


Abstract
There has been considerable interest in the development of PET radioligands that are useful for imaging serotonin transporter (SERT) in the living human brain. For the last decade, 11C-(+)McN5652 has been the most promising PET agent for studying SERT in humans. However, this agent has some limitations. Recently, a new promising SERT PET radioligand, 3-11C-amino-4-(2-dimethylaminomethylphenylsulfanyl)benzonitrile, has been reported. We recently reported the synthesis of a new 18F-labeled SERT PET radioligand, N,N-dimethyl-2-(2-amino-4-18F-fluorophenylthio)benzylamine (4-18F-ADAM), which may have advantages over 11C-labeled radioligands. The purpose of this study was to evaluate this newly developed 18F-labeled PET radioligand as a promising agent for studying SERT in the living human brain. Methods: This agent was evaluated by studying its in vitro binding to different monoamine transporters, its in vivo biodistributions in rats, its integrity and pharmacologic profiles in rat brain, and its distribution in a female baboon brain. Results: In vitro binding assays showed that 4-F-ADAM displayed high affinity to SERT sites (inhibition constant = 0.081 nmol/L, using membrane preparations of LLC-PK1 cells expressing the specific transporter) and showed more than 1,000- and 28,000-fold selectivity for SERT over norepinephrine transporter and dopamine transporter, respectively. Biodistribution of 4-18F-ADAM in rats showed a high initial uptake and slow clearance in the brain (2.13%, 1.90%, and 0.95% injected dose per organ at 2, 30, and 60 min after intravenous injection, respectively), with the specific binding peaking at 2 h after injection (hypothalamus/cerebellum = 12.49). The uptake in blood, muscle, lung, kidney, and liver was also initially high but cleared rapidly. The radioactivity in the femur increases with time for 4-18F-ADAM, indicating that in vivo defluorination may occur. In vivo metabolism studies in rats showed that 4-18F-ADAM was not metabolized in rat brain (>96% of radioactivity was recovered as parent compound at 1 h after injection). However, it metabolized rapidly in the blood. Less than 7% of the radioactivity recovered from plasma was the parent compound, with the majority of radioactivity in the plasma not extractable by ethyl acetate. Blocking studies showed significant decreases in the uptake of 4-18F-ADAM in the brain regions (hypothalamus, hippocampus, and striatum) where SERT concentrations are high when rats were pretreated with (+)McN5652 (2 mg/kg 5 min before intravenous injection of 4-18F-ADAM). However, changes in the uptake of 4-18F-ADAM in these brain regions were less significant when rats were pretreated with either methylphenidate or nisoxetine. The baboon study showed that uptake of 4-18F-ADAM in the midbrain peaked at ∼1 h after injection and then declined slowly. The ratios of the radioactivity in the midbrain to that in the cerebellum (where the concentration of SERT is low) at 2 and 3 h after injection were 3.2 and 4.2, respectively. Conclusion: 4-18F-ADAM is suitable as a PET radioligand for studying SERT in the living brain. Further characterization of this new radioligand in humans is warranted.
Abnormalities in the serotonin transporter (SERT) have been implicated in several neurologic and psychiatric disorders, such as the parkinsonian disorder, depression, suicide, schizophrenia, drug addiction, and eating disorders (1,2). In addition, SERT is the primary target for widely prescribed antidepressant agents (3). In order to study these neurologic and psychiatric disorders, and the mode of action of antidepressant agents in humans, it is desirable to have high-affinity and high-specificity SERT radioligands for imaging SERT with either SPECT or PET (4).
Several radioligands have been developed and evaluated for PET. These include 18F-labeled paroxetine and fluoxetine and 11C-labeled cyanoimipramine, citalopram, sertraline, and fluoxetine (4). All these radioligands have been found not to be ideal agents for PET studies of SERT because of their low specific-to-nonspecific binding ratios in vivo. For the last decade, 11C-(+)McN5652 (Fig. 1) (5) has been the most promising PET agent for studying SERT in humans (6–10). However, this agent has high nonspecific binding and has only moderate signal contrast in human PET studies (7–9). Additionally, its pharmacokinetics are not optimal because of the short half-life of 11C. Labeling this compound with 18F does not improve its imaging properties (11,12).
