


Abstract
Nonivasive assessment of programmed cell death is currently an attractive research topic for the follow-up of tumor therapy and myocardial infarction. Apoptosis imaging with 99mTc-HYNIC-annexin V (HYNIC is hydrazinonicotinamide) is based on the binding of the tracer to externalized phosphatidylserine residues. Concurrently with the externalization of phosphatidylserine, a series of caspases are activated after the onset of apoptosis. These enzymes were chosen as an alternative target for apoptosis imaging. Methods: Ten radiolabeled peptides containing the DEVDG sequence, selective for downstream caspases such as caspase-3, were synthesized and evaluated for their uptake kinetics using an apoptosis test system. The molecular requirement of the peptides for being accepted as caspase substrate was studied using a competitive enzyme assay and matrix-assisted laser desorption/ionization mass spectrometry. Results: Within this series of peptides, radioiodinated Tat49–57-yDEVDG-NH2 (7) and Tat57–49-yDEVDG-NH2 (8) were favorably taken up by apoptotic cells (12.54% ± 1.18% and 12.63% ± 1.17% after 10-min incubation, respectively) as compared with the controls (7.50% ± 0.92% and 8.04% ± 0.28%). The enhanced uptake is interpreted as the interaction of the labeled peptide or fragment with activated caspases. Proof of caspase substrate specificity of peptide 7 and YDEVDG-NH2 (2) was substantiated. The former peptide was shown to have a stronger competition with the fluorescent Z-DEVD-R110 for caspase-3 than peptide 2. In addition, mass spectrometry revealed only fragmentation for peptide 7. Conclusion: It could be demonstrated that peptides consisting of DEVDG and Tat sequence are caspase substrates with enhanced uptake and retention in apoptotic cells. Current efforts are focused on alternative radioisotopes that include radiometal complexes to further improve these characteristics.
Apoptosis is an active, energy-dependent mechanism for the elimination of cells that have been injured, infected, or immunologically recognized as harmful or superfluous (1). This mechanism controls the fate and life expectancy of cells that play important roles in a large number of disorders, including myocardial ischemia or infarction (2), organ rejection after transplantation (3), and neurodegenerative diseases (4).
Scintigraphic imaging with radiolabeled annexin V, a human 36-kDa protein, is currently the most promising technique to detect apoptosis in vivo (5). This agent allows the imaging of an early event in apoptosis, involving the externalization of phosphatidylserine from the inner to the outer leaflet of the plasma membrane (6). The high affinity of annexin V for cells with exposed phophatidylserine (7) is the basis for detecting apoptosis in vivo (5).
Two 99mTc-annexin V compounds have been studied in humans, differing in the bifunctional chelators (8–10). In one case, annexin V was derivatized with the monodentate 1-imino-4-mercaptobutyryl side chain (99mTc-i-AnxV) (11) and, in the second case, the protein was modified with a bis(mercaptoacetyl)diaminopentanoyl group (99mTc-BTAP-AnxV) (10). Recently, a 99mTc-annexin V derivative has been described in which the protein was modified with hydrazinonicotinamide (99mTc-HYNIC-annexin V) (5,12). The investigation of 99mTc-HYNIC-annexin V for human application revealed favorable biodistribution characteristics (11). In addition, 99mTc-HYNIC-annexin V is easily obtained by a kit-formulated preparation (5).
An alternative target in the apoptotic pathway that is potentially useful for the in vivo detection of apoptosis is the appearance of activated caspases (13–15). All known caspases have a cysteine located in the active site and cleave peptide bonds on the carboxyl side of an aspartate residue (termed as P1). Caspases are divided into two classes based on the length of their N-terminal prodomains. Caspases-1, -2, -4, -5, -8, and -10 have long prodomains, whereas caspases-3, -6, -7, and -9 have short prodomains. The caspases with long prodomains play a relevant biologic role in the cleavage of downstream caspases. Downstream caspases (short prodomain) are the foot soldiers of apoptosis, operating at the downstream end of the cascade to cleave substrates (16). Recognition of at least 4 amino acids counted from the N-terminus of the cleavage site is a necessary requirement for efficient catalysis. The preferred tetrapeptide recognition motif differs significantly among caspases, which is an explanation for the diversity of their biologic functions (17). During the execution phase of apoptosis, caspase-3 is responsible for the proteolysis of a large number of substrates either wholly or in part. These substrates contain a DXXD motif similar to that originally described for PARP (poly(adenosine diphosphate-ribose) polymerase) (18).
In an early investigation, radioiodinated Z-VAD-fmk, a pan-caspase inhibitor used as an experimental tool in apoptosis research, was tested with apoptotic Morris hepatoma cells (MH3924Atk8), resulting in a distinct uptake preference but an insufficient cell uptake (19). Enzyme inhibitors have the intrinsic disadvantage of binding-site saturation. Substrates that leave the active enzyme site after cleavage lead to a signal enhancement. One prerequisite, however, which has to be met, is the retention of the cleaved imaging agent.
To obtain information about the uptake and retention in apoptotic and normal control cells, we evaluated ten radioiodinated peptides with nonadherently growing Jurkat cells. The peptide sequence used here was selective for downstream caspases such as caspase-3. One part of the cells was triggered for apoptosis and the other fraction was used as control cells. Before performing these experiments, both cell suspensions were characterized using flow cytometry to determine the apoptotic cell fraction. The kinetics of the cell uptake was measured as a function of time. Proof of caspase fragmentation was given by a competitive caspase-3 assay and matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS).
MATERIALS AND METHODS
All commercially available chemicals were of analytic grade and were used without further purification. 131I-Iodide was obtained in 0.02 mol/L NaOH solution with a specific radioactivity of 90 GBq/μmol at the time of delivery (Amersham International). High-performance liquid chromatography (HPLC) was performed with Lichrosorb 60 RP Select B columns (250 × 4, 5 μm for analytic runs; and 250 × 10, 10 μm for preparative runs; Merck KGaA) using ultraviolet (SPD-10A VP; Shimadzu) and γ-detection (Bioscan). Mass spectroscopy was performed on the MALDI time-of-flight mass spectrometers MALDI III (Kratos-Shimadzu) and Reflex II (Bruker Daltonik). Flow cytometry data were recorded on a Dako Galaxy flow cytometer. The fluorescence measurements were performed with a fluorescence plate reader (Fluoroscan Ascent; Labsystems, Merlin Diagnostika). Radioactive uptake measurements were performed on a Cobra II auto γ-counter (Canberra Packard).
Chemistry
Peptide Syntheses.
The peptides summarized in Table 1 were prepared by solid-phase peptide synthesis using N-α-Fmoc–protected amino acids (Calbiochem-Novabiochem GmbH) and standard O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) coupling chemistry. All peptides were synthesized as C-terminal amides and purified by HPLC at a flow rate of 3.7 mL/min using 0.1% trifluoroacetic acid (TFA) in water (A) and 0.1% TFA in acetonitrile (B) as eluants by a linear gradient of 0% B to 40% B over 30 min. The purity of the products was confirmed by analytic HPLC, and the identity of each peptide was confirmed by MS.
