


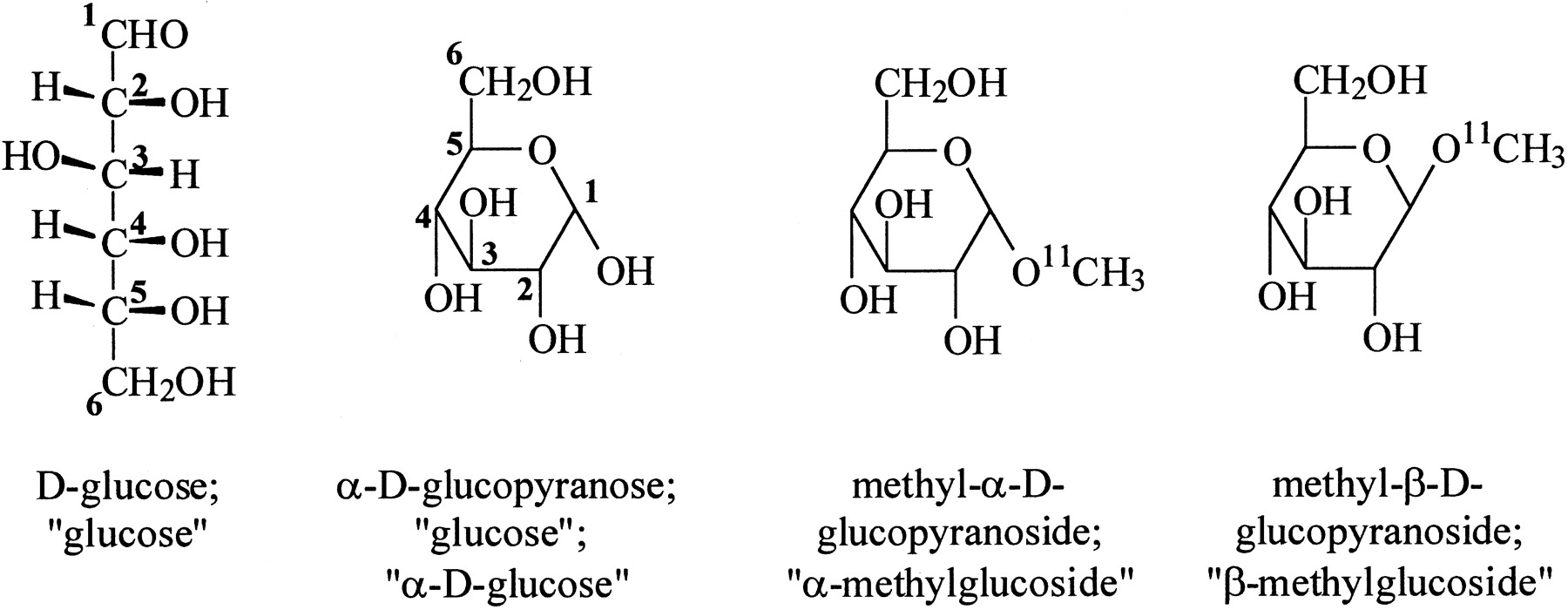
In a preclinical research article in this issue of The Journal of Nuclear Medicine, Bormans et al. (1) examine the uptake of 11C-labeled d-glucose analogs α-methyl-d-glucoside and β-methyl-d-glucoside in the mouse kidney. It appears that both of these compounds are taken up efficiently from the glomerular filtrate by tubular epithelial cells. However, β-methyl-d-glucoside is present in kidney at 30 min to a much greater extent than α-methyl-d-glucoside, suggesting that glucose transport from the tubular epithelial cells to the blood is slower for β-methyl-d-glucoside and that this constitutes a trapping mechanism that would allow PET imaging of the first transport step of glucose reabsorption in kidney. The article by Bormans et al. prompts discussion of several issues. To understand the significance of this work it is helpful to review the basic chemistry, biochemistry, and physiology of d-glucose. The structures of glucose and of the labeled compounds studied by Bormans et al. are shown below:
The leftmost structure is a conventional open-chain representation of the d-glucose molecule, showing the numbering system of the 6 carbon atoms and the stereochemistry of the hydrogen atoms and hydroxyl groups on each atom. The aldehyde function is shown at the top and designated as carbon atom 1 (C1). C6 carries a primary hydroxyl group, and the other 4 carbon atoms (C2–C5) each carry a secondary hydroxyl group. Because of the chirality of the central carbon atoms, d-glucose is 1 of 16 (42) “aldohexose” isomers with this general structure. One of these, l-glucose, is the mirror image of d-glucose. Another conventional representation of d-glucose is shown in the second structure, where the hydrogen atoms on C1–C5 are not shown. In solution, d-glucose exists as an equilibrium between low concentrations of the open-chain form and cyclized forms containing a 6-member (“pyranose”) ring. (Forms containing a 5-member ring are also minor components of the equilibrium mixture but are not shown here.) Formation of the 6-member ring creates a new asymmetric center at C1 and, therefore, 2 distinct isomers (historically termed “anomers”) referred to as α-d-glucopyranose (shown here) and β-d-glucopyranose (where the hydroxyl group on C1 points up instead of down). These anomers of d-glucose can be separated from one another by crystallization, but their fairly rapid interconversion in biologic fluids means that there are no important consequences of this phenomenon for d-glucose itself, even when enzymes and transporters recognize only 1 of the anomers. However, the α-d-glucopyranose and β-d-glucopyranose ring structures can be “locked in” by formation of glucosides. Thus, when glucose is reacted with an excess of methanol under acidic conditions, a mixture of methyl-α-d-glucopyranoside (α-methylglucoside) and β-methylglucoside is produced. Bormans et al. (1) used the very powerful radiomethylating agent, 11C-methyl triflate, in tracer quantities to produce methylglucosides with high specific activity suitable for nuclear medical use (third and fourth structures from the left). These 2 compounds were then separated by chiral high-performance liquid chromatography so that their biologic behavior could be examined separately.
