glycoprotein in shMDR-NIS–expressing cells. (A) Total RNA was isolated from parental HCT-15, HCT/Mock, MN-61, and MN-62 cells, and MDR1 gene was amplified by PCR. GAPDH was used as internal control. PCR products were electrophoresed on agarose gels and then visualized by ethidium bromide staining (top). Amplified DNA levels obtained by densitometry were normalized to GAPDH signals, and relative intensities were expressed as percentage of that observed in control HCT-15 cells (bottom). Results are expressed as mean ± SD of 3 independent experiments. *P < 0.01, as determined by ANOVA. (B) Total cell lysates were prepared from parental HCT-15, HCT/Mock, MN-61, and MN-62 cells. Equal amounts (50 μg of proteins) of cell lysates were separated by 7% SDS–PAGE and then transferred to nitrocellulose membranes. Membranes were immunoblotted with anti–P-glycoprotein or antiactin antibody (top). Protein levels obtained by densitometry were normalized to actin signals, and relative intensities were expressed as percentage of that obtained in control HCT-15 cells (bottom). Results are expressed as mean ± SD of 3 independent experiments. *P < 0.01, as determined by ANOVA.

Expression of NIS protein. (A) Total cell lysates were prepared from HCT/Mock, MN-61, and MN-62 cells. Equal amounts (50 μg of proteins) of cell lysates were separated by 7% SDS–PAGE and then transferred to nitrocellulose membranes. Membranes were immunoblotted with anti-NIS (clone C-2) or anti-actin antibody. Results shown are representative of 3 independent experiments. (B) Immunohistochemical analysis of NIS expression in MN-61 cells. HCT/Mock and MN-61 cells were stained with anti-NIS antibody (clone 44) or isotype-matched control IgG (Ab). Scale bar = 50 μm. DAPI = 4′,6′-diamidino-2-phenylindole.