Peptides Used for Uptake Studies in Apoptotic and Control Cells
Radioiodination of Peptides.
Peptide solution (15 μL; 5 mmol/L in water) was mixed with 25 μL phosphate buffer (pH 7.4) and 20 MBq 131I-iodide. Addition of 5 μL chloramine-T solution (5 mmol/L in water) was followed by 30-s incubation. The reaction was stopped by the addition of 10 μL of a saturated methionine solution, and the mixture was separated by HPLC (20). Quality control of the radioiodinated peptide was performed with reversed-phase HPLC using a gradient starting at 0% and ending at 100% 0.1% TFA in acetonitrile after 30 min. The aqueous phase consisted of 0.1% TFA in water. Under these conditions, the radiolabeled compound was found to be pure and free of unlabeled peptide. The specific activities were >30 GBq/mmol. After the evaporation of the eluant, the 131I-labeled peptide was dissolved in phosphate-buffered saline (PBS) and used for the cell experiments.
99mTc Labeling of HYNIC-Annexin V.
HYNIC-annexin V (Theseus Imaging) was labeled with 99mTc using stannous chloride as a reducing agent and tricine as a coligand. Quality control was performed after 15 min using instant thin-layer chromatography (Pall Corp.) and 0.9% NaCl for development. The radiochemical yields were >95%.
Biology
Cell Cultures.
Exponentially growing human leukemia cells (Jurkat J6) were obtained from the Deutsches Krebsforschungszentrum Heidelberg. The culture medium consisted of RPMI 1640 medium supplemented with 1% 200 mmol/L glutamine and 10% fetal calf serum (all from PAN Biotech GmbH). One day before the experiments were performed, the cells were split 1:4 to achieve logarithmic-phase growth at the time point of the experiment.
Annexin V/Propidium Iodide Assay.
Apoptosis was induced in 106 cells per milliliter using camptothecin concentrations of 1, 10, and 100 μmol/L (Sigma-Aldrich). Samples of the camptothecin-treated cells were harvested after 2, 3, 4, 5, and 6 h, centrifuged for 5 min at 800 rpm, washed with cold PBS, and suspended in 100 μL cold annexin-binding buffer (10 mmol/L N-(2-hydroxyethyl)-piperazine-N′-(2-ethanesulfonic acid), 150 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1.8 mmol/L CaCl2, pH 7.4). After the addition of 1.5 μL annexin V-Alexa Fluor 488 (Molecular Probes) and an incubation period of 15 min, the suspensions were mixed with 300 μL cold annexin-binding buffer. Flow cytometry measurements were performed with mixtures of 100 μL cell suspension, 900 μL cold annexin-binding buffer, and 10 μL propidium iodide solution (250 μg/mL). The fluorescence emissions were adjusted to 530 (FL-I) and >575 nm (FL-III) and measured in triplicate.
After indicated time periods following induction, apoptosis was monitored with annexin V-Alexa Fluor 488/propidium iodide by flow cytometry (two determinations) and Z-DEVD-R110 (EnzChek Caspase-3 Assay Kit; Molecular Probes) by fluorescence-intensity measurements (three determinations).
Caspase Assay.
Apoptotic cells produced as described were taken from the incubation solution after 2, 3, 4, 5, and 6 h and centrifuged for 5 min at 800 rpm. The pellets were washed with 1 mL cold PBS and suspended on ice for approximately 30 min in 50 μL lysis buffer (10 mmol/L Tris, 100 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid [EDTA], 0.01% Triton X-100, pH 7.5). After 30 min on ice, the lysate was centrifuged for 5 min at 5,000 rpm to remove cellular debris. Fifty microliters of the supernatant obtained from each sample were stored at −80°C before analysis. For fluorescence measurements, the supernatants were transferred to a microtiter plate covered with 50 μL substrate working solution (20 mmol/L PIPES [piperazine-N,N′-bis(2-ethanesulfonic acid)], 4 mmol/L EDTA, 10 mmol/L dithiothreitol, 0.2% CHAPS [3-([3-cholamidopropyl]dimethylammonio)-1-propanesulfonate], 50 μmol/L Z-DEVD-R110, pH 7.4). The plates were incubated at room temperature for 60 min, and the fluorescence was measured at 538 nm (excitation, 485 nm).
Cell Uptake Measurements.
The cell uptake measurements were performed with staurosporine-treated cells and Jurkat cell controls. Before commencing the cell uptake measurements, control cells and staurosporine-treated cells were examined by flow cytometry for apoptotic cell fractions. The cell counts were 106 cells/100 μL annexin- binding buffer (99mTc-HYNIC-annexin V experiments) and 5 × 106 cells/350 μL RPMI 1640 medium (radioiodinated peptide experiments). Treated and control cells were incubated at 37°C with 0.16 MBq 99mTc-HYNIC-annexin V (0.371 μg annexin/mL) and 0.2 MBq of the respective radioiodinated peptide. The amount of unlabeled annexin V corresponded to the annexin V concentration used for flow cytometry. The specific activities of the peptides were no carrier added. After 10, 15, 20, 30, and 60 min, the tubes were briefly vortexed and 10-μL samples of the control and apoptotic cells were layered on top of a 400-μL microcentrifuge tube (Roth) containing 350 μL of a 75:25 mixture of silicon oil, density = 1.05 (Aldrich), and mineral oil, density = 0.872 (Acros) (21). Instantaneous centrifugation at 15,000 rpm for 2 min afforded the complete separation of cellular radioactivity from the medium. After freezing the tubes with liquid nitrogen, the bottom tips containing the cell pellet were cut off. The cell pellets and the supernatants were counted in a γ-counter. The cell uptake was calculated as % uptake = 100 × (cpm in pellet)/(cpm in pellet + cpm in supernatant). The experiments were performed in triplicate.
Proof of Caspase-Induced Peptide Cleavage (Competition Experiment).
Stauroporine-treated cells were harvested 3 h after induction, centrifuged for 5 min at 800 rpm, washed in 1 mL cold PBS, suspended in 50 μL lysis buffer, and kept on ice for approximately 30 min. The lysate was centrifuged at 5,000 rpm for 5 min to remove cellular debris. Fifty microliters of the supernatant obtained from each sample were frozen at −80°C until the onset of the competition experiment. Twenty-five microliters of 100 μmol/L Z-DEVD-R110 substrate solution and 25 μL of the synthesized peptide solutions were transferred to the microtiter plate, resulting in final concentrations of 0, 100, 200, and 400 μmol/L. The thawed caspase-3 containing supernatants were added to each well. After an incubation period of 90 min at room temperature, the fluorescence was measured at 538 nm (excitation, 485 nm).
Proof of Caspase-Induced Peptide Cleavage (MALDI-MS Experiment).
Human caspase-3 solution (1.7 μL, 17 units; Calbiochem-Novabiochem GmbH) was mixed with 1 μL of a 1 mmol/L solution of peptide 7 dissolved in caspase reaction buffer. After 1 h at 30°C, the reaction mixture was analyzed by MALDI-MS.
