


Abstract
This study assesses targeting of lung metastases in mice with the radioiodinated thymidine analog 5-[123I/125I]iodo-2′-deoxyuridine (123I-IUdR/125I-IUdR), formulated with varying amounts of tributyltin precursor and injected intravenously. Methods: Six- to 8-wk-old C57BL/6 mice were injected intravenously with B16F10 melanoma cells. Two weeks later, when lung tumors were established, the animals were injected intravenously with 125I-IUdR synthesized using 1, 35, 100, 150, 200, or 250 μg 5-tributylstannyl-2′-deoxyuridine (SnUdR) in the presence of an oxidant. Nontumor-bearing mice were also injected with these formulations and served as control animals. Twenty-four hours later, the animals were killed, and the radioactivity associated with the lungs and other tissues was measured in a γ-counter. The percentage injected dose per gram tissue (%ID/g) and tumor-to-nontumor ratios (T/NT ratios) were calculated. Phosphor imaging was done on lungs from tumor-bearing and nontumor-bearing mice injected with 125I-IUdR formulated with each tin precursor concentration. Scintigraphy was also performed 3 and 24 h after intravenous injection of 123I-IUdR. Results: The %ID/g 125I-IUdR was higher in lungs of tumor-bearing animals than in lungs of control animals. Although the increase in SnUdR present led to a small but statistically significant decrease in the radioactive content of normal lungs, a 3-fold increase was observed in the lungs of tumor-bearing animals with radiopharmaceutical formulated with 100 μg SnUdR (5 μg per mouse). This enhancement in radioactive uptake by the lungs led to approximately 14-fold increases in T/NT ratios. Phosphor imaging (125I-IUdR) of lungs as well as scintigraphy (123I-IUdR) of whole animals substantiated these findings. Conclusion: The formulation for the synthesis of radio-IUdR that leads to the highest %ID/g in tumor and the best T/NT ratio has been identified. Further studies are required to determine the factors responsible for specific enhancement in IUdR tumor uptake.
- 5-[123I/125I]iodo-2′-deoxyuridine
- metastatic lung cancer
- 5-tributylstannyl-2′-deoxyuridine
- tumor targeting
Every year, close to one million new cases of lung cancer are diagnosed; in the United States, it is estimated that approximately 174,000 new cases will be identified in 2004 (1). Most of these patients (∼160,000) will die within 2 y of diagnosis. Although death from this disease occurs at a higher frequency in men than in women, it claims the lives of more women than does breast cancer. The 5-y survival rate of patients with lung cancer is approximately 14% and has not changed over the past several decades (1). Thus, the imperative to improve treatment of this disease is clear.
Auger electron emitters (e.g., 123I, 125I) have been proposed as an attractive alternative to energetic β-emitters (e.g., 131I) for use in cancer therapy. Investigators have found that radiopharmaceuticals such as 5-iodo-2′deoxyuridine (IUdR), labeled with 123I or 125I, are highly toxic to mammalian cells (2–4) and exceedingly efficacious (5- to 7-log cell kill) in the therapy of small-animal malignancies (5–7) when these radionuclides decay in proximity to nuclear DNA.
IUdR is a thymidine (TdR) analog that behaves remarkably like TdR (8). Within the cell, IUdR and TdR are phosphorylated by thymidine kinase to IUdR monophosphate (IdUMP) and TdR monophosphate (dTMP), respectively. dTMP is then further phosphorylated in a stepwise reaction and incorporated into DNA. IdUMP, on the other hand, may either follow a similar fate of phosphorylation and DNA incorporation or be dehalogenated by thymidylate synthetase (TS) to dUMP (9), which is further converted to dTMP via the “de novo” TS-catalyzed reaction. IUdR is extremely stable in vitro but quite unstable in vivo; its overall half-life in the circulation is 5 min in humans and 7 min in mice (10, 11). The notably short half-life of IUdR in vivo, its rapid dehalogenation in the liver, and its cellular uptake only in S phase of the cell cycle are limiting factors in the use of this compound (10, 12). Investigators have attempted to adjust for the rapid dehalogenation by administering the compound intratumorally or in proximity to the tumor and for the cell-cycle specificity by applying continuous or intermittent infusion (5–7, 13, 14). However, obtaining high uptake (percentage injected dose/gram targeted tissue [%ID/g]) of IUdR by tumor cells and high tumor-to-nontumor ratios (T/NT ratios) after intravenous administration remain a challenge for investigators.
In this study, we tested the hypothesis that the intravenous route can be used to administer nucleoside analogs that are broken down by liver enzymes when the targeted tumor is perfused by drug-laden blood before passage of the blood through the liver. Furthermore, we examined the effect of changes in the concentration of 5-tributylstannyl-2′-deoxyuridine (SnUdR) used in the synthesis of radio-IUdR on the in vivo uptake of 123I-IUdR/125I-IUdR by tumor cells and the resulting %D/g and T/NT ratios.
MATERIALS AND METHODS
Preparation of 125I-IUdR
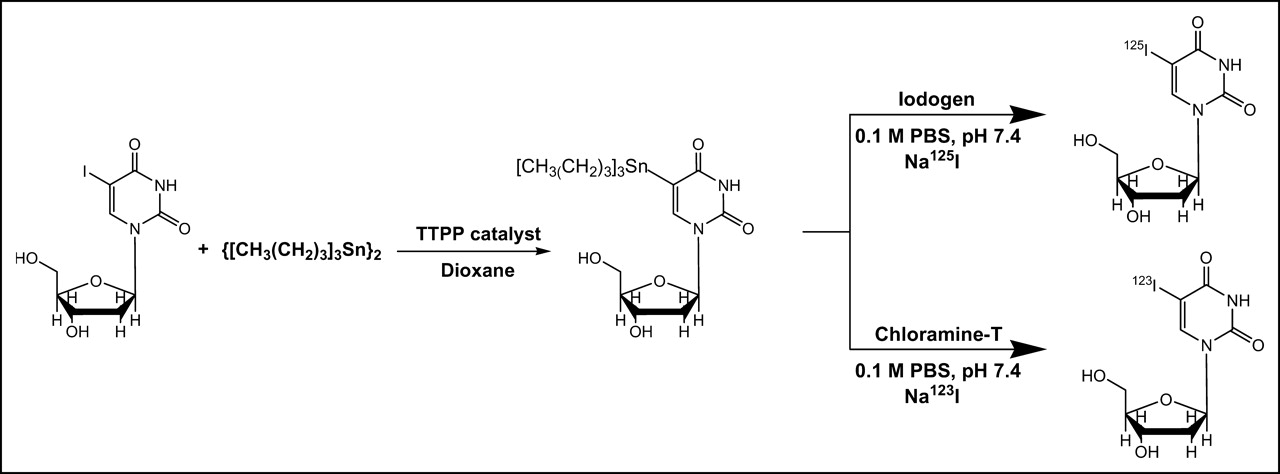
The preparation of 125I-IUdR is shown in Figure 1. A reaction vial was coated with 10 μg 1,3,4,6-tetrachloro-3α,6α-diphenylglycoluril (IODO-GEN; Pierce) and the following were added: 10 μL 0.1 mol/L phosphate buffer (pH 7.4), 1–2 μL dimethyl sulfoxide containing 1, 35, 100, 150, 200, or 250 μg SnUdR synthesized as previously described (15), and ∼37 MBq Na125I in ∼2–5 μL 0.1 mol/L sodium hydroxide (IMS 300; Amersham Biosciences). The reaction mixture was vortexed at ambient temperature for 2 min. The yield and purity of the product were assessed by high-performance liquid chromatography (HPLC) (Waters) using a reversed-phase ZORBAX C18 column (9.4 × 250 mm; Agilent Technology) attached, in series, to an ultraviolet (UV) multiwavelength detector (model 440; Waters) and an inline detector and chart recorder (gamma RAM; IN/US System), and eluted with 10% acetonitrile in water (1 mL/min). The reaction solution was diluted to 2 mL with saline before intravenous injection of 0.1 mL per mouse.