Drug Accumulation and Sensitivity in shMDR-NIS–Expressing Cells
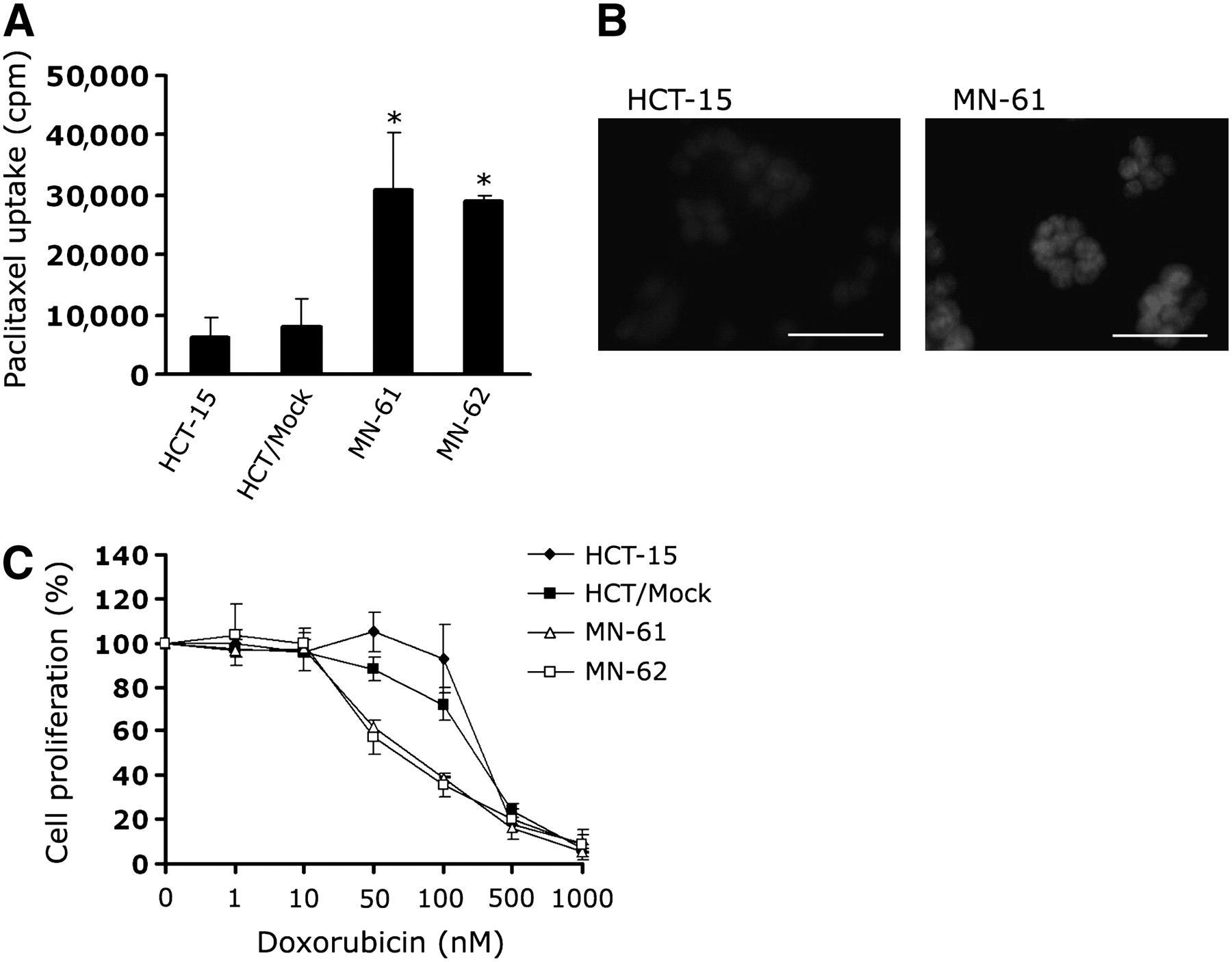
To clarify the functional activity of MDR1 shRNA, we measured the degree of drug accumulation in MN-61 and MN-62 cells. The accumulation of paclitaxel, a P-glycoprotein–transportable compound, was increased by about 3.8- and 2.8-fold in MN-61 and MN-62 cells, respectively, compared with that in parental HCT-15 cells (Fig. 4A). To assess whether shRNA-directed suppression of P-glycoprotein sensitized HCT-15 cells to the anticancer drug, we compared the drug sensitivity of shMDR-NIS–expressing cells with those of parental HCT-15 and HCT/Mock cells by using a cell proliferation assay. The level of accumulation of doxorubicin, another drug that is transported by P-glycoprotein, was also higher in shMDR-NIS–expressing cells (MN-61) than in HCT-15 cells (Fig. 4B). As shown in Figure 4C, sensitivity to doxorubicin was significantly higher in MN-61 and MN-62 cells than in parental HCT-15 cells.
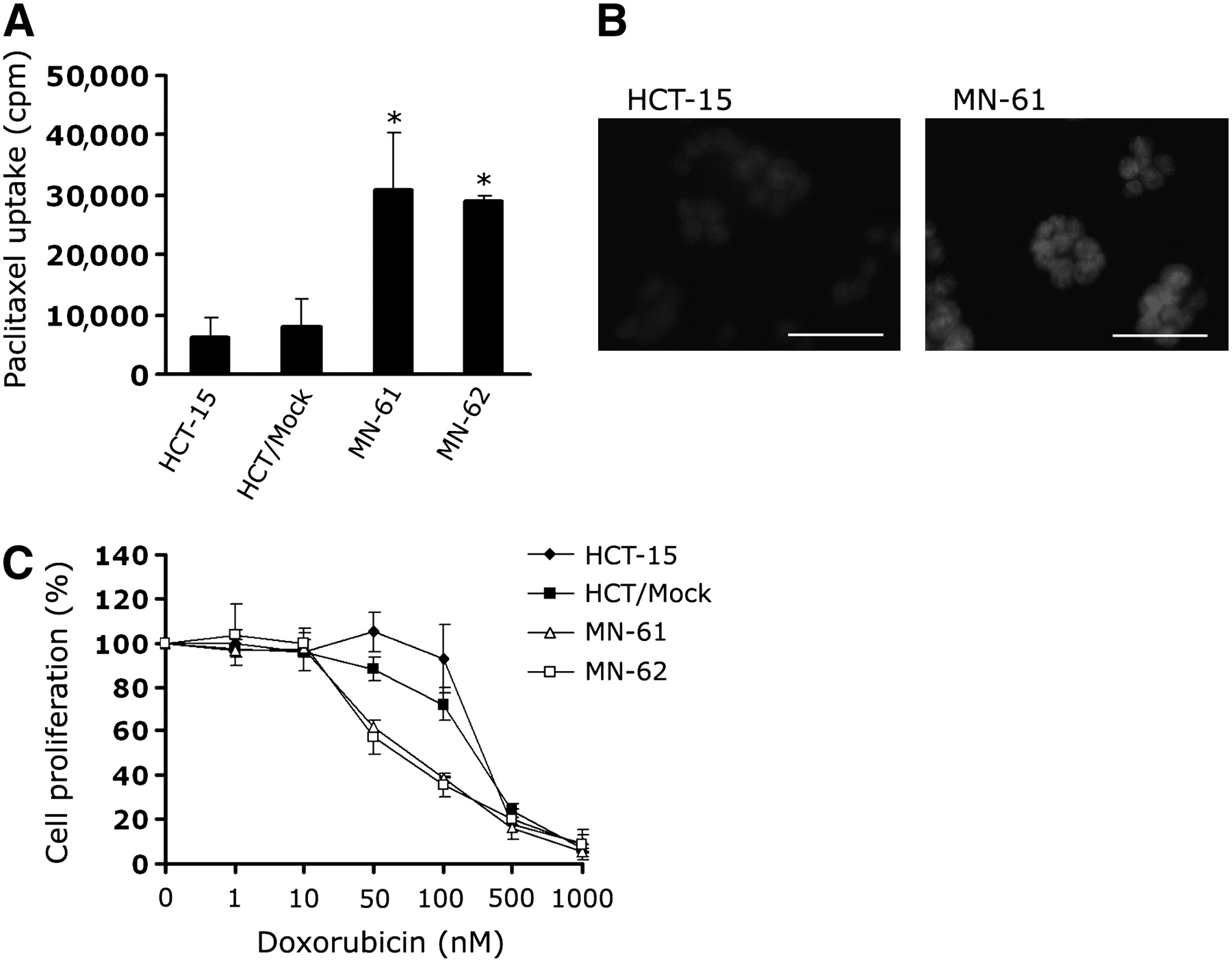
Accumulation of paclitaxel and sensitivity to doxorubicin in shMDR-NIS–expressing cell lines. (A) shMDR-NIS–expressing cells were seeded in 6-well plates, grown for 48 h, and incubated with 50 nM 3H-paclitaxel for 2 h at 37°C. At end of incubation, cells were cooled on ice, washed 3 times with