RESULTS
Establishment of Apoptosis Test Method
For elucidating conditions to obtain optimal counts of apoptotic cells, time-course examinations were performed with campthothecin-treated Jurkat cells. Two commercially available assays were tested, Alexa Fluor 488-tagged annexin V and Z-DEVD-R110, a bisamide derivative of rhodamine 110. The former indicator gave information about externalized phosphatidylserine by flow cytometry, whereas the latter provided information about caspase-3 activity through substrate cleavage-induced fluorescence (22) that was monitored by a fluorescence plate reader. Upon enzymatic cleavage, the nonfluorescent bisamide substrate is converted first into the fluorescent monoamide and then to the more fluorescent R110.
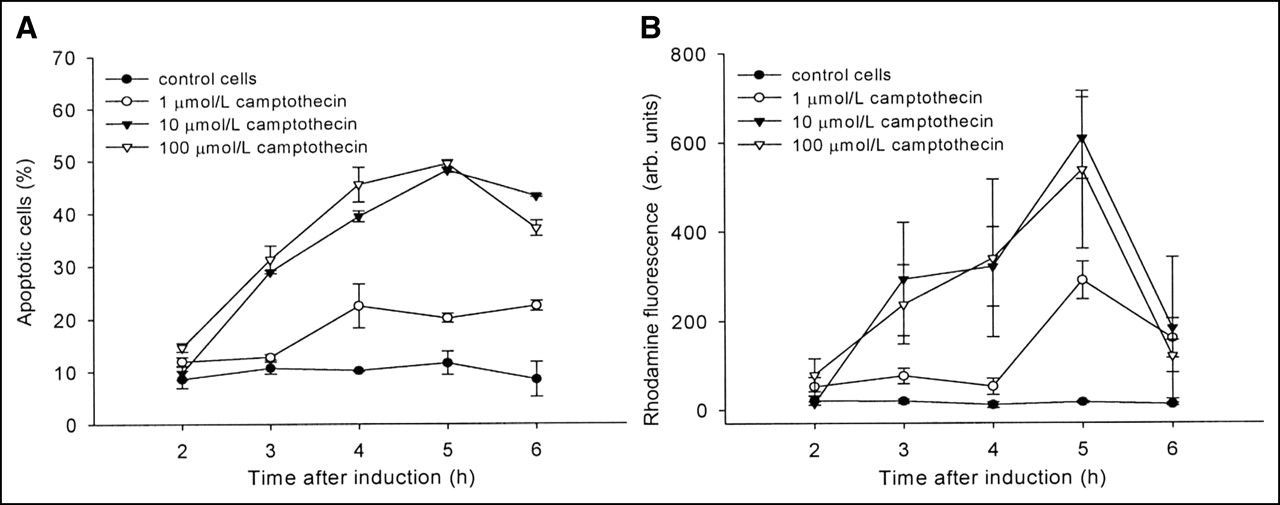
In Figure 1 apoptosis induction of Jurkat cells is shown as a function of time and drug concentration. The apoptotic cell fraction monitored by flow cytometry peaked 5 h after addition of 10–100 mmol/L campthothecin (Fig. 1A). The decline in apoptotic cells at later times reflects the increase in dead cells. Similar results were obtained using the caspase-3 assay with Z-DEVD-R110. The curves in Figure 1B show an almost identical curvature using the same campthothecin concentrations.
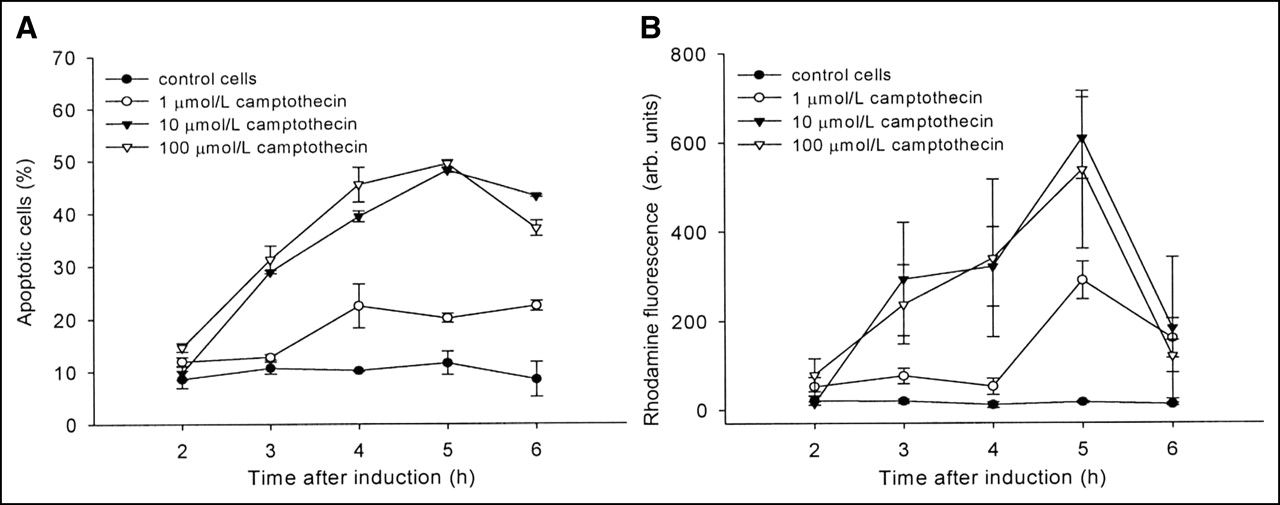
Time-course development of apoptosis obtained with camptothecin-treated Jurkat cells. (A and B) Flow cytometry using Alexa Fluor 488-labeled annexin V showing phosphatidylserine externalization (A) and formation of fluorescent rhodamine 110 mediated through caspase-3 hydrolysis of Z-DEVD-R110 (B) (n = 3). Data are expressed as mean ± SD. arb. units = arbitrary units.
To evaluate the appropriate agent for the induction of apoptosis in Jurkat cells, two compounds were tested: camptothecin and staurosporine. Flow cytometry using Alexa Fluor 488-annexin V (FL-I) and propidium iodide (FL-III) dyed Jurkat cells revealed that smaller amounts of staurosporine induced more apoptotic cells with less time. Figure 2A depicts the flow cytometer printout obtained with 10 μmol/L camptothecin after 4 h, and Figure 2B shows the effect of 1 μmol/L staurosporine after 3 h. The fractions of apoptotic cells indicated in quadrant 1 (Q1) are 38% and 78% for camptothecin and staurosporine, respectively. The meaning of other quadrants is explained in the legend of Figure 2.

Propidium iodide (FL-III)/Alexa Fluor 488-annexin V (FL-I) flow cytometry of Jurkat cells treated with 10 μmol/L camptothecin for 4 h (A) and 1 μmol/L staurosporine for 3 h (B). Q1 = apoptotic cells; Q2 = apoptotic and necrotic cells; Q3 = living cells; Q4 = dead cells.