Pathways used for synthesis of 125I-IUdR and 123I-IUdR. TTPP = tetrakis(triphenylphosphine)palladium; PBS = phosphate-buffered saline.
Preparation of 123I-IUdR
The preparation of 123I-IUdR is shown in Figure 1. To a reaction vial were added 20 μL 0.1 mol/L phosphate buffer (pH 7.4), 1–2 μL dimethyl sulfoxide containing 20 μg SnUdR, 2 μL freshly prepared chloramine-T hydrate (25 μg/μL water), and ∼370 MBq Na123I (MDS Nordion) in 0.01 mol/L sodium hydroxide (pH adjusted to ∼9). The reaction mixture was vortexed at ambient temperature for 10 min. Analysis by reversed-phase C18 HPLC indicated the yield and purity of the product. The reaction solution was diluted to 0.4 mL with saline before intravenous injection of 0.1 mL per mouse.
Tissue Culture
B16F10 is a murine, metastatic, skin melanoma cell line syngeneic with C57BL/6 mice. The cells were routinely maintained in Dulbecco’s modified Eagle medium (DMEM; GIBCO BRL), containing 10% fetal bovine serum, 0.2% gentamicin, and 0.5% penicillin-streptomycin solution, and incubated at 37°C in air containing 5% CO2.
Metastatic Lung Model and Biodistribution Studies
Male C57BL/6 mice, 6- to 8-wk old, were purchased from Charles River Laboratories and inoculated intravenously (tail) with 4 × 105 viable B16F10 cells to establish pulmonary melanoma metastases. Normal mice were used as control animals. Beginning on day 12 (48 h before the injection of radioiodinated compound), all mice were given drinking water containing potassium iodide (0.4 g/L) to block thyroid uptake of 125I. On day 15, when the metastatic foci reached ∼2 × 2 mm diameter, mice (n = 5/group) were injected intravenously with 125I-IUdR (∼18.5 kBq/0.1 mL saline containing 0.05, 1.75, 5.0, 7.5, 10.0, or 12.5 μg SnUdR). Twenty-four hours later, the animals were killed, and the radioactivity associated with the lungs and all other tissues—including blood, heart, bladder, urine, liver, stomach, spleen, small intestine, large intestine, kidneys, testes, thyroid, skin, muscle, bone, tail, and fat—was measured in a γ-counter. The %ID/g and T/NT ratio were calculated. These experiments were repeated 2 or 3 times.
Histology
Lung and other tissues were fixed in 15% formalin buffer and processed for histopathologic studies. The tissue was embedded in paraffin, sectioned (5-μm), and stained with hematoxylin–eosin. Slides were examined under light microscopy to determine the presence of melanoma tumor cells.
Phosphor Imaging
Excised lung tissue collected from control and tumor-bearing mice was subjected to ex vivo radiophosphor imaging, and the uptake of 125I-IUdR (formulated with each of the 6 SnUdR concentrations) was evaluated. Briefly, the formalin-fixed lung tissue was blotted dry on tissue paper and placed on a microscope slide. The slides were placed in a phosphor imaging cassette (Molecular Dynamics, Inc.), which was then closed. Under these conditions, the lung tissues were compressed to a thickness of 1–2 mm. The imaging plates were exposed for 3 h to the decaying 125I present within the lungs (± tumor foci) and the accumulated counts were then processed on a Molecular Dynamics PhosphorImager (Storm 860 Scanner, Scanner Control Version 4.1; Molecular Dynamics, Inc.) to obtain the radiophosphor images.
Scintigraphy
γ-Camera scintigraphy was performed 3 and 6 h after a single intravenous injection of 7.4 MBq 123I-IUdR (additional SnUdR added to achieve 5 μg/mouse) using a standard field-of-view mobile scintillation camera (General Electric Starcam 300 Mobile γ-Camera) equipped with a medium-energy, pinhole collimator. Two groups of animals were injected: control animals (n = 2) and mice bearing 14-d-old B16F10 lung tumors (n = 3). Thyroid uptake was blocked with 0.4% potassium iodide. Animals were anesthetized (80 mg/kg ketamine and 10 mg/kg xylazine) and placed prone (anterior view) on the collimator, and static images were acquired for a fixed time of 10 min with a 128 × 128 matrix and a magnification of 4.
RESULTS
Radiochemistry
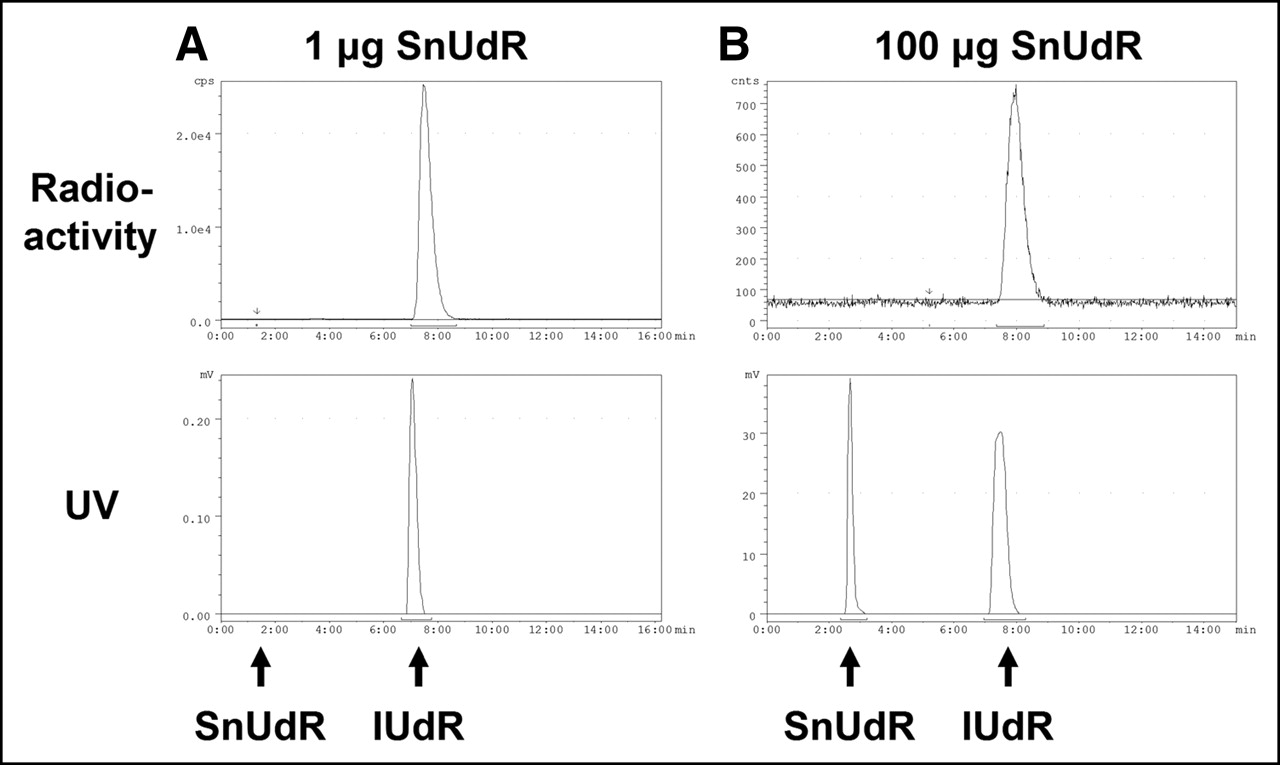
Under the experimental conditions in our studies, the exchange of the tributyltin moiety of SnUdR with 125I (up to 76 MBq) in the presence of IODO-GEN is facile and rapid regardless of the amount of SnUdR (1–250 μg) present. HPLC analysis (Fig. 2) indicates the absence of free 125I (retention time [tR] = 2.5 min) and the comigration of 125I-IUdR (peak tR = 7.5 min) and 127I-IUdR (peak tR = 7.1 min); the ∼30-s delay between 125I-IUdR and 127I-IUdR is a consequence of the connecting sequence of the UV and γ-detectors. As seen in Figure 2 (top), the radiolabeling yield is very high (∼100%; >85% recovery) and, except for the SnUdR peak (tR = 2.4 min) observed when larger amounts of SnUdR were used in the radiosynthesis, no other UV species can be detected (Fig. 2, bottom).