ice-cold PBS, and solubilized with 0.2 mL of 1% SDS. Radioactivity in each sample was determined by scintillation counting. Results are expressed as mean ± SD of 3 independent experiments. *P < 0.01, as determined by ANOVA. (B) HCT-15 or shMDR-NIS–expressing cells (MN-61) were treated with 25 μM doxorubicin for 2 h. At end of incubation, cells were washed 3 times with PBS and then observed by confocal microscopy. Results are representative of 2 similar experiments. Scale bar = 50 μm. (C) Cells (103 per well) were seeded in 96-well plates, doxorubicin dilution series was added in triplicate, and cells were incubated for 4 d under standard culture conditions. Cell viability was assessed with CellTiter 96 Aqueous One solution cell proliferation assay. Results are expressed as mean ± SD of 3 independent experiments.
Functional hNIS Activities in shMDR-NIS–Expressing Cells
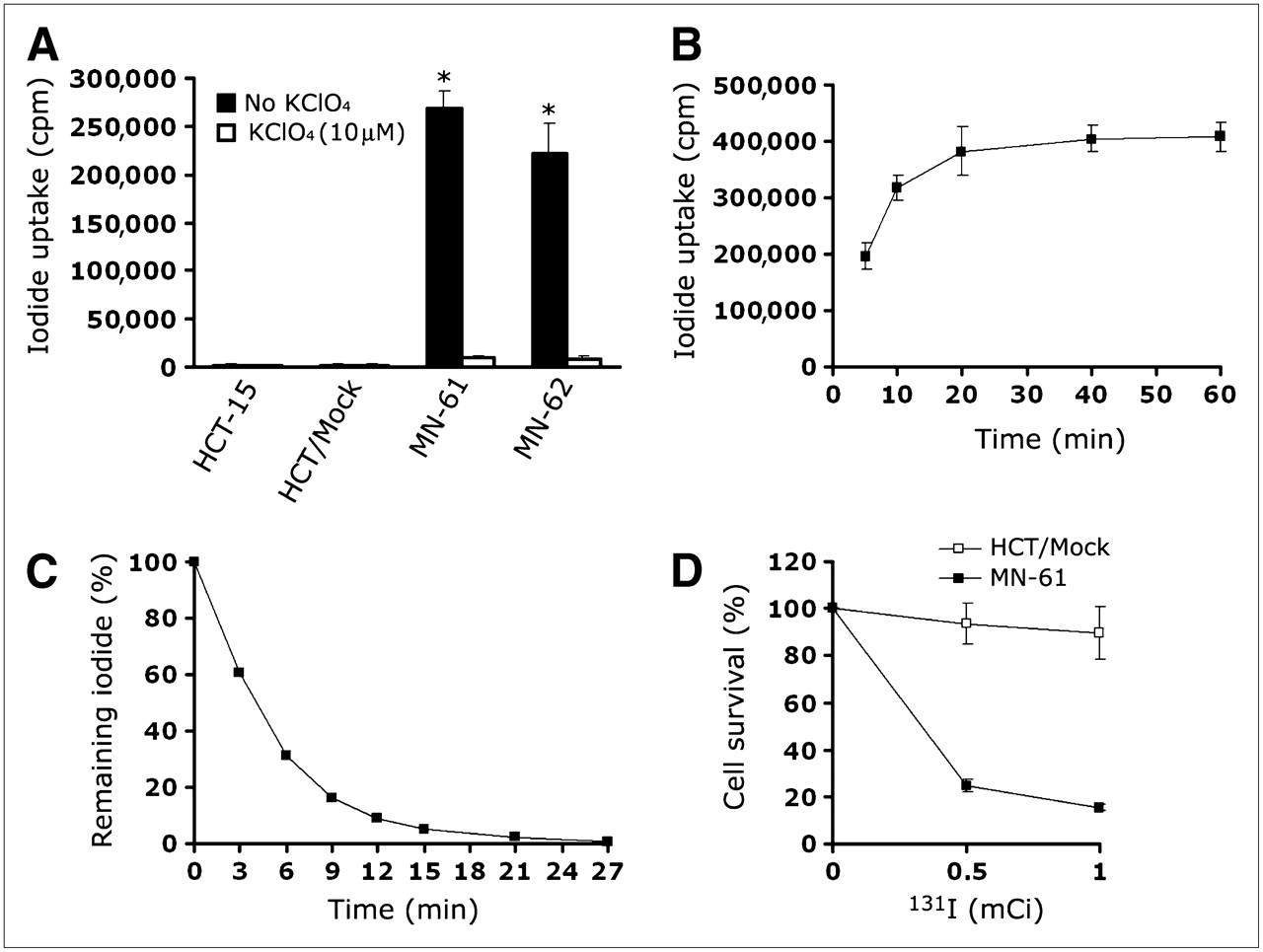
The functional activity of hNIS protein was clearly shown by the measurement of cellular iodide uptake. MN-61 and MN-62 cells showed up to 28.6- and 24.8-fold increases in iodide accumulation, respectively, compared with that in cells coincubated with perchlorate, a competitive NIS inhibitor (Fig. 5A). Parental HCT-15 and HCT/Mock cells did not show an increase in iodide accumulation. Iodide accumulation reached approximately 80% within 10 min and became saturated at 20 min (Fig. 5B), but almost 80%−90% of the accumulated 125I was released into the medium during the initial 10 min (Fig. 5C).