HISTORICAL BACKGROUND
It is almost 30 y since chemists at the Brookhaven National Laboratory first synthesized 2-deoxy-2-18F-fluoro-d-glucose (18F-FDG), which today sustains the field of clinical PET (2). FDG, like glucose, is transported into the brain and other tissues by facilitative (nonenergy linked) carrier molecules. Furthermore, FDG, like glucose, is a substrate for hexokinase isoforms and is converted to FDG-6-phosphate. However, FDG-6-phosphate is unlike glucose-6-phosphate in that, for fundamental structural reasons, it cannot undergo the next step in metabolism of glucose itself, which is conversion to fructose-6-phosphate. Another property that distinguishes FDG from glucose is that FDG is only poorly transported by the energy-linked carriers for glucose that are found, for example, in the small intestine, which harvests dietary glucose, and in the kidney, which recovers the glucose present in the glomerular filtrate. Thus, FDG is excreted in the urine, a circumstance that aids the clearance of untrapped FDG from tissues and so may improve its radiopharmaceutical characteristics (3).
Except in extreme circumstances, such as deep hypoglycemia or the near-absence of transporters, FDG studies are not informative about glucose transport because the phosphorylation step is normally rate determining for tissue glucose consumption. Development of labeled compounds that are specific markers for glucose transport, therefore, represents an opportunity in radiopharmaceutical research.
FDG was developed before the advent of modern molecular biologic techniques, which allow us, for example, to recognize families of biologically active proteins in DNA sequence data, extract messenger RNA with partial complementarity to known genes, generate pure clones of genes coding for interesting proteins, and make mutant and chimeric proteins, cells, and animals for mechanistic studies. Before the genomic era, structure–activity relationships for glucose analogs for accumulation and metabolism in various cells and tissues had been determined, and this information together with physiologic data from tissue preparations provided evidence for multiple transport systems for glucose. For example, facilitative glucose uptake in muscle is sensitive to insulin, whereas glucose uptake in brain or erythrocytes has little or no sensitivity. Clinical observations supported the involvement of at least 2 gene products in energy-linked glucose transport, because distinct syndromes for familial glucose malabsorption and renal glycosuria had been recognized (4). The existence of several transport systems for glucose was not surprising in view of the central importance of glucose in metabolic pathways. The need to maintain plasma glucose levels within tight limits and the tissue-specific requirements for handling glucose logically meant that a multiplicity of transporters would be required for precise control of transport of glucose into and out of many different types of cells.