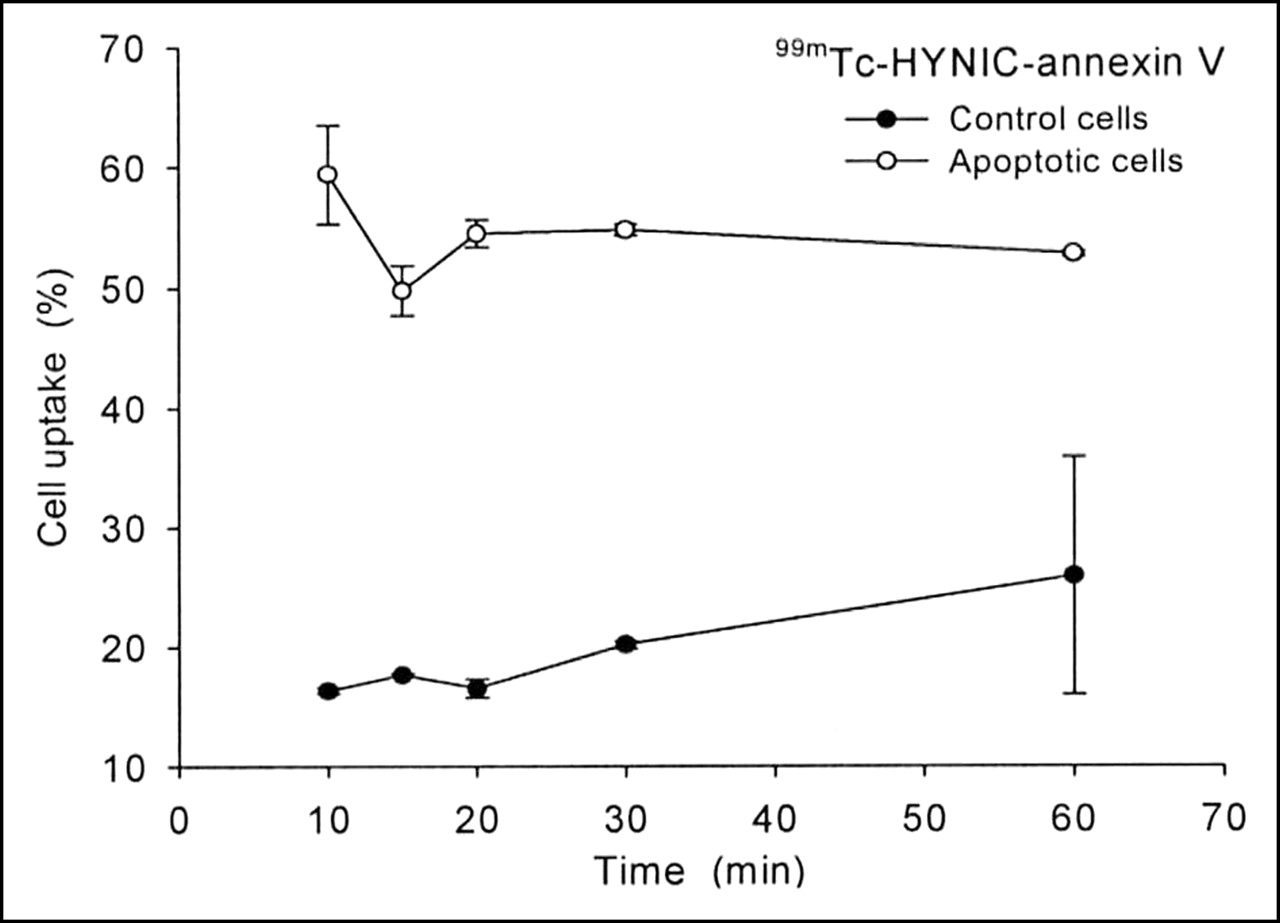
Flow cytometry results obtained with Alexa Fluor 488-tagged annexin V were compared with the radioactive uptake of 99mTc-HYNIC-annexin V in staurosporine-treated and control cells. Figure 3 illustrates the time-course accumulation of 99mTc-HYNIC-annexin V. Twenty minutes after induction, 55% of the radioactivity was measured in apoptotic cells, whereas flow cytometry using Alexa Fluor 488-annexin V revealed 62% after the same time period. In control cells, apoptosis was higher with radiolabeled annexin (15%) as compared with flow cytometry (8%).
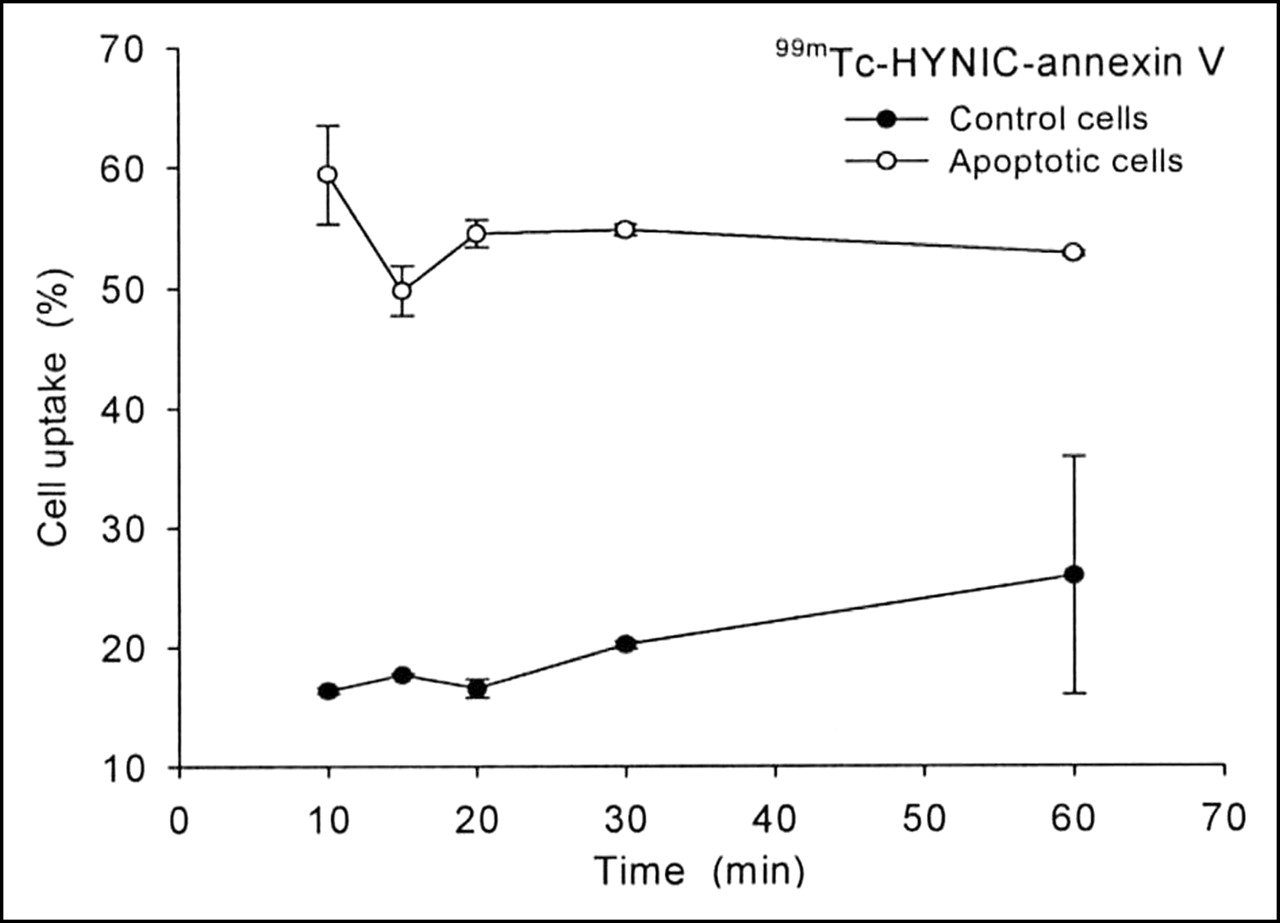
Uptake kinetics of 99mTc-HYNIC-annexin V in apoptotic and control cells. Cells showed 62% apoptosis with Alexa Fluor 488-annexin V measured by flow cytometry (n = 3). Data are expressed as mean ± SD.