Iodide uptake in shMDR-NIS–expressing cells. (A) Iodide accumulation was measured in HCT-15, HCT/Mock, and shMDR-NIS–expressing cells (MN-61 cells and MN-62 cells). shMDR-NIS–transfected HCT-15 cells showed 25-fold increase in iodide accumulation compared with shMDR-NIS–expressing cells coincubated with perchlorate. Parental HCT-15 or vector-transfected HCT-15 cells did not show perchlorate-sensitive iodide accumulation. Results are expressed as mean ± SD of 3 independent experiments. *P < 0.01, as determined by ANOVA. (B) MN-61 cells were incubated with Na125I at 37°C for 10–60 min. At various time points, cells were quickly washed with PBS, and their radioactivity was counted with γ-counter. Results are expressed as mean ± SD of 3 independent experiments. (C) MN-61 cells were incubated with Na125I at 37°C for 30 min, medium was removed at various time points, and its radioactivity was determined. Results are expressed as mean ± SD of 3 independent experiments. (D) Cytotoxicity of 131I. HCT/Mock or MN-61 cells were incubated for 7 h with Na131I at 18.5–37.0 MBq/10 mL in HBSS supplemented with 10 mM NaI. After incubation with radioiodine, cells were trypsinized, seeded in 6-well plates (103 cells per well), and incubated for 10 d. After cell colony development, cells were fixed with methanol and stained with Diff Quick staining kit. Colonies containing more than 10 cells were counted, and cell survival was expressed as percentage of colonies relative to that in untreated control. Results are expressed as mean (remaining percentage of 125I) ± SD of 3 independent experiments.