GLUCOSE ANALOGS THAT ARE TRANSPORTED AND PHOSPHORYLATED
The behavior of radiolabeled glucose analogs toward hexokinase and the prospects for development of alternatives to FDG, particularly single photon-emitting sugars, were discussed in an earlier review (5) and are briefly reiterated and updated here. Early workers (6) recognized that glucose analogs substituted at C1, C5, or C6 with halogens are either unstable or are unreactive with hexokinase for fundamental structural reasons, whereas glucose substituted at C4 with a fluorine atom is a very poor substrate for yeast hexokinase (7). Of the 8 d-aldohexoses, only d-glucose and its C2 epimer d-mannose are good substrates for phosphorylation by hexokinases. 2-Deoxy-d-glucose, 2-deoxy-2-fluoro-d-mannose, and 2,2-difluoro-deoxy-d-glucose are also good substrates and, in fact, are suitable in positron-labeled forms for measurement of local glucose metabolic rates (8–10). Radioiodinated 2-deoxy-2-iodo-d-glucose is chemically unstable (11,12) and, therefore, unsuitable for radiopharmaceutical use. McCarter et al. (13) improved the stability of 2-iodo–substituted glucose and mannose by replacing the hydrogen on C2 by a fluorine atom. However, neither 2-deoxy-2-fluoro-2-iodo-d-glucose nor 2-deoxy-2-fluoro-2-iodo-d-mannose possessed detectable activity with hexokinase from yeast or bovine heart, and 2-deoxy-2-123I-iodo-d-mannose was found, in mouse biodistribution experiments, to lack the properties of a marker for glucose metabolism (10). Glucose derivatives modified at C2 with bulky substituents—for example, 2-O-methyl-d-glucose—are very poor substrates for hexokinase, and 2-deoxy-2-chloro-d-glucose is only a moderately good substrate (6,14). Thus, it does not appear possible to adopt the strategy of preparing glucose derivatives substituted with other iodine-containing groups (e.g., iodoalkyl, iodovinyl, or iodophenyl moieties), which has been successfully used for receptor-binding radiopharmaceuticals, because the enzyme cannot tolerate bulky substituents at C2. It is also likely that the facilitative glucose transport process cannot handle large groups at C2, because 2-deoxy-2-bromo-d-glucose exhibits little uptake into brain (12,15).
LABELED GLUCOSE ANALOGS THAT ARE TRANSPORTED BUT NOT PHOSPHORYLATED
FDG and the other 2-substituted hexoses that are good substrates for hexokinase are also good substrates for facilitative transport. Another positron-labeled sugar, 3-O-11C-methyl-d-glucose, is transported by both facilitative and concentrative transporters but is phosphorylated only very slowly (16). It has thus been used to estimate regional glucose distribution volumes in brain, and its slower plasma clearance kinetics due to tubular reabsorption is an advantage for this purpose. Tritium-labeled 3-O-methyl-d-glucose is commonly used for similar purposes in laboratory experiments. Another glucose derivative substituted at C3, 3-deoxy-3-fluoro-d-glucose, is also well transported (17) but is poorly phosphorylated (8). However, 3-deoxy-3-18F-fluoro-d-glucose appears to be less suitable for PET experiments than 3-O-11C-methyl-d-glucose because several metabolic products are also formed from 3-deoxy-3-fluoro-d-glucose, at least in heart (18). The structural requirements for interaction of carbohydrates with facilitative carriers that were established many years ago on the basis of studies in erythrocytes (19,20) suggest that 3-deoxy-3-iodo-d-glucose or any analog with a larger group at C3 will not be well transported. This expectation is supported by recent studies (21).