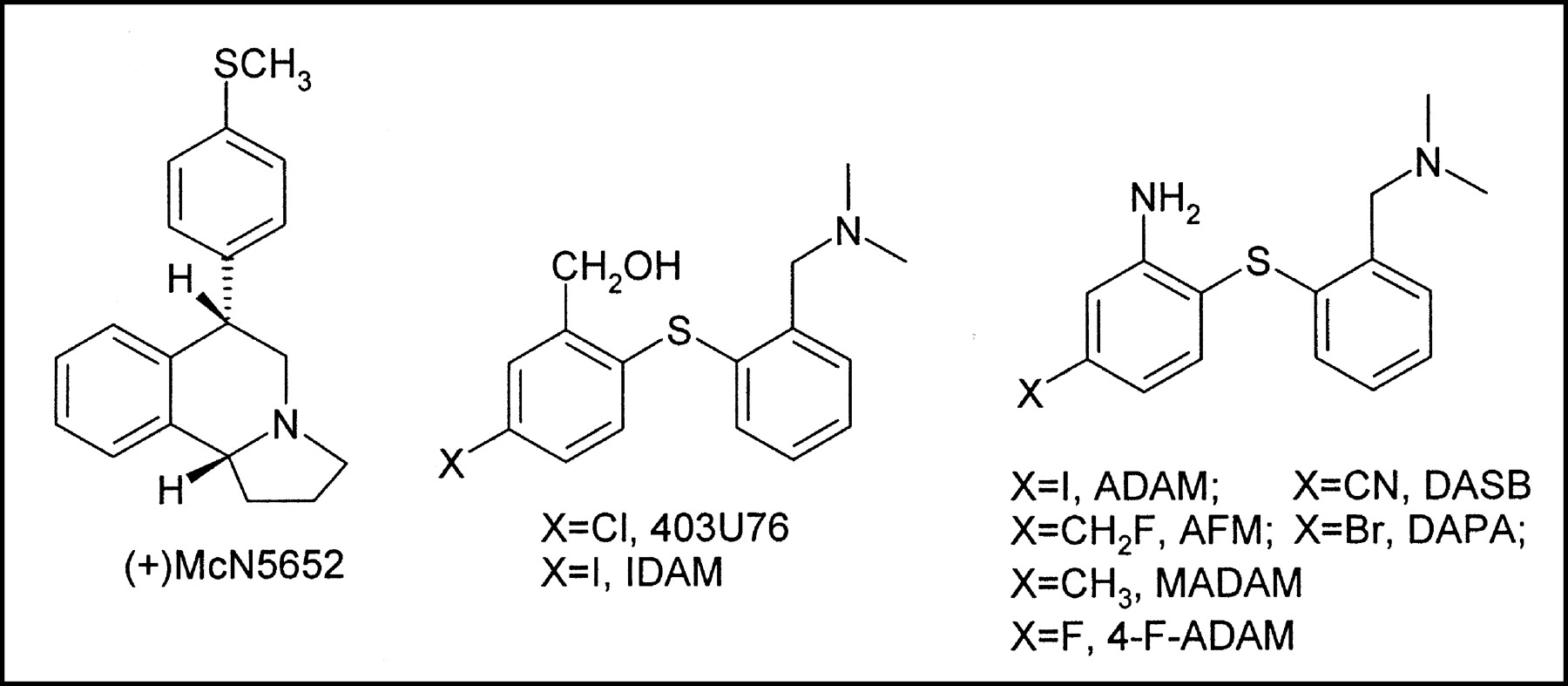
Chemical structures of (+)McN5652, 403U76, IDAM, ADAM, DASB, AFM, DAPA, MADAM, and 4-F-ADAM.
Recently, a new class of potent serotonin reuptake inhibitors, namely the N,N-dimethyl-2-(arylthio)benzylamines, has been reported to possess very high selectivity and affinity for SERT over norepinephrine and dopamine transporter (DAT) binding sites (13,14). Analogs have been labeled with 123I(5-iodo-2-((2-((dimethylamino)methyl)phenyl)thio)benzyl alcohol [IDAM], 2-((2-((dimethylamino)methyl)phenyl)thio)-5-iodophenylamine [ADAM], and 5-iodo-2-(2-dimethylaminomethylphenoxy)-benzyl alcohol)) (15–17) and 11C(11C-IDAM, 11C-ADAM, 11C-N,N-dimethyl-2-(2-amino-4-methoxyphenylthio)benzylamine, 3-11C-amino-4-(2-dimeth-ylaminomethylphenylsulfanyl)benzonitrile [11C-DASB], 11C-N,N-dimethyl-2-(2-amino-4-methylphenylthio)benzylamine [MADAM], 11C-N,N-dimethyl-2-(2-amino-4-fluoromethylphenylthio)benzylamine (11C-AFM), and 11C-5-bromo-2-[2-(dimethylaminomethylphenylsulfanyl)]phenylamine [DAPA] (Fig. 1) (18–26) as SERT SPECT and PET agents. Compound 11C-DASB, in particular, has been found to be highly suitable for probing the serotonin reuptake sites and drug occupancy for serotonin reuptake inhibitors with PET in humans (27,28). However, 18F has some advantages over 11C. First, it has lower positron energy than does 11C (0.635 vs. 0.96 MeV); therefore, there is less spatial resolution smearing. Second, because of its long half-life, 18F enables one to establish a more optimized scanning protocol. Third, 18F is convenient for radiosynthesis. Finally, the radioligands can be transported off site if a cyclotron is not available. We and others have therefore attempted to label this class of compound with 18F. One of the first 18F-labeled compounds, N,N-dimethyl-2-(2-fluoromethyl-4-iodophenylthio)benzylamine (F-IDAM), has a high affinity toward SERT (inhibition constant [Ki] = 0.003 nmol/L) and shows excellent initial brain uptake and binding properties in rats. This compound, however, defluorinates rapidly in baboons and is not the ligand of choice for PET studies of SERT in baboons or humans (29). Another 18F-labeled SERT agent, (2-[2-amino-4-chloro-5-fluorophenyl]thio)-N,N-dimethylbenzenemeth-anamine (18F-ACF), also shows excellent brain uptake and selective localization after intravenous injection in rats (30). Other fluorinated analogs of IDAM and ADAM have also shown high affinity toward SERT (24,25). Two of these compounds, N,N-dimethyl-2-(2-amino-4-fluorophenylthio) benzylamine (4-F-ADAM, or AFA) and AFM (Ki = 4.8 and 1.8 nmol/L, respectively, for SERT expressed in cloned human transporters) (Fig. 1), have been labeled with 11C and showed excellent brain uptake and a high degree of specific binding in rats (24). To improve the quality of PET studies and the feasibility of transporting the radioligands off site, we have labeled 4-F-ADAM with 18F (31). At the same time, 18F-AFM also has been prepared and evaluated in baboons and found to be a potential radioligand for studying SERT using PET (32). We report here the characterization of N,N-dimethyl-2-(2-amino-4-18F-fluorophenylthio)benzylamine (4-18F-ADAM) as a useful agent for studying SERT in the brain.