Next, we investigated the therapeutic effectiveness of 131I in MN-61 and HCT/Mock cells by using a clonogenic assay. The viability of MN-61 cells decreased in correlation with an increase in the 131I concentration (Fig. 5D). In contrast, the percentage of viable HCT/Mock cells remained at almost 100% even when these cells were incubated with various concentrations of 131I. These results indicated that transduction with the dual expression vector could induce the expression of a functional hNIS gene and that, although the iodide efflux rate was rapid, the amount of accumulated 131I was sufficiently high to selectively kill MN-61 cells.
Combination Therapy with 131I and Doxorubicin
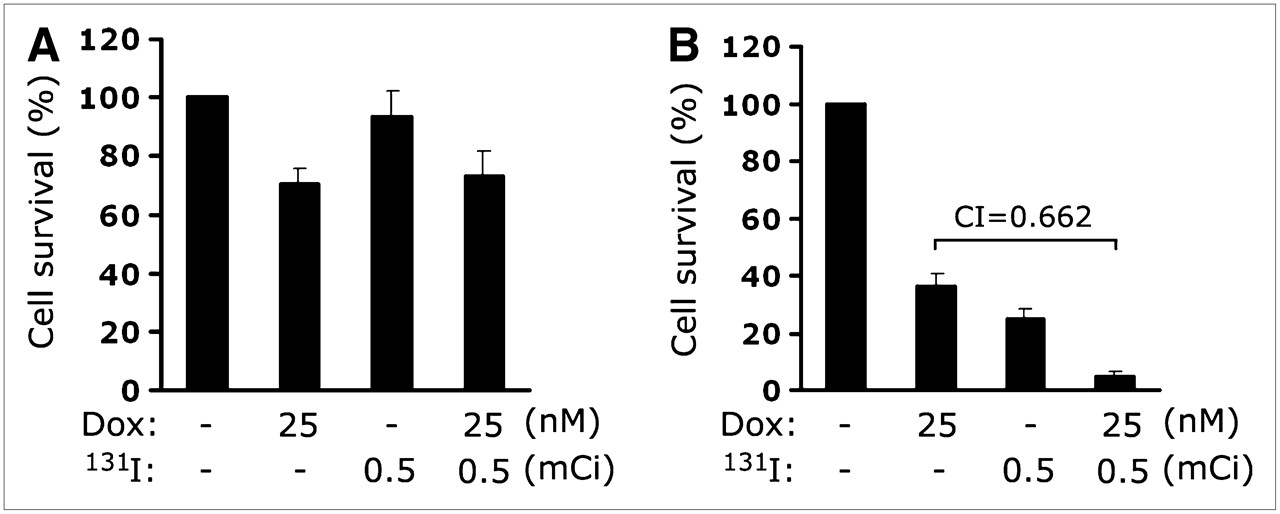
The effects of combination therapy with 131I and doxorubicin in vitro were estimated from the survival of HCT/Mock and MN-61 cells in a clonogenic assay. 131I at 18.5–37.0 MBq and 25–50 nM doxorubicin were administrated at concentrations that resulted in 20%−40% cell survival when either drug was given alone. When MN-61 cells were treated with 131I at 18.5 MBq and 25 nM doxorubicin for 10 d, cell survival decreased to 4.9% ± 1.9% compared with that obtained with 131I treatment (24.7% ± 3.2%) or doxorubicin treatment (36.3% ± 4.4%) alone (Fig. 6B). In contrast, HCT/Mock cells did not show a therapeutic effect of the combination of 131I and doxorubicin (Fig. 6A). To determine whether this combination had a synergistic or additive effect, we calculated a combination index as previously described (18,19). The combination of 131I at 18.5 MBq and 25 nM doxorubicin had a 95% killing effect, with a combination index of 0.662, suggesting that this combination was synergistic.