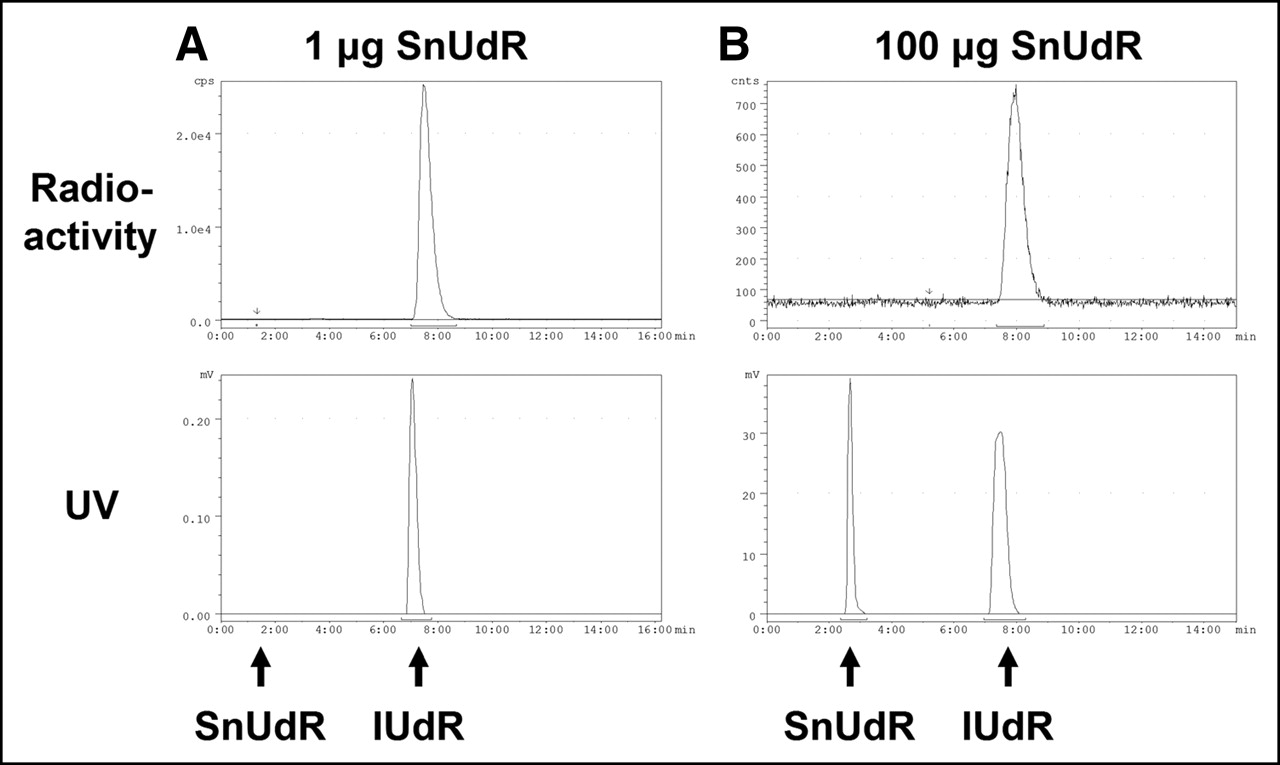
HPLC analysis of 125I-IUdR synthesized with varying amounts of SnUdR (127I-IUdR coinjected with 125I-IUdR). (A) SnUdR formulation: 1 μg. (B) SnUdR formulation: 100 μg. (Top) Radioactivity trace: 125I-IUdR tR = 7.5 min. (Bottom) 254-nm UV trace: 127I-IUdR tR = 7.1 min, SnUdR tR = 2.4 min.
The radiosynthesis of 123I-IUdR is inefficient in the presence of IODO-GEN. However, when chloramine-T is used and the reaction mixture (SnUdR and Na123I) is vortexed for 10 min (compared with 2 min for 125I), a radiolabeling yield of ∼100% is also obtained. HPLC analysis indicates the absence of free 123I (tR = 2.5 min) and the comigration of 123I-IUdR (peak tR = 7.5 min) and 127I-IUdR (peak tR = 7.2 min), with a small SnUdR peak at ∼2.6 min (data not shown).
Biodistribution
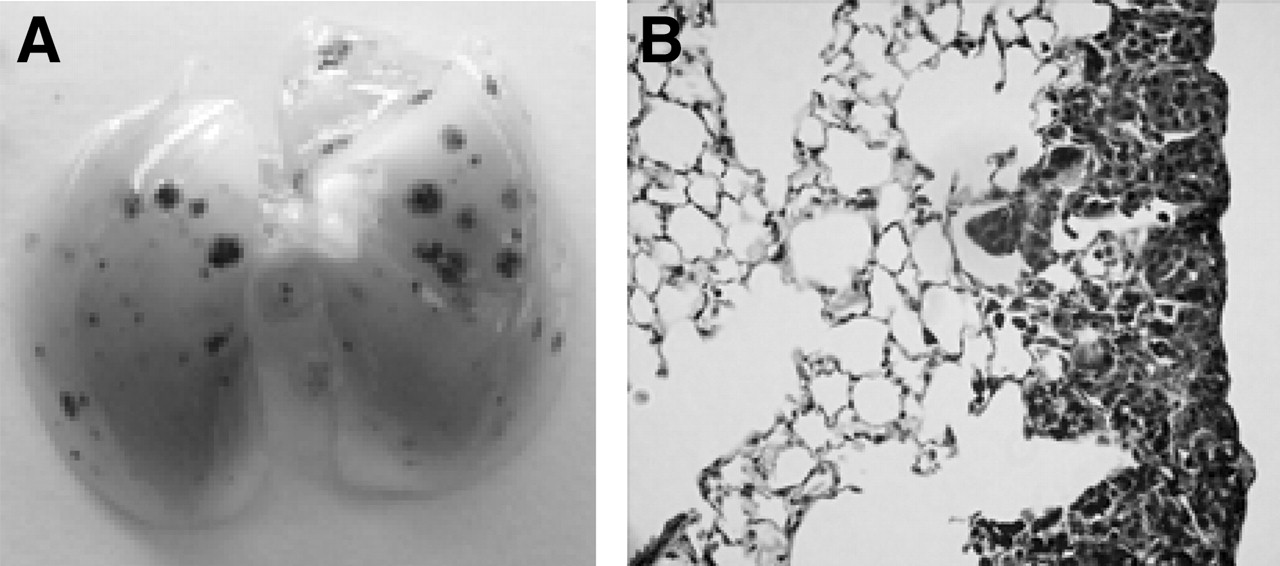
The intravenous injection of B16F10 tumor cells into mice leads to the development of dark brown visible (1–2 mm) tumor foci in the lungs over a 2-wk period (Figs. 3A and 3B), and the animals die within an additional 2 wk. Although this metastatic cell line was selected for its ability to localize and grow in the lungs, small tumor foci are nevertheless occasionally seen within other organs (e.g., the kidneys, brain, and liver). It has also been reported (and confirmed in our studies) that most of the metastases are confined to the thin subpleural connective tissue layer of the lungs (16).