MATERIALS AND METHODS
Synthesis of No-Carrier-Added (NCA) 4-18F-ADAM
NCA 4-18F-ADAM was synthesized as previously reported (31). Briefly, the precursor, N,N-dimethyl-2-(2,4-dinitrophenylthio)benzylamine, in dimethyl sulfoxide was added to dried potassium 18F-fluoride/Kryptofix 222 (Merck) (K-18F/K2.2.2), and the solution was heated at 120°C for 15 min. The intermediate, N,N-dimethyl-2-(2-nitro-4-18F-fluorophenylthio)benzylamine, was purified with a C18 Sep-Pak (Waters) and reduced with Cu(OAc)2-NaBH4 in EtOH. The final product was purified with high-performance liquid chromatography (C18, 10 × 250 mm, Luna [2] [Phenomenex]; CH3CN:0.1 mol/L HCO2NH4 [30:70] containing 0.3%, by volume, of acetic acid; 5 mL/min). The fraction containing 4-18F-ADAM was collected, evaporated to dryness, formulated in saline, and filtered through a 0.22-μm filter (Millipore) into a multiinjection vial.
In Vitro Binding Assay
The in vitro binding assays were performed in LLC-PK1 cell membrane homogenates that express SERT, DAT, and norepinephrine transporter (NET), respectively, as described previously (16). Typically, aliquots of membrane suspensions (100 μL, corresponding to 30–40 μg of protein) were mixed with 50 mmol/L Tris-HCl (pH 7.4), 120 mmol/L NaCl, and 0.1% bovine serum albumin (all from Sigma); 0.4 nmol/L 125I-IPT or 125I-IDAM; and 8–10 concentrations (10−10 to 10−5 mol/L) of 4-F-ADAM. Nonspecific binding was defined with 10 μmol/L 2β-carbomethoxy-3β-(4-fluorophenyl)tropane for DAT and NET assays and 1 μmol/L IDAM for SERT assays. Incubation was performed for 1 h at room temperature, and the bound ligand was collected on glass fiber filters presoaked with 1% polyethylenimine (Sigma) and counted in a γ-counter (model 5000; Packard). The results of competition experiments were subjected to nonlinear regression analysis using EBDA software (Elsevier-BIOSOFT).
Measurement of Partition Coefficient (log P)
The log P of 4-18F-ADAM was determined by a published method (16). Basically, 3 g each of 1-octanol and phosphate buffer (pH 7.4, 0.01 mol/L) were pipetted into three 10-mL test tubes containing 4-18F-ADAM (about 800,000 cpm). The test tubes were capped, stirred with a vortex mixer for 5 min at room temperature, and then centrifuged. Two weighed samples (2 g each) from the organic and aqueous layers were counted in a well counter. Samples from the 1-octanol phase were repartitioned 2 more times or until consistent log P values were obtained. The log P was calculated as follows: (counts/g in octanol)/(counts/g in buffer). The measurement was repeated 3 times.
Biodistribution of 4-18F-ADAM in Rats
The animal experiments were performed according to a protocol approved by the University of Pennsylvania Institutional Animal Care and Use Committee. The methods for evaluation of 4-18F-ADAM in rats are identical to those for evaluation of 18F-ACF (30). Male Sprague–Dawley rats weighing 280–310 g were used for each biodistribution study. While the rats were under ether anesthesia, 0.2 mL of a saline solution containing 1,480 MBq of 4-18F-ADAM was injected into the femoral vein. The rats were allowed to awaken and then were reanesthesized and euthanized by cardiac excision at 2, 30, 60, 120, or 240 min after injection. Organs of interest were removed, blotted to remove adhering blood, and placed in tared counting vials. The radioactivity of each sample was measured in a NaI detector, and the sample was weighed. The percentage dose per gram or per organ was calculated by comparing the counts in tissues with the counts in aliquots of the injected tracer that had been measured at the same time and diluted 100 times.
Regional brain distribution in rats was measured after an intravenous injection of 4-18F-ADAM. Samples from different brain regions—cortex (frontal plus occipital), striatum, hippocampus, cerebellum, and hypothalamus—were dissected, weighed, and counted. The percentage dose per gram of tissue was calculated by comparing tissue counts with the counts of the diluted initial dose as described above. The specific binding ratio in each region was obtained by dividing the count in the region by that in the cerebellum. The cerebellum was considered devoid of SERT and was used as the background region.
Pharmacologic profile experiments were performed on male Sprague–Dawley rats (230–250 g). The rats were pretreated with monoamine transporter inhibitors ((+)McN5652 for SERT sites, methylphenidate for DAT sites, and nisoxetine for NET sites, respectively; 2 mg/kg in 250 μL of saline) 5 min before intravenous injection of 4-18F-ADAM (592 kBq in 200 μL of saline) in the femoral vein. The brain regions were dissected 2 h after isotope injection and processed as described above.
In Vivo Metabolism of 4-18F-ADAM in Rats
Three male Sprague–Dawley rats (280–310 g) were injected in the femoral vein with 18.5 MBq of 4-18F-ADAM in 200 μL of saline. At 1 h after injection, the rats were euthanized. A sample of blood and the brain were rapidly removed and homogenized in 10 volumes of 50 mmol/L Tris buffer (pH 7.4). The homogenates were then extracted with ethyl acetate (3 × 2 mL). The organic layers were evaporated to near dryness and were analyzed by thin-layer chromatography (silica gel, CH2Cl2:MeOH, 9:1 [v/v]). The spots on the thin-layer chromatography plates were counted using a Phosphor Imager SI (Molecular Dynamics).