Effect of combination of 131I and doxorubicin on survival of MN-61 cells. MN-61 (B) or HCT/Mock (A) cells were incubated for 7 h with Na131I at 18.5 MBq/10 mL in HBSS supplemented with 10 mM NaI. After incubation with radioiodine, cells were trypsinized and seeded in 6-well plates (103 cells per well), 25 μM doxorubicin was added, and cells were incubated for 10 d. After cell colony development, cells were fixed with methanol and stained with Diff Quick staining kit. Colonies containing more than 10 cells were counted, and cell survival was expressed as percentage of colonies relative to that in untreated control. Results are expressed as mean ± SD of 3 independent experiments.
124I Imaging of Tumor Xenografts In Vivo and Quantification
At 8–12 wk after the inoculation of HCT/Mock and MN-61 cells, tumor diameter reached up to ∼10 mm, and whole-body imaging was performed by small-animal PET after the intraperitoneal injection of 124I at 18.5 MBq. In contrast to control HCT/Mock tumors, which showed negligible uptake of 124I, MN-61 tumors showed prominent uptake, indicating functional NIS expression (Figs. 7A and 7B). Physiologic uptake was also observed at the sites of the thyroid and stomach, in which NIS is normally expressed (Fig. 7B). Regions of interest were drawn on tumors expressing the shMDR-NIS construct, and 124I activity in tumors was quantified at selected time points (Fig. 7C). The accumulation of 124I in tumors was maintained for up to 3 h; the percentage injected dose per gram (%ID/g) at 1 h was 7.2, and that at 3 h was 7.3. However, a higher signal-to-noise ratio was achieved at 3 h than at 1 h because of decreased nonspecific background activities. By 15 h, the accumulation had decreased to 1.2 %ID/g.
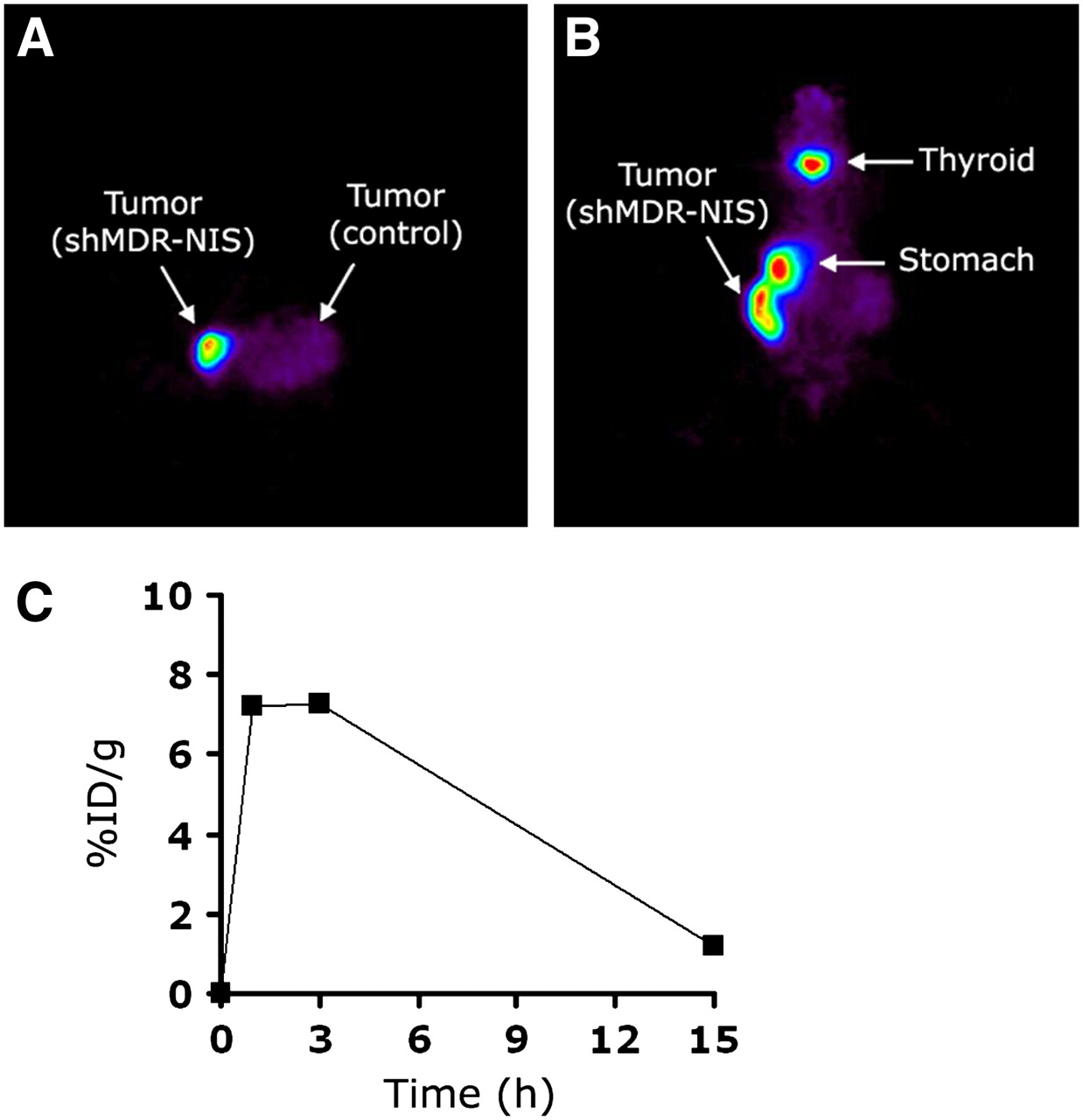
In vivo tumor images of tumor-bearing nude mouse at 3 h after injection of 124I. (A and B) Mouse was subcutaneously transplanted with shMDR-NIS–expressing cells (MN-61) in right flank and control cells (HCT/Mock) in contralateral left flank. Transverse (A) and coronal (B) small-animal PET images clearly visualized shMDR-NIS–expressing tumor in right flank. (C) 124I accumulation in shMDR-NIS–expressing cells (MN-61) plotted over time. Uptake of 124I in tumor lesions was maintained for up to 3 h after injection but, by 15 h, had decreased to 1.2 %ID/g.
DISCUSSION