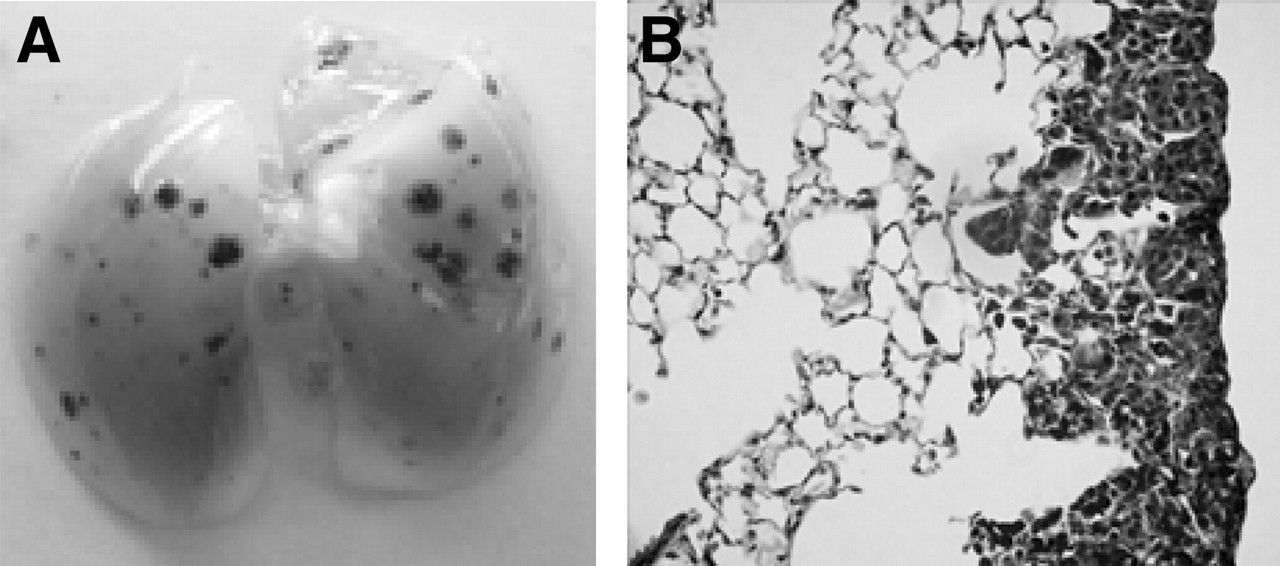
Lung from mouse bearing 14-d-old B16F10 lung tumor metastases. (A) Photograph of lung indicates melanin-filled, tumor-cell foci. (B) Section (5 μm) shows tumor growth.
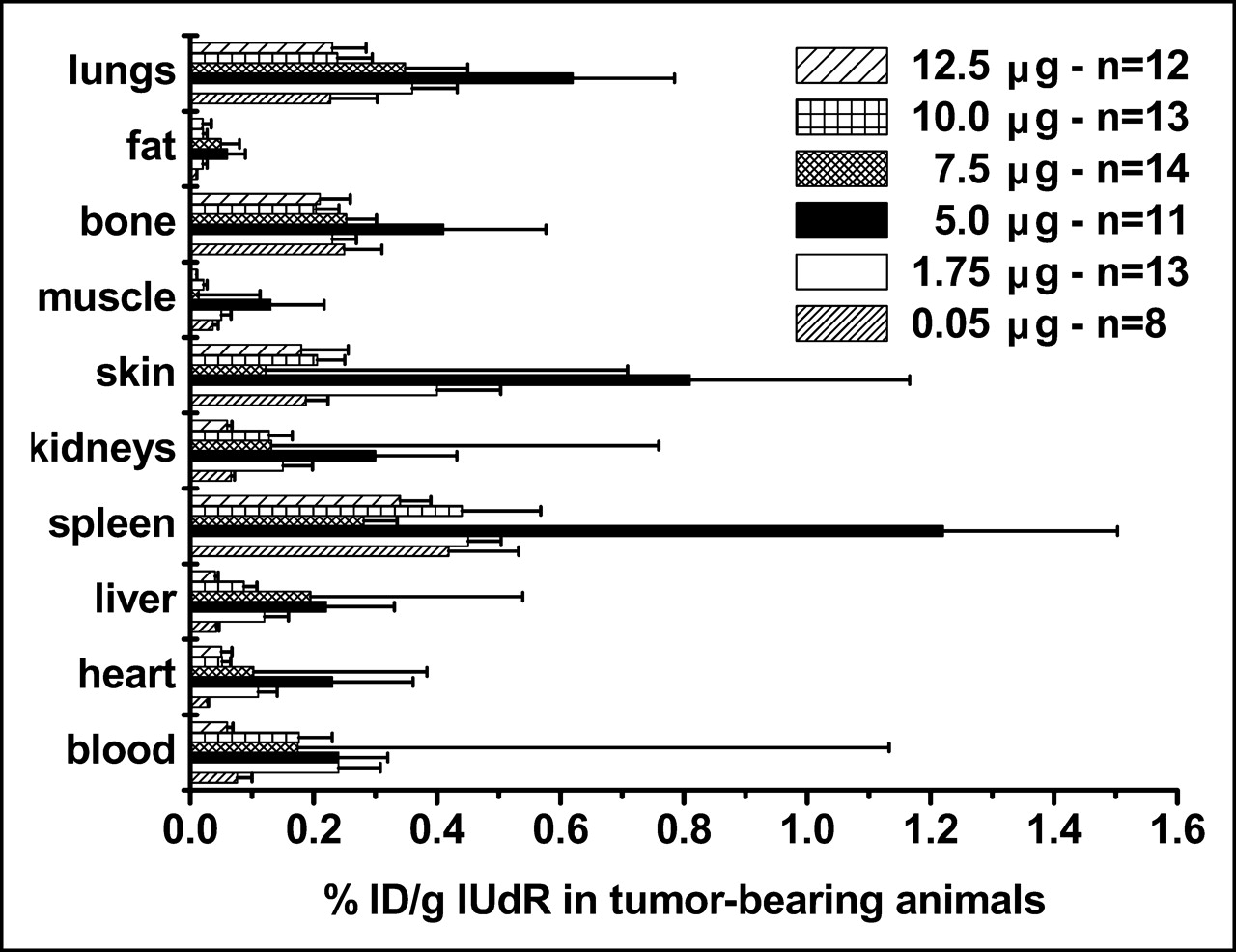
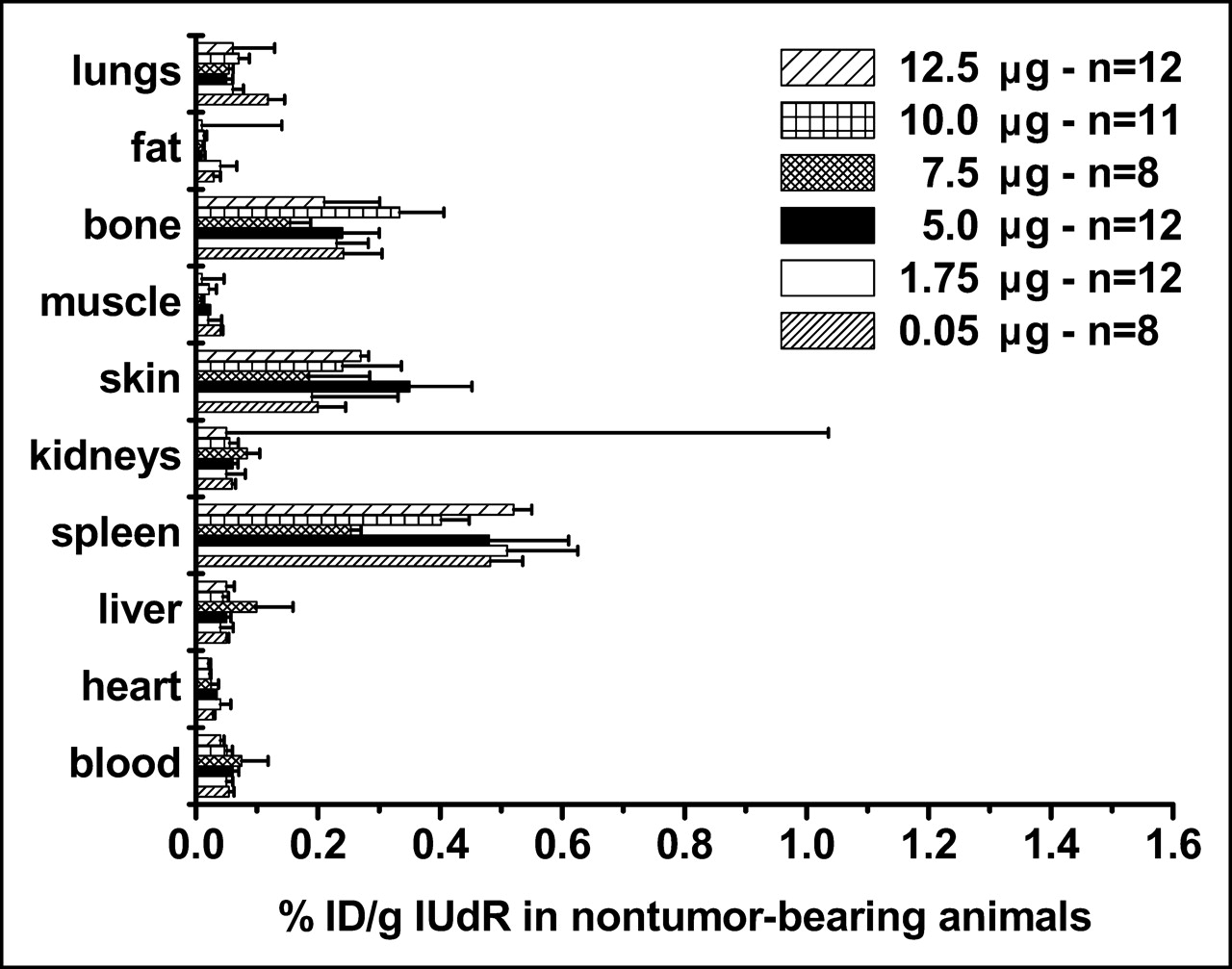
We compared the %ID/g in tumor-bearing and control animals using 125I-IUdR formulations made with 6 concentrations of SnUdR (molecular weight = 517) in the presence of ∼37 MBq Na125I. Since this radionuclide is available at the no-carrier level (81.5 TBq/mmol), the 125I-IUdR synthesized used ∼0.25 μg of the added SnUdR. In