Uptake Kinetics of Radiolabeled DEVDG Peptides
Table 1 summarizes the amino acid sequences of 10 peptides investigated in this study. These peptides were synthesized by standard solid-phase synthesis techniques and analyzed by HPLC and MALDI-MS. All peptide sequences included one tyrosine in various positions that was labeled by electrophilic radioiodination using chloramine-T as an oxidant.
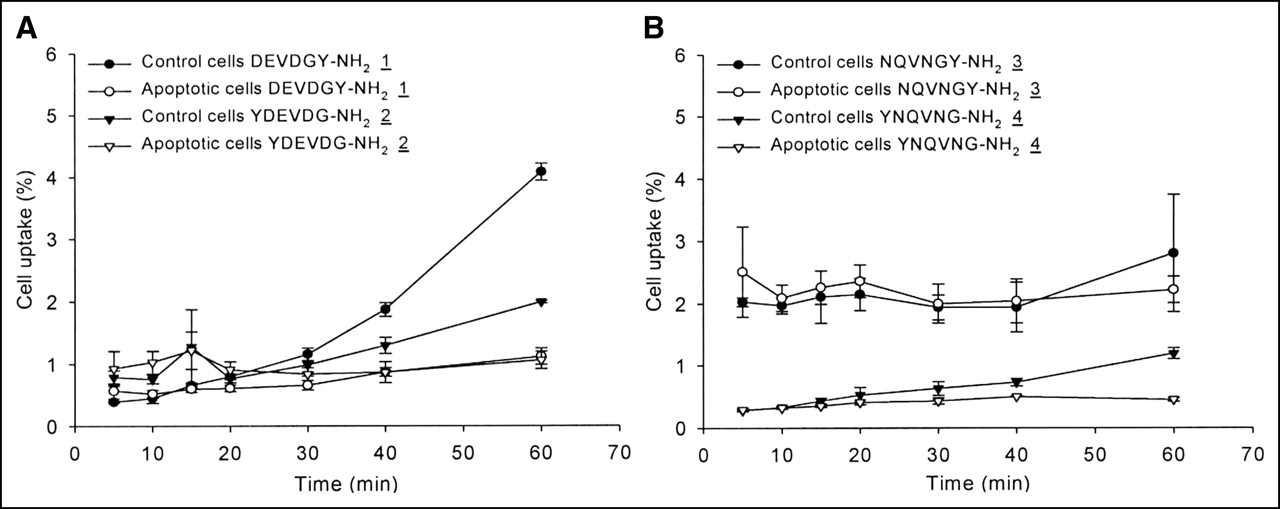
The uptake of radiolabeled peptides 1–4 as a function of time is shown in Figure 4. In general, the absolute uptake values of peptides 1–4 proved to be rather low, ranging between 0.3% and 4.0% of the total dose. Differences between apoptotic and control cells were not observed within the first half of the experiments. At later times, the control cells preferentially accumulated peptide 1 and, to a lesser degree, peptides 2, 3, and 4.

Uptake in apoptotic and control cells of 131I-labeled DEVDGY-NH2 (1) and YDEVDG-NH2 (2) (A) as well as NQVNGY-NH2 (3) and YNQVNG-NH2 (4) (B) (n = 3). Data are expressed as mean ± SD.
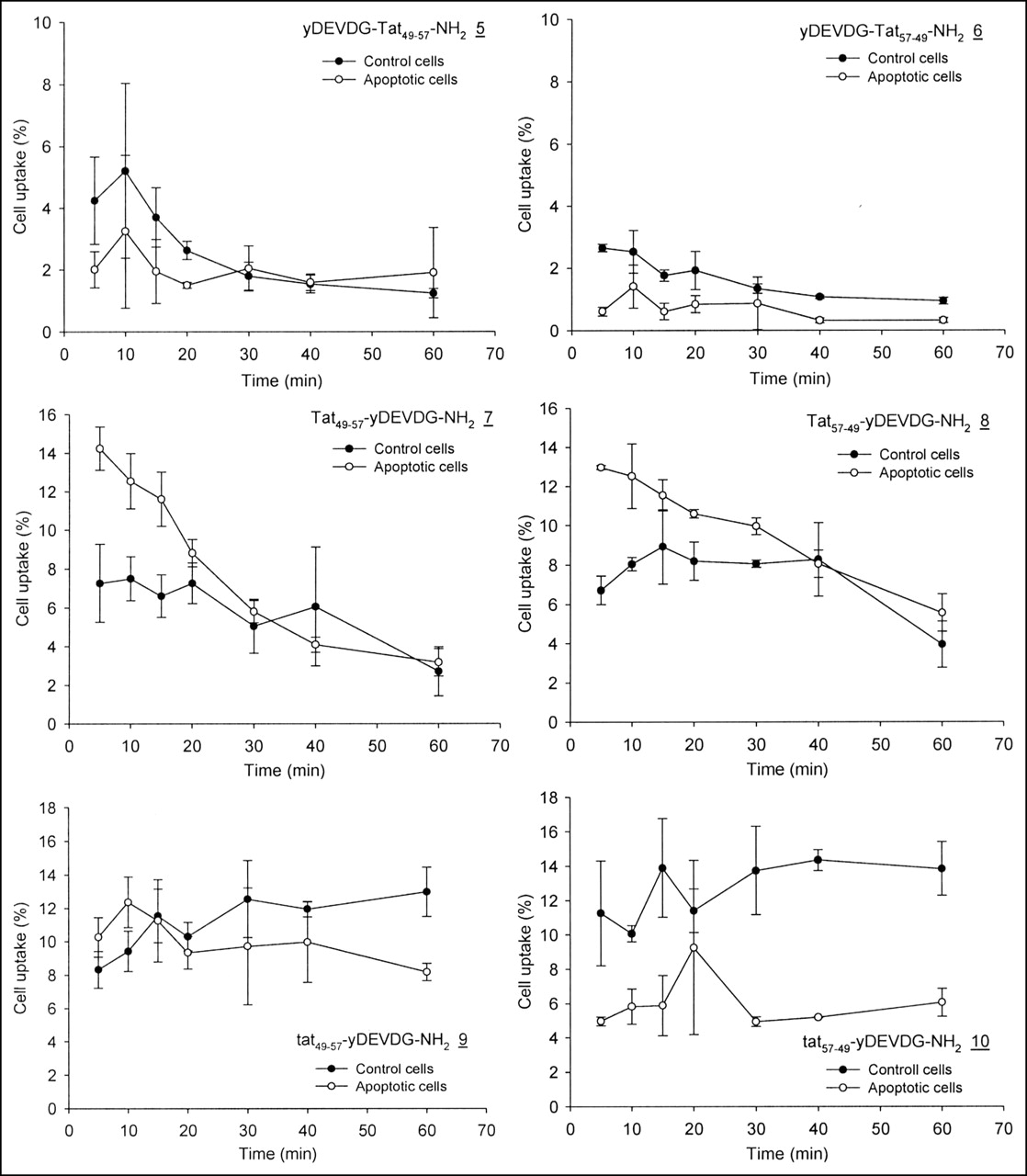
With the exception of Tat49–57-yDEVDG-NH2 (7) and Tat57–49-yDEVDG-NH2 (8) similar characteristics were observed for the DEVDG peptides containing Tat sequences. Figure 5 demonstrates at almost all times higher uptake values of peptides 5, 6, 9, and 10 in control cells. For peptides 7 and 8, the accumulation of radioactivity commenced at 14% and decreased with time to about 9% before they merged with the controls after 30 min.