PET Study on a Baboon
The PET study was performed on a female baboon (Papio anubis) with her ovaries removed. After overnight fasting, the baboon was immobilized with an intramuscular injection of 1 mL of a 0.9%:0.3% (w/v) mixture of alfaxalone:alfadolone acetate (Saffan; Pittman-Moore) per kilogram of body weight so that peripheral cannulas could be inserted. During scanning, anesthesia was maintained by intravenous infusion of 20% Saffan diluted with saline, at a rate of 40–80 mL/h as needed. Passive inhalation of oxygen (1.5 L/min) was also maintained to keep the oxygen saturation > 95%. An additional intravenous line in a superficial brachial vein was used for hydration (0.9% physiologic saline solution, 5 mL/kg/h), as well as for radioligand administration. A cylindric polycarbonate positioning device equipped with a customized foam head-holder enabled reproducible placement of the animal in the PET scanner.
PET was performed using the G-PET brain scanner developed at the University of Pennsylvania (33). This scanner uses gadolinium orthosilicate crystals incorporated into an Anger-logic detector and operates only in the 3-dimensional or volume imaging mode (no septa) to maximize sensitivity. The scanner has a transverse field of view of 25.6 cm, an axial field of view of 25 cm, and a spatial resolution of 3.7 mm in full width at half maximum.
The baboon was injected (intravenous bolus) with 106 MBq of 4-18F-ADAM. Imaging was initiated in a dynamic mode by acquiring twelve 15-min images over a period of 3 h. The data were acquired and reconstructed with a fully 3-dimensional iterative image reconstruction algorithm (row-action maximum-likelihood algorithm) with system attenuation correction incorporated in the algorithm (34). Corrections for scatter and randoms were applied immediately before image reconstruction, using a background subtraction technique based on tail-fitting of the emission sinogram data outside the body (33). Attenuation correction was based on a singles transmission scan obtained using a point source of 137Cs singles γ-rays (662 keV). This source rotates around the field of view in 45 s. The transmission data were acquired immediately after the emission scan using energy discrimination to separate the transmission events (662 keV) from the emission events (511 keV). The transmission scan was reconstructed and tissue threshold was applied before attenuation correction was performed, so as to preserve measured attenuation variations in the lung region and lung/tissue wall, while minimizing noise from measured soft-tissue density fluctuations (35).
Guided by a baboon atlas (36), we drew regions of interest on the midbrain, striatum, occipital cortex, frontal cortex, skull, and cerebellum of the PET images. Mean counts per pixel were calculated and corrected for the effects of decay.
RESULTS
N,N-Dimethyl-2-(2-amino-4-fluorophenylthio)benzylamine was prepared in a multistep synthesis as reported (31). Briefly, reaction of 2-chloro-5-fluoronitrobenzene with 2-thio-N,N-dimethylbenzamide gave N,N-dimethyl-2-(2-nitro-4-fluorophenylthio)benzamide in 60% yield. Reduction of N,N-dimethyl-2-(2-nitro-4-fluorophenylthio)benzamide with BH3-THF complex gave N,N-dimethyl-2-(2-nitro-4-fluorophenylthio)benzylamine and N,N-dimethyl-2-(2-nitro-4-fluorophenylthio)benzylamine·BH3. Reduction of N,N-dimethyl-2-(2-nitro-4-fluorophenylthio)benzylamine with SnCl2 gave N,N-dimethyl-2-(2-amino-4-fluorophenylthio)benzylamine in 30% yield.
NCA 4-18F-ADAM was synthesized by nucleophilic substitution of the corresponding nitro- precursor with K-18F/K2.2.2 followed by reduction with Cu(OAc)2-NaBH4 in EtOH and purification with high-performance liquid chromatography in ∼5%–10% yield. The radiochemical yield was not optimized. The radiochemical purity was >98%, and the specific activity was 22.2 GBq/μmol (31). The log P of 4-18F-ADAM was determined to be 2.73 (n = 3).
In vitro binding of 4-F-ADAM to monoamine transporters expressed in LLC-PK1 cells showed high affinity toward SERT (Ki = 0.081 nmol/L, Table 1). Binding affinities toward DAT and NET were more than 1,000-fold lower (2,267 and 117 nmol/L for DAT and NET, respectively), suggesting that 4-F-ADAM is a highly potent and selective ligand for SERT.
Selectivity of Ligands for Monoamine Transporters (Ki, nmol/L)
Table 2 shows the distribution of radioactivity in various rat tissues at 2, 30, 60, 120, and 240 min after the injection of 4-18F-ADAM. The initial brain uptake was high (2.13% ± 0.33% injected dose per organ, or 1.08% ± 0.18% injected dose per gram at 2 min after injection). The radioactivity washed out from the brain slowly, with 1.90%, 0.95%, and 0.41% injected dose per organ at 30, 60, and 120 min, respectively. Similar to other lipophilic compounds, uptake of 4-18F-ADAM in the muscle, lung, kidney, and liver was initially high but cleared from these organs rapidly. The clearance from blood was also rapid. The activity in the femur increases with time.
Biodistribution of 4-18F-ADAM in Rats
Radioactivity in all brain regions was high and similar at 2 min after injection. However, the washout rate from these regions was different, with cerebellum (region with low SERT binding sites) being the fastest (Table 3). The ratio of specific binding to nonspecific binding increased with time, reaching a peak at 2 h after injection. The ratios of hypothalamus (region high in SERT binding sites) to cerebellum were 5.57 and 12.49 at 1 and 2 h, respectively, after injection (Table 4).