tumor-bearing mice, the highest %ID/g is obtained in most organs and tissues when IUdR is made with 100 μg SnUdR (5 μg/mouse) (Fig. 4). A notable increase in radioactivity is also observed when the 125I-IUdR solution contains 1.75 and 7.5 μg SnUdR per mouse. No such increases in %ID/g are observed in the tissues and organs of nontumor-bearing mice and, in general, these values are lower (Fig. 5).
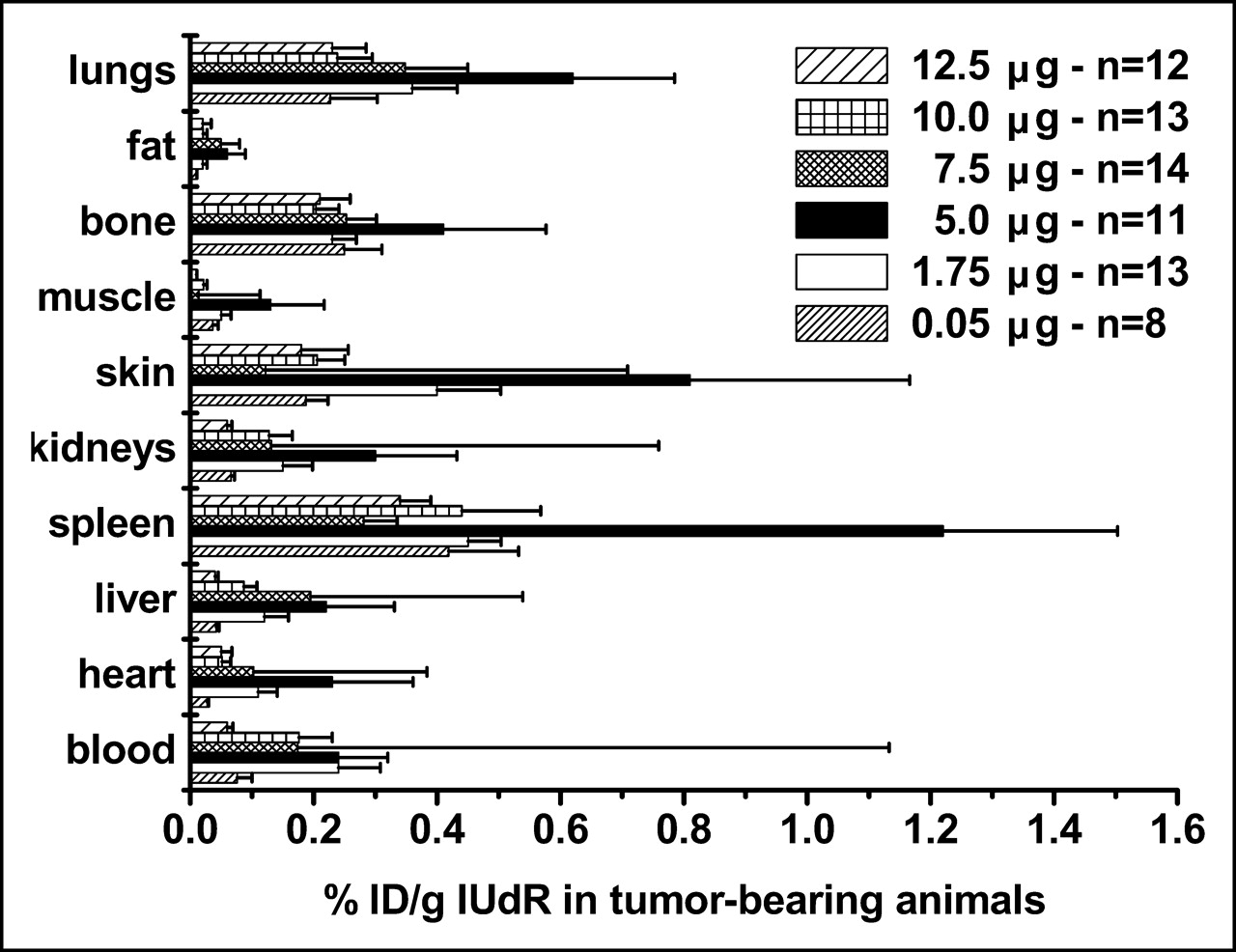
Biodistribution of radioactivity in B16F10 tumor-bearing mice (n = 8–14) 24 h after intravenous injection of 125I-IUdR preparations containing varying amounts of SnUdR (%ID/g, mean ± SEM).