The C6 position is also tolerant in terms of facilitative transport of glucose analogs, and 6-deoxy-d-glucose, 6-deoxy-6-fluoro-d-glucose, and 6-deoxy-6-iodo-d-glucose all appear to be transported. The data of Koumanov et al. (22) are consistent with facilitated transport of 6-deoxy-6-123I-iodo-d-glucose into the heart and brain. Although these authors concluded that this radioiodinated sugar was promising for clinical studies, its uptake in brain and heart was lower than the uptake of labeled 3-O-methyl-d-glucose.
CURRENT UNDERSTANDING OF GLUCOSE TRANSPORT
Since the first report of the cloning of a mammalian glucose transporter in 1985 (23), molecular biologic techniques have helped to confirm the existence of 2 very different families of mammalian glucose transporters, known as SGLTs and GLUTs (24). One family comprises molecules that are sodium glucose cotransporters (SGLTs) that are believed to have 14 transmembrane domains. These couple the movement of sodium down a gradient with the pumping of glucose up a gradient. The high Na+ outside gradient used to drive concentrative glucose transport is maintained by the sodium, potassium adenosine triphosphatase in the basolateral membrane. The prototype, SGLT1, is responsible for transport of glucose out of the small intestine across the brush border membrane of intestinal epithelial cells, but is thought to normally play a relatively minor role in reabsorption of glucose in the kidney. SGLT1 moves 2 sodium ions into epithelial cells per glucose molecule and so can pump glucose against a high concentration gradient. A related transporter, SGLT2, is found in the first (S1 and S2) part of the proximal kidney tubule, where it pumps glucose from blood filtrate through the apical membrane of epithelial cells. SGLT2 is believed to exhibit a Na+-to-glucose ratio of 1:1 and pumps glucose from a relatively high glucose concentration—initially, the plasma concentration of about 5 mmol/L. It is believed to be responsible, in euglycemic individuals, for the bulk of glucose reabsorption. Further along the tubule, in the S3 domain, SGLT1 further reduces the glucose concentration to the near-zero level found in the fully formed urine. This picture of sequential operation of SGLT2 and SGLT1 is logical because it avoids the use of the more energetically expensive SGLT1 for the bulk of the glucose transport. There is evidence for at least one more member of the SGLT family in kidney (25).
The other family of glucose transporters is composed of facilitative transporters whose prototype is termed GLUT1 and which is capable only of carrying glucose down a gradient. The GLUT molecules have amino acid sequences consistent with 12 transmembrane domains. GLUT1 is responsible for glucose transport into erythrocytes and through the blood–brain barrier and is upregulated in many tumors. A syndrome resulting from GLUT1 deficiency has been recognized (26) and can involve spasticity, developmental delay, and microcephaly. A recent article (27) describes the use of 18F-FDG PET in diagnosis of this syndrome. GLUT2 is found in tissues that include the liver and pancreatic β-cells. Deficiency of GLUT2 leads to a form of glycogen storage disease known as Fanconi–Bickel syndrome (28) that involves hepatic glycogen accumulation and a distinctive tubular nephropathy. These disorders reflect roles of GLUT2 in transporting newly synthesized glucose out of hepatocytes and in transporting glucose through the basolateral membranes of tubular and intestinal epithelial cells. Its affinity for glucose is lower than that of GLUT1, but it has a broader substrate specificity and also transports other sugars such as fructose and glucosamine. GLUT3 has a high affinity for glucose and is the major form found in neurons. GLUT4 is the insulin-sensitive transporter found in muscle tissue. GLUT1–GLUT4 are collectively termed class I facilitative glucose transporters. At least 8 other members of the GLUT family have been identified (24). They include GLUT5, which is responsible for transport of fructose at the apical membrane of small intestinal epithelial cells. Thus, the absorption of dietary fructose relies entirely on facilitative transport, unlike the situation for glucose, but both sugars use different transporters at the apical and basolateral poles of the epithelial cells.