Uptake in apoptotic and control cells of yDEVDG-conjugated Tat sequences (5–10) (n = 3). Data are expressed as mean ± SD.
Proof of Caspase Specificity
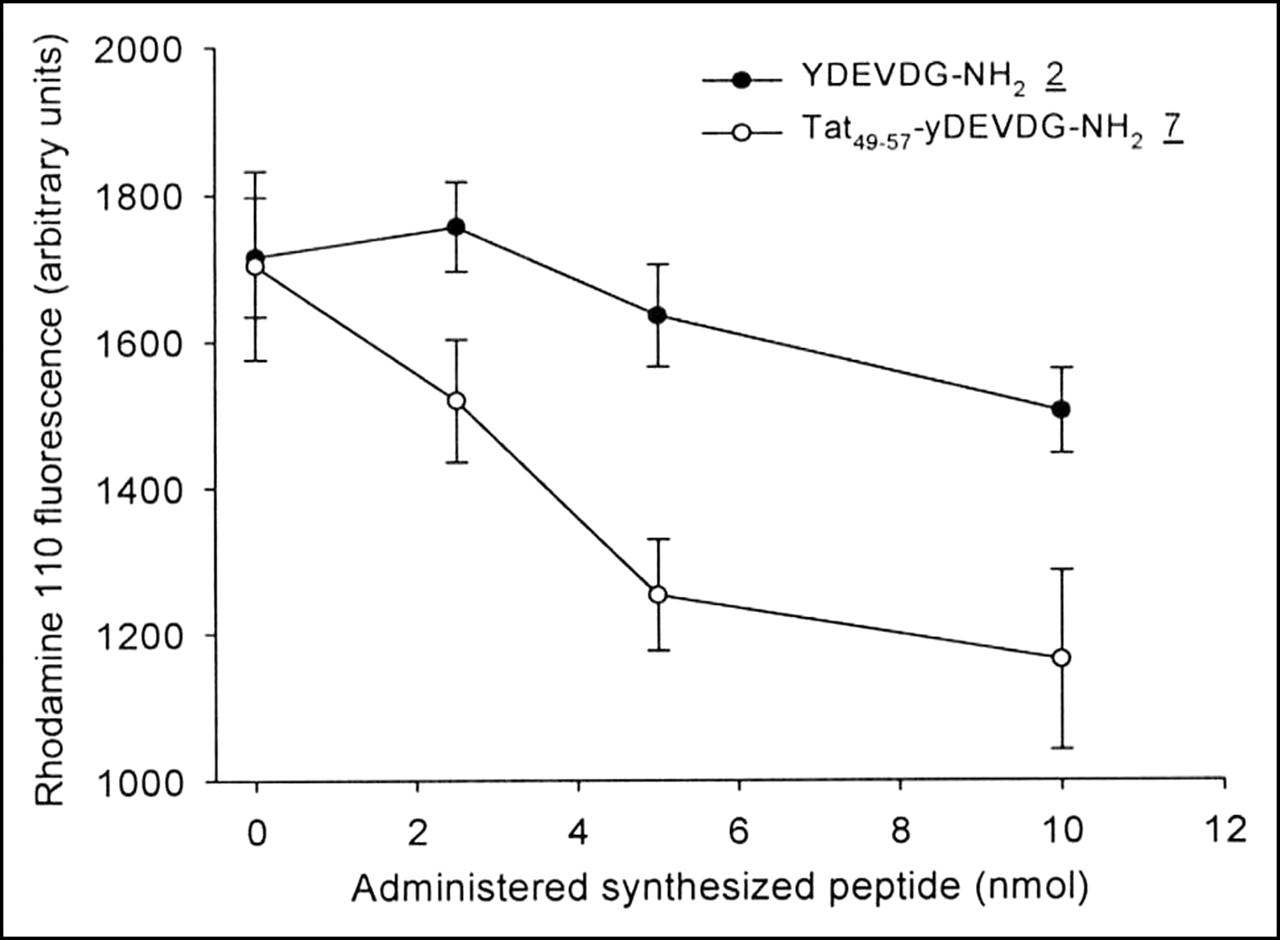
Proof of suitability for serving as caspase substrates was obtained using peptides 2 and 7 in a competitive caspase assay with Z-DEVD-R110. The competition experiments were performed with cell lysates received from apoptotic cells. Because of the presence of competing peptides intervening with activated caspases, the formation of fluorescent dye originating from Z-DEVD-R110 was influenced. Figure 6 shows the decline of the Z-DEVD-R110 fluorescence signal with increasing amounts of YDEVDG-NH2 (2) and Tat49–57-yDEVDG-NH2 (7). The latter peptide showed a stronger competitive effect than peptide 2.

Rhodamine 110 formation through caspase-mediated hydrolysis of Z-DEVD-R110 with increasing amounts of peptides 2 and 7 (n = 3). Data are expressed as mean ± SD.