NIS expression could lead to a novel gene strategy for radioiodine therapy in thyroid diseases (20). Transfer of the NIS gene into a variety of tumors, including melanoma, colon carcinoma, ovarian adenocarcinoma, and lung and prostate cancers, confers radioiodide uptake capacity (21–23). Although NIS-based radiotherapy is effective for a wide range of cancers, several problems remain, such as rapid washout of radioiodide from cells and limited retention of radioiodide within cells (24). Many strategies have been proposed to enhance the antitumor effects of NIS-based radioiodide therapy; these include combination with various reagents or gene therapy. Several antineoplastic drugs, such as cisplatin, doxorubicin, and 5-fluorouracil, are already known to have radiosensitizing effects and are used widely in the field of external radiotherapy for various cancers (25–27). Chemoradiotherapy has the advantage of enhancing the sensitivity of tumor cells to ionized radiation at a concentration lower than the optimal cytotoxic range (28). It has the potential disadvantage, however, of increased toxicity because chemotherapy may inhibit the repair of radiotherapy-induced sublethal damage in normal tissue. In the present study, to minimize the extratumoral side effects of chemotherapy, we developed a dual expression vector system in which NIS gene expression is combined with MDR1 shRNA expression. We investigated the feasibility of the combination of NIS-based radioiodide therapy and RNAi-based gene therapy with our novel vector system in vitro and performed 124I small-animal PET of nude mice bearing tumor xenografts expressing both the NIS gene and MDR1 shRNA.
RNAi relies on the sequence-specific interaction between siRNA and mRNA. The degradation of long double-stranded RNA to siRNA is mediated by a double-stranded RNA–specific enzyme, RNase III dicer. siRNA is incorporated into a nuclease complex known as the RNA-induced silencing complex (RISC), in which unwinding of the duplex siRNA takes place. The antisense strand binds in a highly sequence-specific manner to target mRNA, which is then endonucleolytically cleaved and degraded (29). Many studies have used siRNA as an experimental tool to dissect the cellular pathways that lead to uncontrolled cell proliferation and cancer. To develop siRNA for cancer therapy, several researchers have investigated the effects of siRNA in cancer models (30–32). In the present study, we developed a novel dual expression vector system combining the NIS gene with MDR1 shRNA to allow an effective combination of 131I with doxorubicin, which is a P-glycoprotein–transportable compound. Four MDR1-specific target sequences were tested according to published recommendations (8–10,33), and the most effective sequence was chosen. The introduction of this vector increased sensitivity to the anticancer drug in HCT-15 colon cancer cells, thereby allowing the effective use of low-dose doxorubicin and resulting in synergistic cytotoxicity in combination with 131I.