The complete picture of sugar transport in some tissues, perhaps most tissues, can be complex. Thus, recent immunohistochemical studies in rodents have demonstrated the presence of at least 6 members of the GLUT family in the brain. GLUT1 is found in endothelial cells and controls entry of glucose into the brain, whereas GLUT3 and GLUT4 are expressed in a regionally specific manner in neurons (29). Glia contain GLUT1, but with a distinctive pattern of posttranslational glycosylation (30). Poppe et al. (31) reported messenger RNA for SGLT1 in brain. To make things even more complicated, a very recent article has suggested that, in addition to the facilitative GLUT isoforms and concentrative Na+-linked glucose transporters, brain contains a novel concentrative H+-linked glucose transporter (32).
As mentioned above, the reabsorption of glucose is completed by its passage through the basolateral membranes of tubule cells via the GLUT2 carrier. It appears that methyl-d-glucosides accumulated by epithelial cells can also be reabsorbed into the blood, because the fraction of the injected 11C appearing in urine is <5% of that appearing in urine after administration of methyl-l-glucosides (Table 2 in Bormans et al. (1)). Thus, the methyl-d-glucosides seem to be substrates for GLUT2. However, there is some confusion on this point in the literature. It is generally accepted that methyl-d-glucosides are not substrates for GLUT1 (33), and the small degree of uptake by brain and erythrocytes reported by Bormans et al. supports this view. It is possible that species differences in GLUTs or tissue-specific posttranslational modification (34) could account in part for this confusion. Because β-11C-methylglucoside was retained by kidney to a much greater extent than α-11C-methylglucoside (kidney-to-blood ratio of about 8 compared with about 2 at 30 min, and radioactivity remaining in the kidneys of 31% and 8% injected dose, respectively), the suggestion of Bormans et al. that β-11C-methylglucoside is a much poorer substrate for GLUT2 than the α-glucoside is compelling.
The demonstration by imaging plate autoradiography that, after intravenous administration of a mixture of α- and β-11C-methyl-d-glucosides, 11C is localized in the kidney cortex and outer medulla is consistent with retention in tubular cells. Judging by the tissue distribution data, this represents almost exclusively β-11C-methyl-d-glucoside, though it would be useful if this were confirmed by autoradiography after administration of pure β-11C-methyl-d-glucoside rather than a mixture of anomers. From a technical point of view, this is an interesting and challenging example of ex vivo autoradiography using 11C. Similar experiments do not appear to have been conducted using longer-lived isotopes, though the autoradiographic measurement of uptake of α-14C-methyl-d-glucoside in explants of fetal and newborn mouse kidney was recently reported (35).
Henry et al. (36) showed that methyl 6-deoxy-6-iodo-d-glucoside is transported into rat brain, erythrocytes, and neonatal cardiomyocytes (though to a lesser extent than the 3-O-methylglucose), implying that this iodine-substituted methyl glucoside analog is a substrate for both GLUT1 and GLUT4. Although it would seem unlikely, it appears that the change in structure at C6 compensates for the change in structure at C1, in terms of ability to be transported by GLUT isoforms. It is interesting that these workers did not find a similar difference in kidney uptake between α- and β-methyl-6-deoxy-6-iodo-d-glucoside.
PROSPECTS FOR NEW RADIOPHARMACEUTICALS FOR IMAGING SUGAR TRANSPORT
PET and SPECT markers for the process of glucose transport might be useful in early diagnosis of neurologic diseases where transport is compromised, such as Alzheimer’s disease (37) or Huntington’s disease (38). Additionally, because GLUT1 is well expressed by many tumors in tissues where a lower expression of a different isoform is normally predominant (39), a transported analog, especially if GLUT1 selective, might be a useful tumor marker. Issues that would have to be resolved include competition between radiotracer and glucose for uptake in target tissue and uptake into erythrocytes that would lower tissue-to-background ratios, at least for extracerebral tissues. A general problem with radiotracers that are substrates for facilitative transporters is the absence of any trapping mechanism. According to Bormans et al. (1), trapping of β-methyl-d-glucoside in the kidney is provided by the fact that there is a greater inward energy-linked flux into tubule cells than the outward facilitative flux.