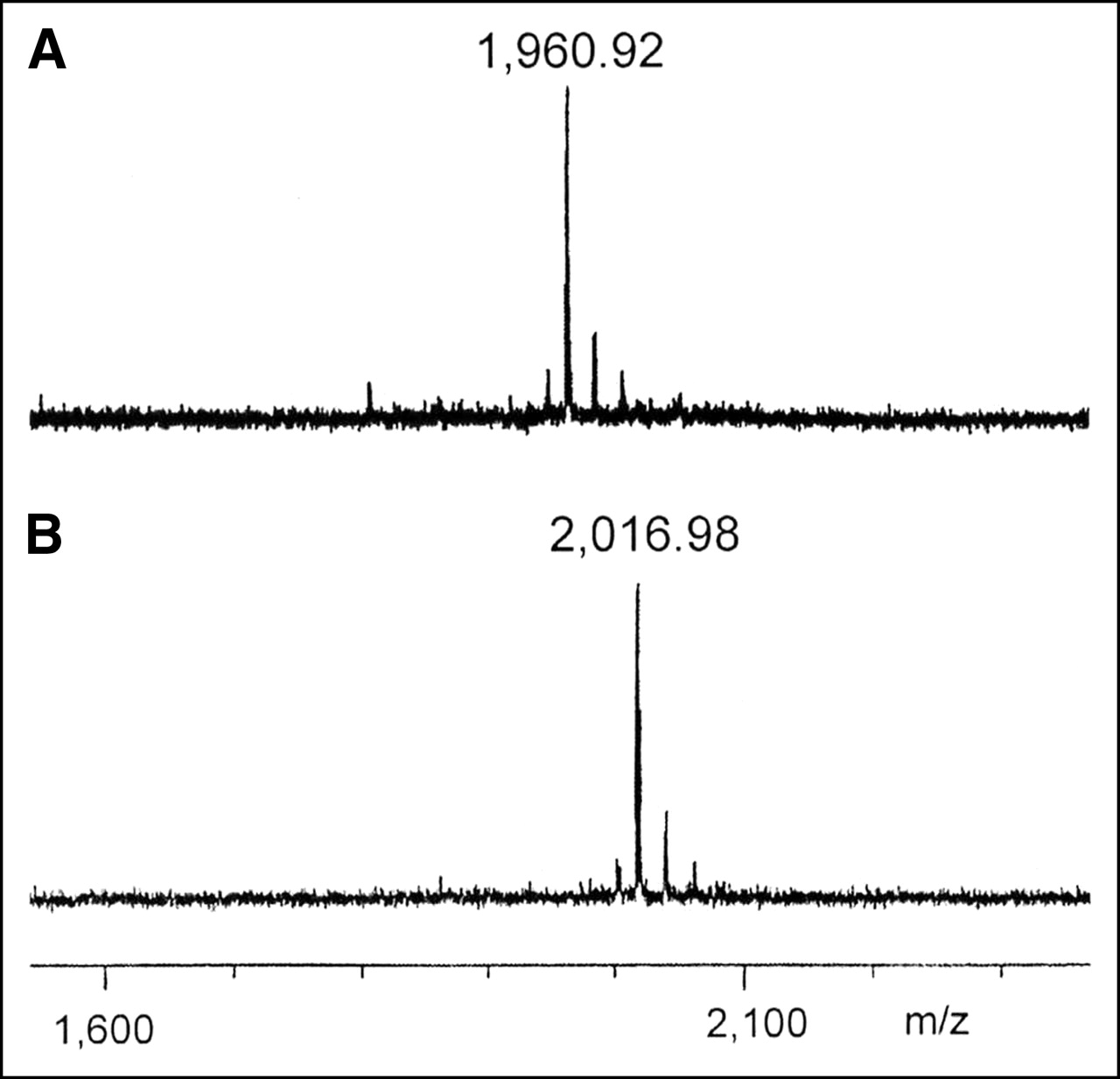
Immediate evidence for the substrate compatibility was obtained using MALDI-MS to detect caspase-catalyzed peptide fragments. No fragmentation was observed using peptide 2, whereas with peptide 7 the expected fragment was detected. The mass difference between fragment and parent molecule of 56 indicates the hydrolytic loss of the C-terminal G-NH2 (Fig. 7).

MALDI mass spectra of peptide 7 incubated with (A) and without (B) commercial caspase-3 (m/z represents molecular mass/charge).
DISCUSSION
Apoptosis is a dynamic cellular process that does not affect all tumor cells after cytostatic drug treatment. The onset of this phenomenon is an individual cellular process and apoptotic cells are rapidly eliminated in vivo, which makes it difficult to evaluate the potency of a new apoptotic imaging agent in tumor tissue. Therefore, a test system was established that can reliably be used to investigate the uptake and kinetics of new radiolabeled compounds in control and apoptotic cells. Nonadherently growing Jurkat cells were chosen, which allowed the analyses of cells before commencing the experiments. In addition, the use of cell suspensions permitted sampling in a time-controlled manner and evaluation of free and cellular radioactivity by a 1-step gradient centrifugation.
The dynamics of apoptosis required the determination of the optimal time period after which uptake measurements are best performed. Time-course measurements of externalized phosphatidylserine and caspase-3 activation in the apoptotic cell fractions were performed (Fig. 1). Because of the coherent occurrence of these effects, the rapid performance of flow cytometry assay was favored, which simplified the characterization of the cells before uptake measurements.
Besides camptothecin, we used staurosporine for the initiation of apoptosis. Camptothecin is a quinoline-based and staurosporine is an indole-based alkaloid. Whereas the former is a DNA topoisomerase I inhibitor, the latter represents an inhibitor of kinase proteins. As shown in Figure 2, staurosporine proved to be more effective in stimulating apoptosis. The cell uptake measurements with the series of radioiodinated peptides were, therefore, performed with staurosporine.
The flow cytometry data correlated well with cell uptake experiments performed with 99mTc-HYNIC-annexin V in staurosporine-treated and control cells (Fig. 3). The high binding values of 99mTc-HYNIC-annexin V at the beginning of the experiment and the steady course over 1 h are due to the unhindered access of this agent to externalized phosphatidylserine-binding sites on apoptotic cell membranes. The slight, but steady, increase of radioactivity may reflect unspecific binding or diffusion of its metabolite into the cells.
Using the same technique, two radiolabeled peptides comprising the DEVDG sequence (1 and 2) were evaluated for their differential uptake in control and apoptotic cells. DEVDG was used because it is a sequence accepted by caspase-3 (23). To examine the influence of the label position, 3-131I-iodotyrosine was placed either on the N- or at the C-terminal end. In addition, the influence of the free carboxylic acid groups on the uptake behavior of DEVDG was examined by replacing aspartic acid (D) and glutamic acid (E) with asparagine (N) and glutamine (Q), yielding NQVNG (3 and 4). The results of the cell uptake studies are summarized in Figures 4A and 4B.
Throughout the experiments, the early uptake values of radiolabeled peptides 1–4 were similar in control and apoptotic cells. Beyond 30 min, control cells started to increase peptide uptake. The divergent uptake characteristic might be related to the compromised cell membrane structure during apoptosis, which impeded cell membrane transport of these peptides.