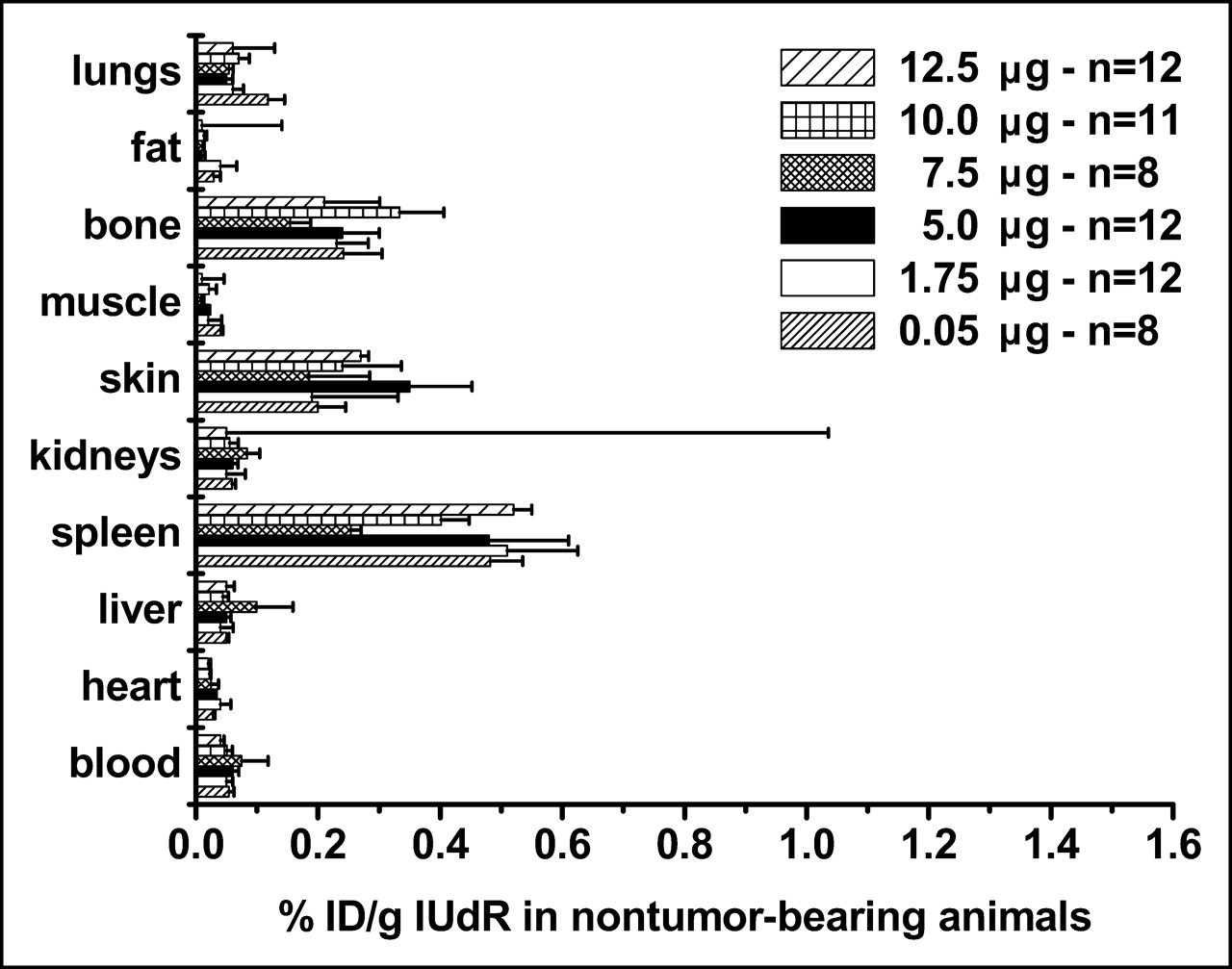
Biodistribution of radioactivity in control (nontumor-bearing) mice (n = 8–12) 24 h after intravenous injection of 125I-IUdR preparations containing varying amounts of SnUdR (%ID/g, mean ± SEM).
The presence of tumor foci is always associated with significant increases in radioactive content (%ID/g) (Fig. 6). The differences in uptake are a function of the amount of SnUdR present. Inexplicably, there is a gradual and highly significant increase (compared with values obtained with the 1-μg formulation) in uptake of 125I-IUdR that peaks 3-fold at the 100-μg SnUdR formulation; this is followed by a decrease in the %ID/g as the amount of SnUdR is increased to 250 μg (Fig. 6). Since the coadministration of >1 μg SnUdR also leads to a significant decrease (P = 0.036) in the radioactive content of normal lungs, a similar rise and fall in T/NT ratio is also observed (Fig. 7). Consequently, the highest T/NT ratio (13.6) is seen at the 100-μg SnUdR formulation and the lowest ratio (1.9) is seen at the 1-μg SnUdR formulation, approximately a 7-fold difference. It is important to note that the %ID/g in the lungs of tumor-bearing mice underestimates the true %ID/g since most of the weight reflects that of normal lung tissue and not the small masses of tumor foci. Consequently, the absolute T/NT ratios would be expected to be much higher than those reported in Figure 7.

Uptake (%ID/g) of 125I within lungs of B16F10- melanoma-bearing (2-wk-old tumors) and control mice after intravenous injection of 125I-IUdR synthesized with varying amounts of SnUdR.

T/NT ratios (mean ± SEM) calculated from lung data in Figure 6.
Phosphor Imaging
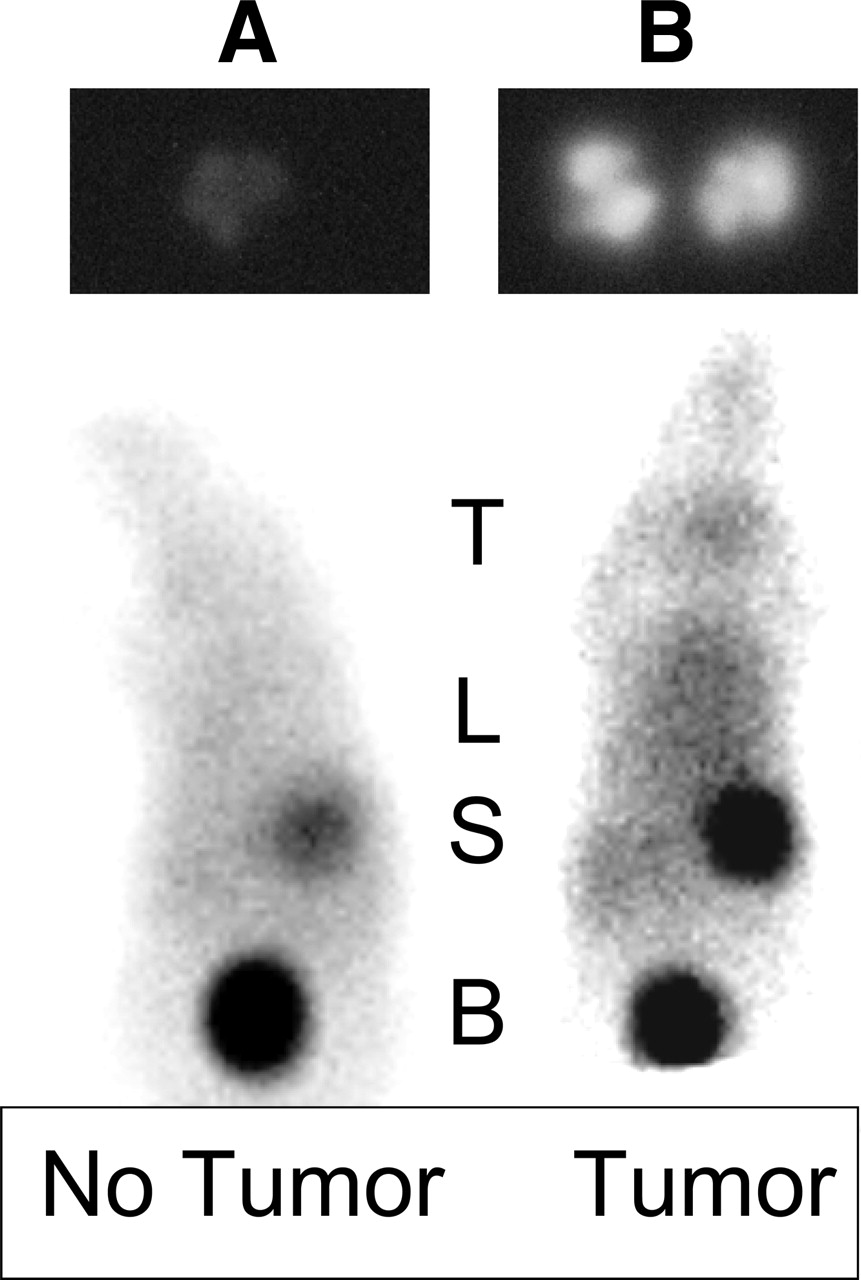
Lungs from control (nontumor-bearing) and tumor-bearing mice injected with IUdR formulated with 6 concentrations of SnUdR were assessed by phosphor imaging. Again, minimal activity is observed in nontumor-bearing lungs (Fig. 8A, top) and the greatest uptake of 125I-IUdR occurs when the radiopharmaceutical is formulated with 100-μg tin precursor—that is, 5 μg SnUdR per mouse (Fig. 8B, top).