The combination of doxorubicin and radioiodine could be a useful strategy for cancer therapy, but the molecular mechanism of this synergy is presently unknown. Doxorubicin acts as an intercalating agent by binding to DNA and inhibiting chain elongation but also interferes with topoisomerases and induces breaks in DNA (34,35). In addition, doxorubicin is also known to be an activator of the p53 pathway (36), which acts as a transcriptional factor and induces the expression of several proteins related to cell cycle arrest and apoptosis. Indeed, previous studies showed that treatment with adriamycin induced the expression and activation of p53 (37). Thus, it is possible that the sequential addition of doxorubicin may accelerate radiotherapy-induced apoptosis through the increased activation or expression of p53. Furthermore, a previous study showed that the expression of p53 was positively correlated with hNIS activity in a dual expression vector expressing hNIS and the p53 gene (11). More recently, it was reported that doxorubicin enhanced the expression of a transgene under the control of the CMV promoter in anaplastic thyroid carcinoma cells (38). Thus, treatment with doxorubicin may enhance hNIS activity through p53 upregulation as well as activation of the CMV promoter. Although our results showed that treatment with 131I and then doxorubicin was effective in cancer therapy, doxorubicin pretreatment before radioiodide therapy may be also effective in treating cancers that are resistant to conventional chemotherapy or radiotherapy. Our dual expression vector could be a useful therapeutic tool in a combination of NIS-based radiotherapy and chemotherapy.
For safe and efficient gene therapy in clinical applications, technology that allows noninvasive monitoring of the level and distribution of vector-mediated gene expression in vivo is required. The NIS gene is well known for its many advantages as an ideal reporter gene, the well-understood biodistribution of its ligands, and its nonimmunogenic properties in humans. Ligands of this reporter gene include 123I, 131I, and 99mTc-pertechnetate as γ-tracers and 124I for PET. In the present study, small-animal PET with 124I revealed a clear image of shMDR-NIS–expressing tumors in an in vivo tumor xenograft model. The uptake of 124I in tumors could also be quantified at selected time points successfully. Therefore, radiologic imaging of NIS derived from our dual expression vector could be a highly effective method for monitoring targeted sites in a noninvasive manner.
The ultimate success of gene therapies will depend on gene transfer vectors that facilitate the expression of a specific gene at therapeutic levels in cancer cells without eliciting cytotoxicity. Although the data presented here appear to be promising, the lack of specificity conferred by nonspecific promoters may be problematic. MDR1 gene products are present not only in cancer cells but also in various normal tissues, such as the endothelium of blood vessels in the brain (39) and hematopoietic progenitors found in normal bone marrow (40). Although the level of MDR1 expression in these tissues is relatively low, functional P-glycoprotein expression may be important in limiting potential toxicity after exposure to anticancer drugs. Thus, a targeted delivery system in which shRNA is selectively expressed in cancer cells should be developed. Furthermore, our results were obtained in stable transfected cells with a high transduction efficiency and easy drug administration. Our findings will need to be confirmed with clinically relevant models in vivo.
CONCLUSION
We have developed a dual expression vector system containing the NIS gene and MDR1 shRNA and tested its therapeutic effects in colon cancer cells. A combination of 131I and low-dose doxorubicin was more effective in killing colon cancer cells than 131I or doxorubicin alone. Moreover, 124I small-animal PET imaging revealed a clear image of the in vivo tumor xenografts, and accumulated 124I activity could be quantified over time. Although several problems need to be resolved for further applications, the present study suggests the possibility of a new strategy of RNAi-based gene therapy accompanied by NIS-based radioiodide therapy and imaging in colon cancer cells.
Acknowledgments
We thank Dr. Je-Yeol Cho (Kyungpook National University, Daegu, Republic of Korea) for the kind donation of the hNIS expression construct. The production of 124I was supported partially by the QURI project of MOST (Ministry of Science and Technology) and by a project titled “Development of Diagnosis and Therapy for Intractable Disease Using Radioisotopes” of KOSEF (Korea Science and Engineering Foundation). This study was supported by grant RTI04-01-01 from the Regional Technology Innovation Program of The Ministry of Knowledge Economy (Advanced Medical Technology Cluster for Diagnosis and Prediction at Kyungpook National University); by grant 0720550-2 from the National R&D Program for Cancer Control, Ministry of Health & Welfare, Republic of Korea; and by the Brain Korea 21 Project in 2008.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication January 22, 2008.
- Accepted for publication May 7, 2008.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}