An additional observation was that the position of 3-131I-iodotyrosine exhibits an uptake-directing role. The comparison of peptides 2 and 4 with N-terminal bound 3-131I-iodotyrosine and peptides 1 and 3 with the corresponding C-terminal position reveals that the C-terminal bound 3-131I-iodotyrosine favored the cell uptake to some degree.
From these results it becomes evident that the DEVDG or NQVNG sequences alone are insufficient for gaining uptake differences between apoptotic and control cells opposite to the observed ones. The main reason for this shortcoming is probably the lack of cell membrane permeability of these peptides. Therefore, cell membrane transporter sequences were taken into consideration. Two groups of cell-penetrating peptides have been described. MAP (model amphipathic peptide [X-KLALKLALKALKAALKLA-amide]) (24) and transportan (25) belong to the first group, whereas Tat (26) and penetratin (27) belong to the second group. The difference in cargo delivery efficiency between these 2 groups of cell-penetrating peptides is explained by the ability of the amphipathic transportan–MAP group of peptides to enter and exit cells, in contrast to the Tat/penetratin group of peptides, which are trapped in the cells (28).
Due to the trapping characteristic of Tat, the DEVDG sequence was conjugated to Tat49–57 (RKKRRQRRR), including the label d-3-131I-iodotyrosine (y). Tat48–60 (GRKKRRQRRRPPQ) encompasses the whole basic region and nuclear localization signal of the HIV transactivating factor protein Tat (29). The sequence lacking N-terminal glycine and C-terminal proline-proline-glutamine was taken up by Jurkat cells (26) and was, therefore, selected for these experiments. Penetratin, which was also considered for this purpose, contains methionine and is, consequently, less appropriate because of its sensitivity against chloramine-T.
The yDEVDG sequence was coupled to the N-terminal (5) as well as to the C-terminal (7) end of Tat49–57. The same synthesis was performed with the inverse sequence of the Tat fragment Tat57–49 (6 and 8). When incubating apoptotic and control cells with Tat conjugates 7 and 8, a general higher cellular uptake was observed as compared with peptides 5 and 6, indicating yDEVDG on the C-terminus of Tat to be more favorable for cell uptake (Fig. 5). With peptides 7 and 8, the uptake preference was, for the first time, reversed, meaning that apoptotic cells retained more radioactivity than the control cells. This observation met our working hypothesis that the interaction of a radiolabeled peptide with an activated caspase would lead to a retention advantage over cells not containing the activated enzyme. The prerequisite for this assumption is the recognition of the offered peptides as caspase substrates (vide infra).
The uptake values of the apoptotic cells were well above those of the controls. The decrease in cellular radioactivity indicates the efflux of either the intact peptide or a fragment carrying the label. In contrast to the results obtained with radiolabeled (30) and fluorescence tagged Tat49–57 and Tat57–49 sequences (26), the Tat sequence order seems to play a minor role here with respect to the uptake and retention characteristics of Jurkat cells.
The Tat conjugates 7 and 8 were also transformed to tat49–57-yDEVDG (9) and tat57–49-yDEVDG (10) by replacing l- with d-amino acids in the Tat sequence. As observed with peptides 1–6, this modification resulted again in a favored uptake into normal Jurkat cells (Fig. 5).
Although the cell membrane of apoptotic cells does not give preference to facilitated transport, the differences observed with peptides 7 and 8 can be related to the interaction with the activated enzymes. To obtain experimental proof for this assumption, the caspase-3 substrate properties of YDEVDG-NH2 (2) and Tat49–57yDEVDG-NH2 (7) were investigated with two different approaches.
Assuming that the peptides 2 and 7 are caspase-3 substrates, Z-DEVD-R110 could be used to monitor the degree of enzyme competition by measuring the depletion of the fluorescence signal. The result of this experiment shown in Figure 6 demonstrates the suppression of R110 formation as a consequence of increasing amounts of the two peptides, which identifies especially peptide 7 as a superior caspase-3 substrate.
An additional experiment applied MALDI-MS to show caspase-mediated fragmentation products. Commercially available caspase-3 (Fig. 7) and apoptotic cell lysate containing natural caspase-3 (data not shown) were incubated with peptide 7, indicating the expected fragmentation. MALDI-MS detectable fragmentation of peptide 2 was not detected under these conditions, a result correlating with the less-effective competitive characteristics of this compound shown in Figure 6.
CONCLUSION
We conclude that the cellular transport of peptides is hampered by morphologic changes of the apoptotic cell membrane, causing the observed uptake handicap for most of the DEVDG-containing peptide derivatives. Tat49–57-yDEVDG-NH2 (7) and Tat57–49-yDEVDG-NH2 (8) seem to represent sequences that avoid this drawback. They were found to exhibit a significant uptake difference in favor of apoptotic cells. The enhanced retention is interpreted to represent the interaction of the labeled peptides (7 and 8) or fragments thereof with activated caspases. Preliminary proof that the peptides interact with these enzymes was demonstrated with a competitive enzyme assay and MALDI-MS. They are, therefore, considered as lead structures for the synthesis of other peptide derivatives. Current efforts are focused on the preparation and evaluation of peptides with alternative labels, which include radiometals complexed with peptide-integrated chelators. In comparison with peptide fragments, which comprise 3-131I-iodotyrosine as the label, it is expected that radiometal tags are better retained in apoptotic cells, especially if their complex comprising parts alter their charge after fragmentation. Another mechanism, which would ideally meet the apoptosis-induced metabolic trapping concept, should be taken into consideration if caspase-induced fragmentation would lead to the release of the radiometal by transcomplexation. Weak complexes such as 111In-oxine, for example, are known to undergo spontaneous transcomplexation in, for example, platelets. Combined with peptide sequences evaluated in this study, this characteristic may result in an improved trapping mechanism.
Acknowledgments
We thank Dr. Martina Schnölzer (Deutsches Krebsforschungszentrum), who kindly performed the MALDI-MS measurements of the caspase-3 fragmented peptides. Samples of HYNIC-annexin V labeling kits were generously provided by Dr. Jean-Luc Vanderheyden (Theseus Imaging). Financial support was obtained from the Tumorzentrum Heidelberg–Mannheim and the European Union (FP6, NoE EMIL).
Footnotes
Received Oct. 6, 2004; revision accepted Jan. 23, 2005.
For correspondence or reprints contact: Michael Eisenhut, PhD, Deutsches Krebsforschungszentrum, Abt. Radiopharmazeutische Chemie–E030, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany.
E-mail m.eisenhut{at}dkfz.de
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- A PET Imaging Strategy to Visualize Activated T Cells in Acute Graft-versus-Host Disease Elicited by Allogenic Hematopoietic Cell Transplant
- In Vivo Small-Animal PET/CT of EphB4 Receptors Using 64Cu-Labeled Peptide
- The future of imaging: developing the tools for monitoring response to therapy in oncology: the 2009 Sir James MacKenzie Davidson Memorial lecture
- Multimodality Cardiovascular Molecular Imaging, Part I
- Nuclear Imaging Probes: from Bench to Bedside
- Mortals Turn Me On...