Regional Brain Distribution of 4-18F-ADAM in Rats
Ratios of Tissue-to-Cerebellum vs. Time After 4-18F-ADAM Injection in Rats
Pharmacologic profiles of the binding of 4-18F-ADAM in rat brain regions were assessed by pretreating rats with various monoamine transporter inhibitors ((+)McN5652 for SERT, methylphenidate for DAT, and nisoxetine for NET). Table 5 showed that there were significant decreases in the uptake of 4-18F-ADAM in the brain regions (hypothalamus, hippocampus, and striatum) where SERT concentrations are high when rats were pretreated with (+)McN5652 (2 mg/kg 5 min before intravenous injection of 4-18F-ADAM). However, there were less significant changes in the uptake of 4-18F-ADAM in these brain regions when rats were pretreated with either methylphenidate or nisoxetine. The ratios (Table 6) of SERT-rich regions to cerebellum were significantly decreased when rats were pretreated with (+)McN5652, suggesting that in vivo uptake of 4-18F-ADAM in rat brain was specific to SERT binding sites.
Effects of Pretreatment with Monoamine Transporter Inhibitor on Specific Binding of 4-18F-ADAM in Rats
Tissue-to-Cerebellum Ratios After Pretreating Rats with Monoamine Transporter Inhibitors