Despite this problem, some work has continued in this area. Brunet-Desruet et al. (40) synthesized 2 radioiodinated acetal derivatives of glucose, but these were not transported by either GLUT1 or GLUT4. Xu et al. (41) have developed glucose analogs related to the antibiotic nojirimycin that contain a 6-member ring with a nitrogen atom instead of an oxygen atom. These compounds bind to enzymes involved in glycosylation reactions and are potential tumor-imaging agents but were not transported into brain.
HIGH-AFFINITY NONTRANSPORTED RADIOLIGANDS FOR GLUCOSE TRANSPORTERS
It was suggested over a decade ago that high-affinity ligands could be developed for GLUT isoforms (42,43). Experience with neurotransmitter receptor and transporter radioligands suggests that dissociation constants in the nanomolar range would be required. Several compounds are known to interact with glucose transporters with much higher affinities than glucose, including phloretin, phlorizin, forskolin analogs, and cytochalasin B (20,42). These compounds and others, such as 2,3-dioxoindole (44), might be leading compounds for development of a more potent inhibitor. Potential advantages of this approach over using reversibly transported substrates would be less competition with glucose and the possibility of more stable uptake. A disadvantage is that a radioligand would be sensitive to the concentration of transporter, rather than flux through the transporter, as with a transported analog, and might be insensitive to factors that regulate transporter flux. Nevertheless, subtype-specific high-affinity radioligands capable of binding in vivo to glucose carriers might have wide applicability.
CONCLUSION
Future studies with α- and β-methyl-d-glucosides should include experiments with cells transfected with GLUT2 to seek support for the hypothesis that the β-compound accumulates in kidney because it is a poor substrate for efflux via this GLUT isoform. Also, if it is true that most of the glucose in glomerular filtrate is reabsorbed through SGLT2, then experiments should be conducted in oocytes expressing this carrier rather than SGLT1, although it is recognized that this is technically more difficult (45). In addition, although Bormans et al. (1) report the biodistribution of 11C-methylglucosides in the mouse, their studies of the transport through SGLT1 were done with the human cloned transporter. Though there is significant homology between mouse and human genes (35), it is at least possible that α- and β-methyl-d-glucosides exhibit different relative kinetics with mouse and human SGLT1. Another question is the possible role of facilitative transporters other than GLUT2 and Na+-linked transporters other than SGLT1 and SGLT2. Several secondary questions should also be addressed, such as whether alteration of blood glucose concentrations would affect the transport of β-methyl-d-glucosides from the tubular cells. Nevertheless, Bormans et al. should be congratulated for an imaginative study using a readily prepared positron-emitting tracer that raises new questions and has potential clinical applicability. Their article illustrates the use of sophisticated techniques, including chiral separation, autoradiography with 11C, and electrophysiologic measurements of a specific transporter artificially expressed in oocytes, to explain tissue distribution data.
Acknowledgments
This work was conducted at the Brookhaven National Laboratory under contract DE-AC02-98CH10886 with the U.S. Department of Energy and supported by its Office of Health and Environmental Research.
Footnotes
Received Mar. 25, 2003; accepted Mar. 26, 2003.
For correspondence or reprints contact: S. John Gatley, PhD, Medical Department, Brookhaven National Laboratory, Building 490, Upton, NY 11973.
E-mail: gatley{at}bnl.gov
REFERENCES
In this issue




{kind=link}
Jump to section
- Article
- HISTORICAL BACKGROUND
- GLUCOSE ANALOGS THAT ARE TRANSPORTED AND PHOSPHORYLATED
- LABELED GLUCOSE ANALOGS THAT ARE TRANSPORTED BUT NOT PHOSPHORYLATED
- CURRENT UNDERSTANDING OF GLUCOSE TRANSPORT
- PROSPECTS FOR NEW RADIOPHARMACEUTICALS FOR IMAGING SUGAR TRANSPORT
- HIGH-AFFINITY NONTRANSPORTED RADIOLIGANDS FOR GLUCOSE TRANSPORTERS
- CONCLUSION
- Acknowledgments
- Footnotes
- REFERENCES
- Figures & Data
- Info & Metrics