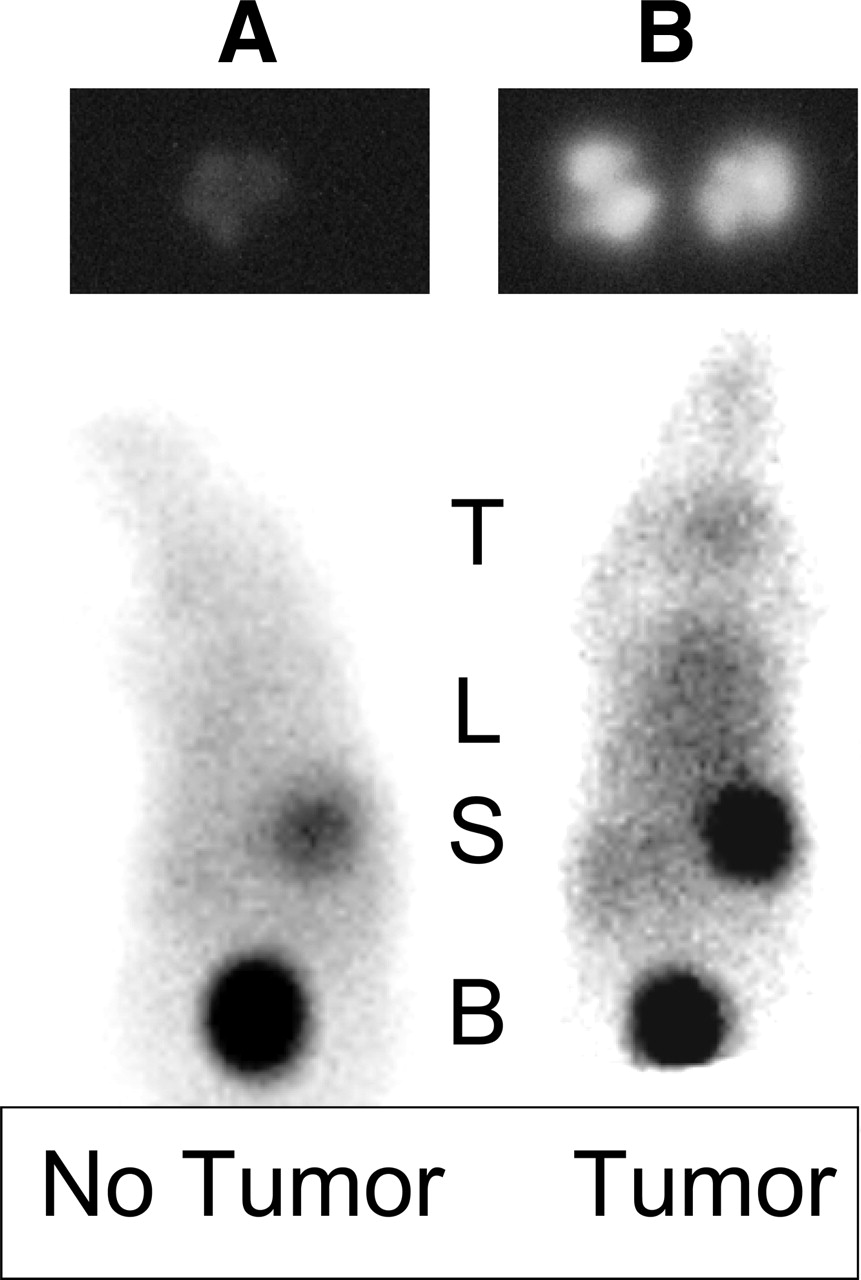
Images of control (A) and tumor-bearing (B) mice. (Top) Phosphor images (3-h exposure) of lung of control mouse (left) and tumor-bearing mouse (right) injected 24 h earlier with 185 kBq 125I-IUdR preparation containing 5 μg SnUdR. (Bottom) Scintigraphic images of control mouse (left) and tumor-bearing mouse (right) injected 3 h earlier with 7.4 MBq 123I-IUdR preparation containing 5 μg SnUdR. T = thyroid; L = lung (± tumor foci); S = stomach; B = bladder.
Scintigraphy
Scintigraphy was performed 3 and 24 h after intravenous injection of 123I-IUdR in control and tumor-bearing mice. In the normal mice (Fig. 8A, bottom), radioactivity is evident in the stomach and bladder, demonstrating dehalogenation of the radiopharmaceutical and excretion of free radioiodine. In some instances, radioactivity is also observed within the thyroid despite our attempts to block radioiodine uptake by addition of potassium iodide to the drinking water 48 h before radiopharmaceutical injection. In tumor-bearing animals, in addition to the above organs, intense radioactivity is evident within the lung region (Fig. 8B, bottom).
DISCUSSION
IUdR is a thymidine analog that was first synthesized and reported in 1959 (17). Within a year, various investigators described its uptake and DNA incorporation in mammalian DNA-synthesizing normal and tumor cells in vitro (18) and its pharmacology in normal and tumor-bearing animals (19, 20) and in humans (20). These and later studies demonstrated that IUdR has a very short half-life in the circulation (10, 11) and is rapidly degraded and dehalogenated mainly in the liver and excreted via the kidneys/bladder (9, 11).
IUdR was radiolabeled with 131I (10), 125I (2, 21–23), and 123I (24, 25), and the potential of the radiolabeled analogs (in imaging and therapy) was examined in tumor-bearing animals. It soon became evident, however, that the absolute uptake by tumors and the T/NT ratios were low when this radiopharmaceutical was administered systemically. To bypass the rapid breakdown, ensure the availability of radiolabeled IUdR molecules to dividing cancerous cells, and enhance the tumor uptake and T/NT ratios, investigators have relied on (a) locoregional administration where feasible (e.g., intraperitoneal, intratumoral, intrathecal), (b) appropriate dose fractionation, or (c) metabolic modulation. These approaches lead to excellent tumor imaging and therapy (5- to 7-log cell kill), in both various animal tumor models (5–7, 26) and patients (13, 14, 27).