In vivo metabolism studies in rats showed that there were no metabolites in rat brain 1 h after injecting 4-18F-ADAM. More than 96% of the radioactivity in the brain was the unchanged parent compound. However, only ∼7% of radioactivity in the plasma existed as the parent compound. The majority of radioactivity in the plasma was not extractable by ethyl acetate.
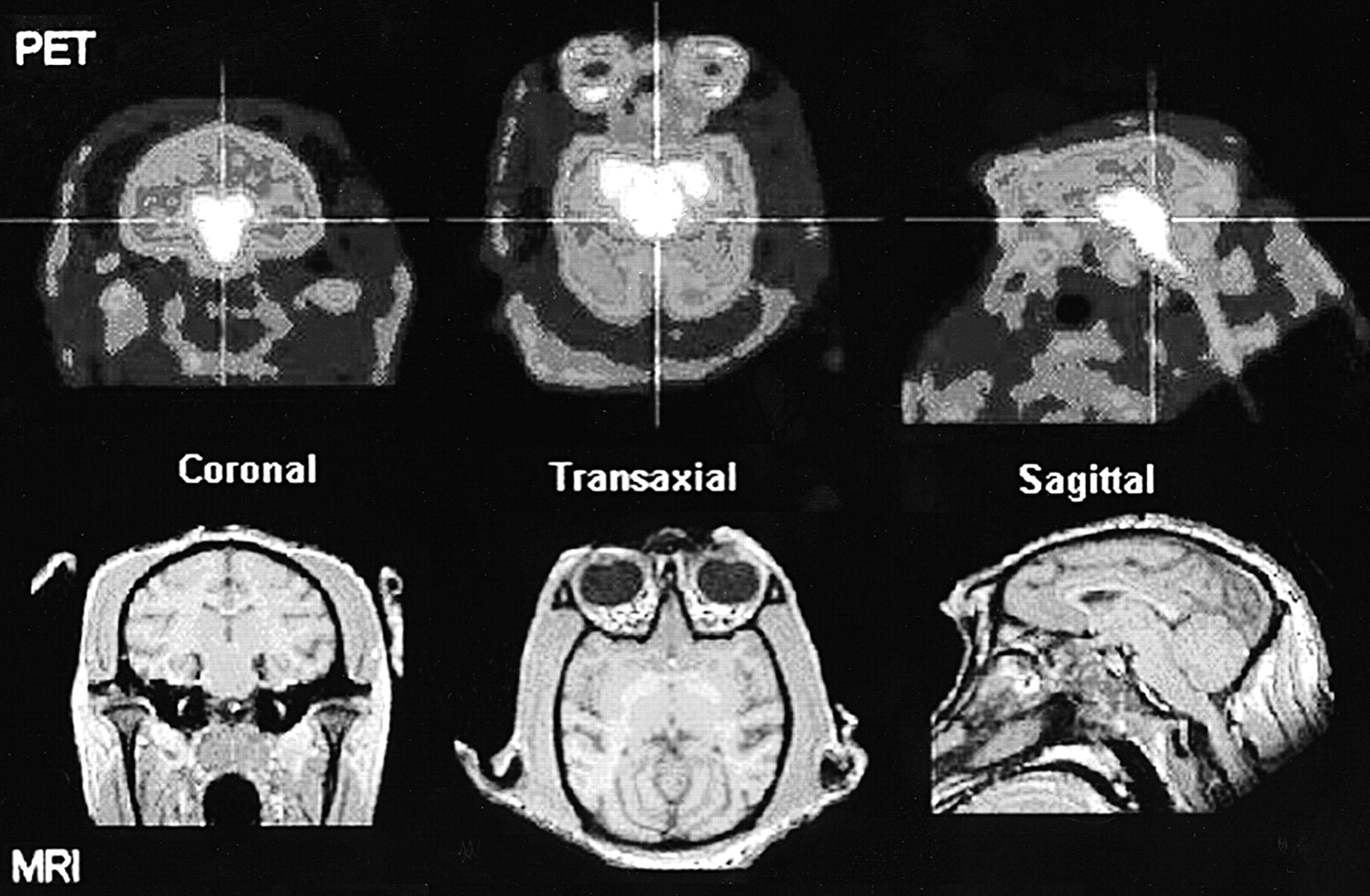
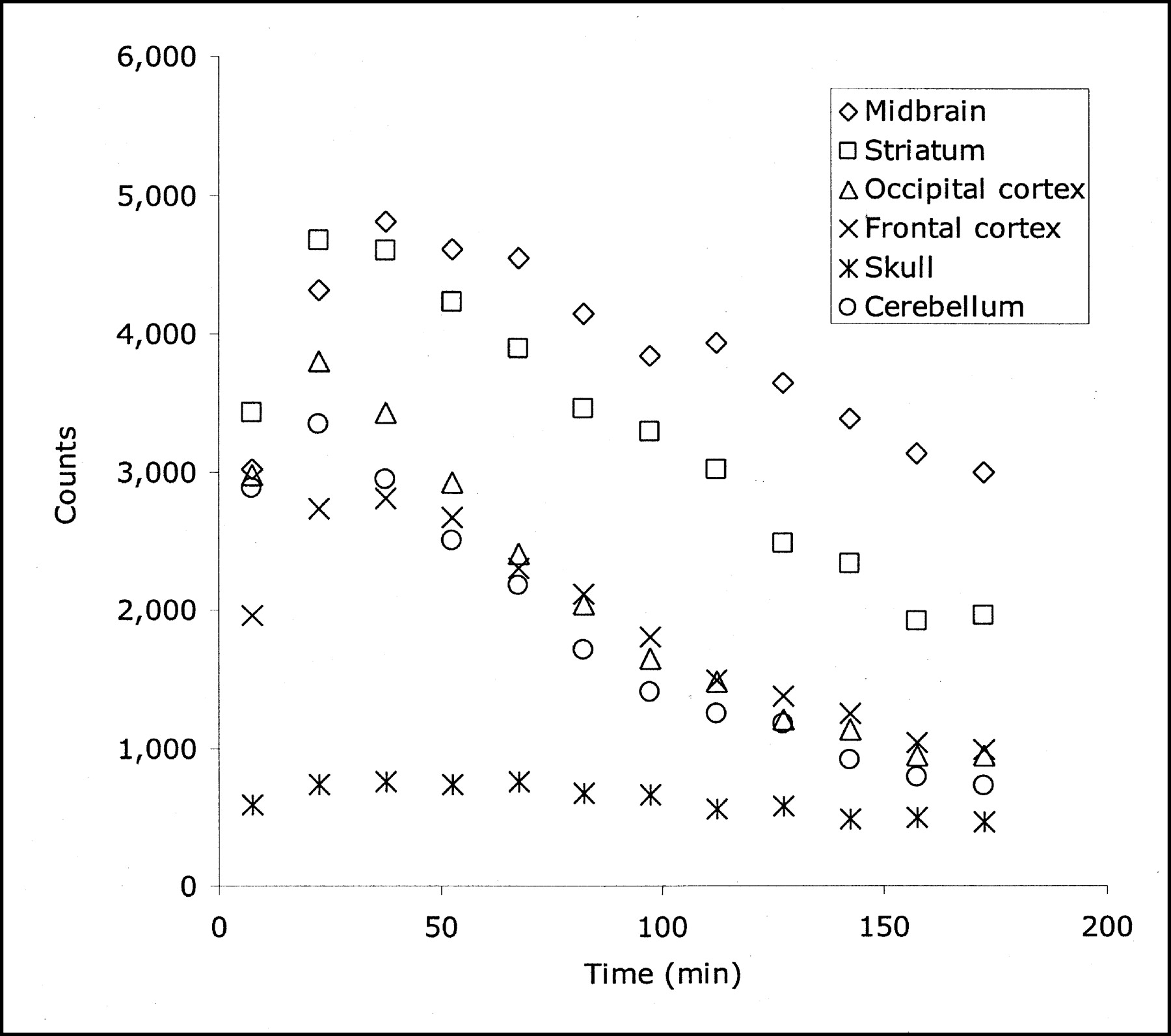
The distribution of radioactivity in the baboon midbrain after injection of 4-18F-ADAM is depicted in Figure 2. The top row of this figure shows PET scans taken at the level of the midbrain in 3 orthogonal views 90–120 min after radioligand injection. The bottom row of this figure shows the corresponding MR images at the same level. The time course of the radioactivity in the midbrain, striatum, occipital cortex, frontal cortex, skull, and cerebellum is shown in Figure 3. The uptake of 4-18F-ADAM in the midbrain peaks at ∼1 h after injection and then declines slowly. The ratios of the radioactivity in the midbrain to that in the cerebellum at 2 and 3 h after injection were 3.2 and 4.2, respectively (Fig. 4).
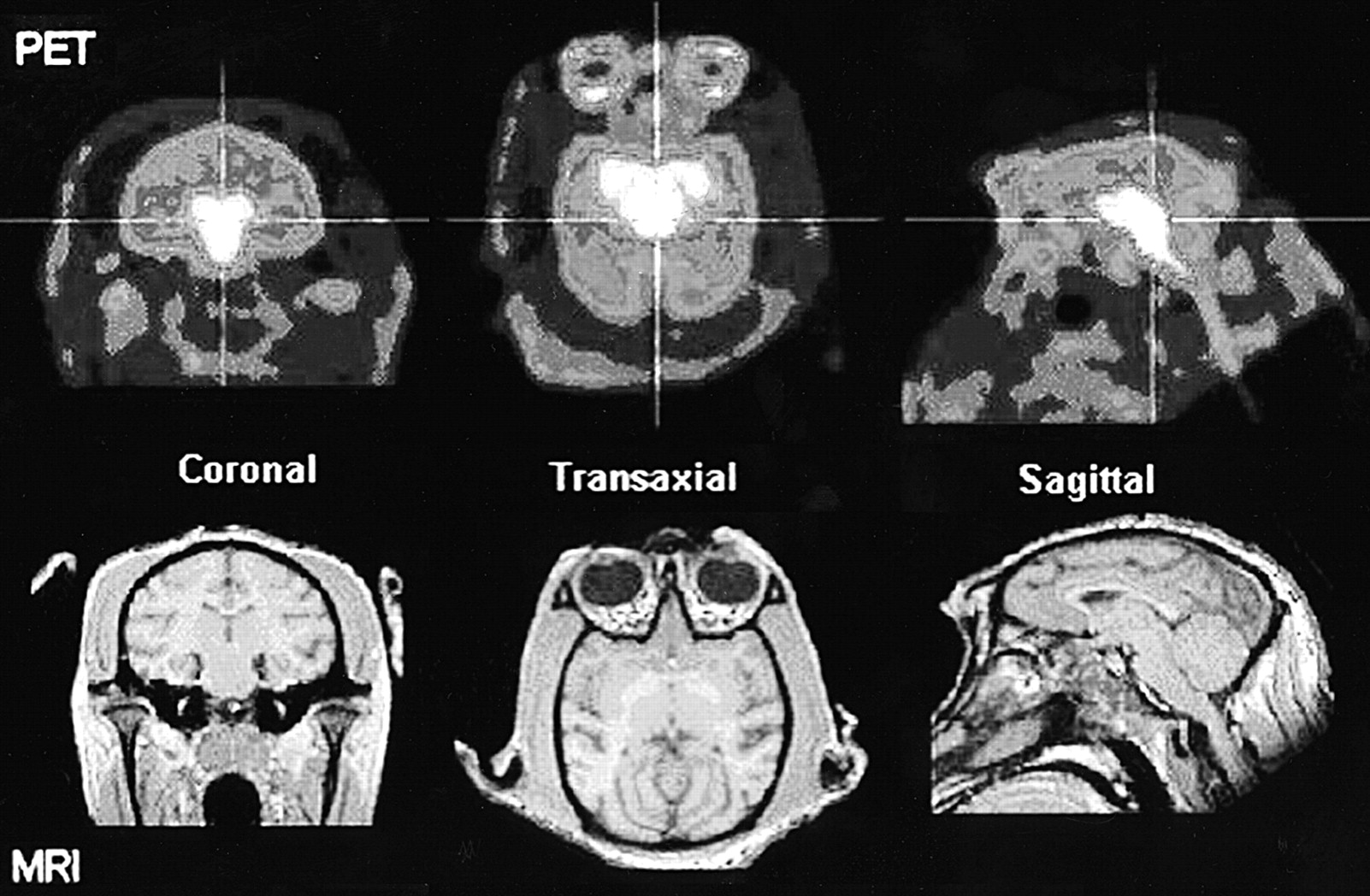
PET images of 4-18F-ADAM in baboon brain and MR images of baboon brain at same level as PET images. PET scan displays specific binding of 4-18F-ADAM to SERT in midbrain region at 90–120 min after radioligand injection. MRI scan reveals anatomic information about midbrain region.
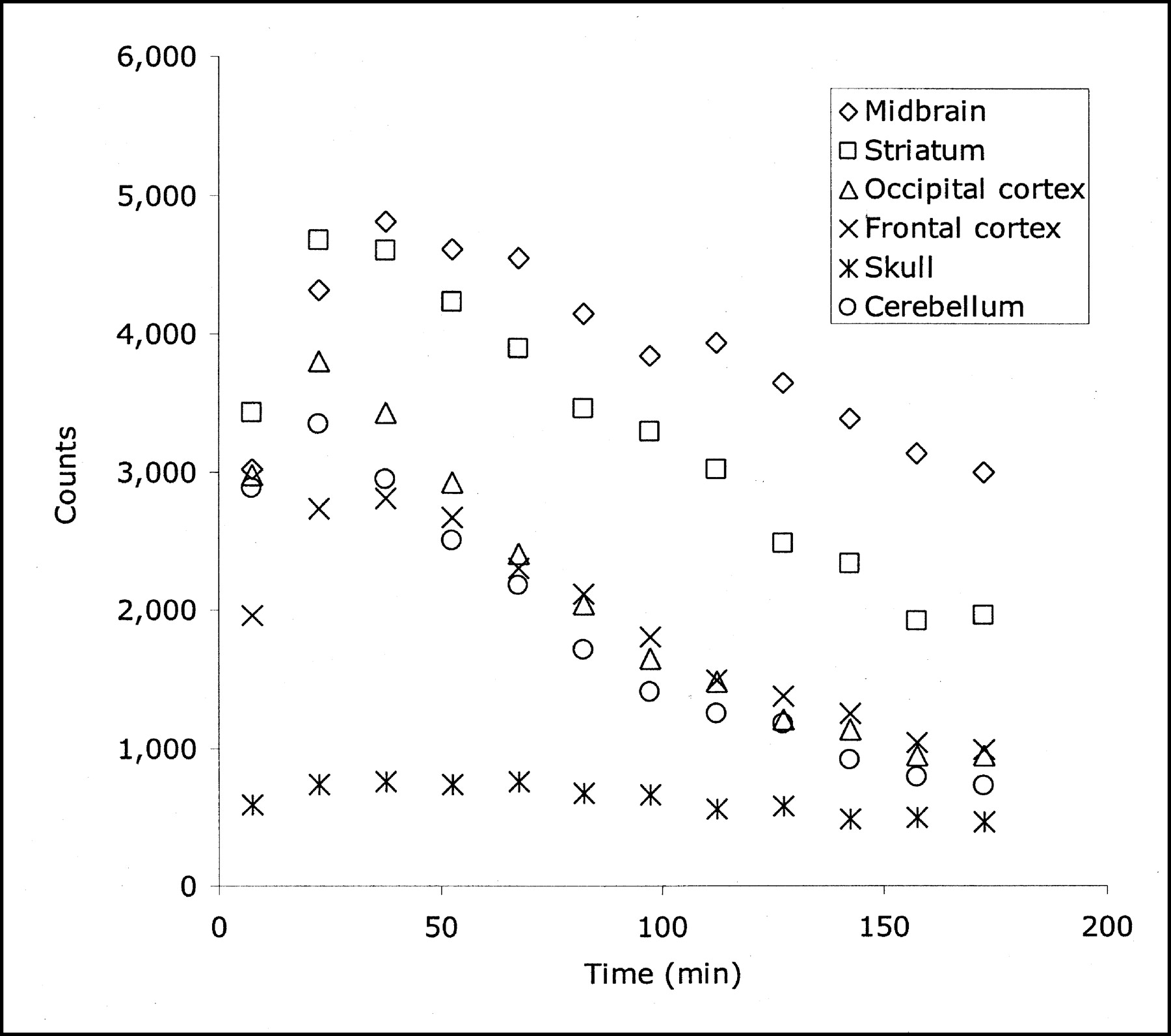
Time–activity curve of 4-18F-ADAM in baboon brain. Curve shows that uptake of 4-18F-ADAM in midbrain region peaks at ∼30–40 min after injection and that defluorination of 4-18F-ADAM may not occur in baboon.