The concept of locoregional administration of radio-IUdR has been pursued mainly as a consequence of the failure of such molecules to arrive intact at a tumor (passage through the liver leads to very rapid enzymatic degradation). We hypothesized in 1996 that the intravenous route could be used to administer nucleoside analogs that are broken down by liver enzymes when the targeted tumor is perfused by drug-laden blood before its passage through the liver. The approach relies on the fact that after a drug is injected intravenously, it will travel first to the heart and then to the pulmonary arteries, perfuse the lungs, travel back through the pulmonary veins to the heart, and, finally, through the aorta to the rest of the body. We theorize that an agent such as IUdR will be chemically intact during its first pass through the lung after intravenous injection and, as such, will be taken up by DNA-synthesizing tumor cells that are within the lung. Inherent to the absolute success of such an approach are 3 main assumptions. (i) The target is within an area that can be easily and directly accessed. (ii) Once within the vicinity of the tumor-containing tissues, the agent (a) freely diffuses throughout the tumor, (b) is taken up selectively by cancerous but not by noncancerous cells, (c) is very rapidly taken up (passively/actively) by cancerous cells, and (d) is retained indefinitely by cancerous cells. (iii) When the agent has diffused out of the target area, it is either converted quickly into an inactive (i.e., nontoxic) form or excreted rapidly from the body. IUdR is an agent that meets all of these requirements when injected intravenously into animals bearing lung cancer. (i) Since the tumor cells within the lungs are perfused by blood being oxygenated, the nucleoside present within the blood will also reach these tumor cells and be taken up by DNA-synthesizing cells. (ii) Being a low-molecular-weight molecule, IUdR diffuses readily within tissues to reach dividing tumor cells. (iii) Being a TdR analog, IUdR is taken up selectively by dividing cancerous cells located within areas of nondividing cells. (iv) IUdR is taken up by DNA-synthesizing cells at very high rates (10,000–20,000 molecules per second). (v) IUdR is indefinitely retained after DNA incorporation. (vi) The majority of cells within the lungs are nondividing and will not incorporate IUdR into their DNA.
To test our hypothesis, we had examined previously the uptake of radiolabeled IUdR injected intravenously into mice bearing lung LS174T human adenocarcinoma metastases. Despite the fact that most of the lung was devoid of tumor, a substantial amount of radioactivity was present within the lungs of tumor-bearing animals (12.9% ± 1.4% ID/g and 48.8% ± 14.7% ID/g, respectively, in 2- and 3-wk-old tumors). Consequently, very high T/NT ratios were obtained (range of T/NT2-wk ratios, 38–937; range of T/NT3-wk ratios, 322–11,037). Unfortunately, we have not been able to reproduce these high uptakes. In the current studies with B16F10 melanoma, the injection of no-carrier, pure 125I-IUdR preparations synthesized with minimal quantities of SnUdR (1 μg) and 37–74 MBq 125I routinely results in a disappointingly modest 2-fold increase in localization of radioactivity within the lungs of tumor-bearing animals (compared with lungs from control animals)—that is, the %ID/g in lung uptake increases from 0.1 to 0.2. Whereas these values are substantially lower than those we had observed (13%–49% ID/g), they nevertheless are promising since a very small percentage of the weighed tumor-bearing lung reflects the true mass of tumor foci. For example, if we consider that the total tumor weight represents 20% of the weighed lungs, then the true %ID/g within the tumor will be 2% with a corresponding T/NT ratio of 20. If these expectations were to materialize in humans, radio-IUdR might play a significant role in the scintigraphic detection of lung metastases in patients.
In an attempt to clarify this situation, we have investigated whether changing the radiolabeled IUdR formulation would affect the %ID/g and T/NT ratio. These experiments were initiated in part as an attempt to reproduce our earlier results but also as a consequence of recent in vitro studies in which the rate of DNA incorporation of 125I-IUdR in 3 human glioblastoma cell lines (LN229, U251, and U87) was enhanced by coincubation with 10 μmol/L unlabeled IUdR (28, 29). These findings are counterintuitive and do not agree with previous observations in which a decrease (rather than an increase) in 125I-IUdR/131I-IUdR DNA incorporation was seen after in vitro incubation of various types of mammalian cells in the presence of unlabeled IUdR (8, 30). However, pretreatment with IUdR also led to an in vivo increase in 125I-IUdR uptake in LN229, U251, and U87 tumors (29). The authors postulated that these effects might be a consequence of metabolic modulation of the de novo thymidine synthetic pathways (28, 29). Such in vitro and in vivo enhancement in radio-IUdR uptake by DNA-synthesizing mammalian cells had been demonstrated previously in the presence of certain antimetabolites (7, 14, 31–40). With preexposure of tumor cells in vitro to methotrexate (MTX)—a dihydrofolate inhibitor that blocks the intracellular methylation of dUMP to dTMP (and therefore the accumulation of TdR triphosphate pools) and prevents the dehalogenation of 125I-IdUMP (33)—a >8-fold increase in 125I-IUdR uptake occurs (7). Fluorodeoxyuridine (FUdR), a thymidylate synthetase inhibitor, also potentiates the uptake of IUdR by mammalian cells (32, 35, 37). Investigators have examined the possible use of such modulators to enhance radio-IUdR uptake by tumors in mice, rats, and humans, and the results have been encouraging. We have shown that coadministration of MTX with radio-IUdR in rats bearing intrathecal tumors (7, 39) produces (a) significant increases in the radio-IUdR incorporation in tumor cells without any corresponding increase in normal tissue uptake, (b) a higher T/NT ratio, and (c) a concomitant and substantial enhancement of therapeutic efficacy (from 4-log cell kill in the absence of MTX to 7-log cell kill when MTX is coadministered). Other investigators have also observed substantial improvements in tumor targeting and scintigraphic imaging after FUdR–123I-IUdR or FUdR–125I-IUdR administration (33, 34, 37, 38, 40). The use of such metabolic modulators in patients with liver metastases from colorectal cancer produces a marked enhancement of 123I-IUdR uptake by tumor metastases (14).
The current experiments indicate that increasing the amount of SnUdR in the formulation from 1 μg (<0.05 μg/mouse) to 100 μg (∼5 μg/mouse) produces a 3-fold increase in the radiolocalization of 125I-IUdR within the lungs of tumor-bearing mice without any corresponding decrease in normal lung tissues (Fig. 6). However, further increases in the amount of SnUdR in the formulation cause a decrease in lung-associated radioactivity in tumor-bearing animals. Though the reason for this observation is still unclear, the T/NT ratios for all of these SnUdR concentrations are highly favorable (1.9–13.6) (Fig. 7) and lead to clear differences in the images obtained (Fig. 8). Additional studies are necessary to clarify the mechanism(s) involved in this enhancement of radio-IUdR uptake by DNA-synthesizing tumor cells. However, it seems likely that the coinjected SnUdR may be inhibiting/modulating the de novo thymidylate synthetic pathways.
CONCLUSION
The formulation of 123I-IUdR/125I-IUdR influences the %ID/g uptake and T/NT ratios in various tissues and in tumor cells. The highest %ID/g 123I-IUdR/125I-IUdR and most favorable T/NT ratio are obtained when the radiopharmaceutical injectate contains 5 μg tin precursor. Further studies are necessary to identify the factors responsible for specific enhancement in tumor uptake of radio-IUdR.
Acknowledgments
This work was supported in part by National Institutes of Health grants 1 R01 CA89648 and 5 T32 CA09536.
Footnotes
Received Sep. 20, 2004; revision accepted Dec. 15, 2004.
For correspondence or reprints contact: Amin I. Kassis, PhD, Department of Radiology, Harvard Medical School, 200 Longwood Ave., Armenise Building, Boston, MA 02115.
E-mail: amin_kassis{at}hms.harvard.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.