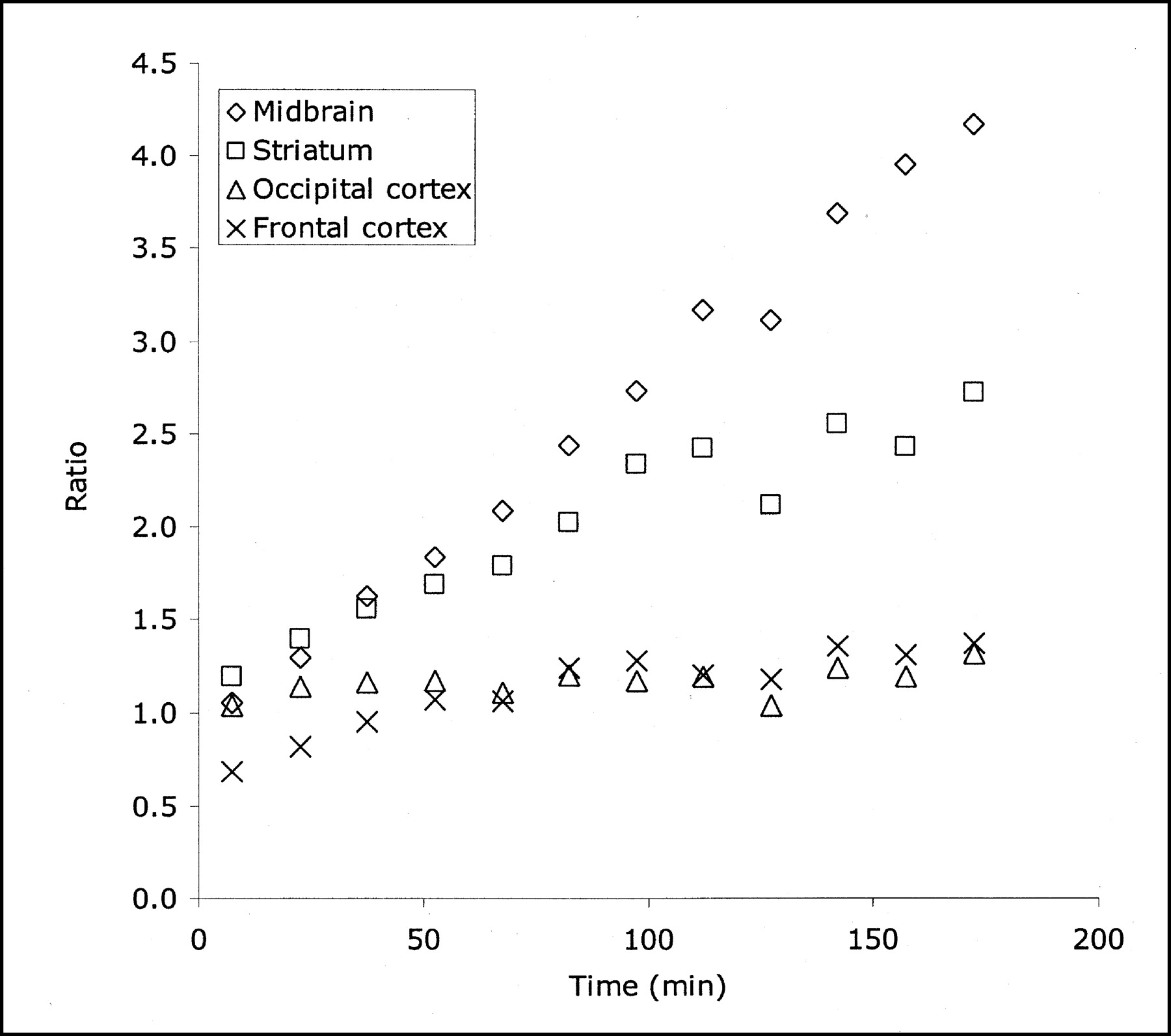
Ratio of different brain regions to cerebellum increases with time in baboon brain after injection of 4-18F-ADAM.
DISCUSSION
Recently, diarylsulfides have been developed as SERT radioligands for SPECT (15–17) and PET (18–26). All these PET radioligands are labeled with 11C, and one of the radioligands, 11C-DASB, has been evaluated in baboons (37) and in humans (27,28). The results show that 11C-DASB may offer some advantages over 11C-(+)McN5652 (37). However, because of its relatively long half-life (110 min) and stable carbon–fluorine bond, a 18F-labeled radiopharmaceutical offers some advantages over its 11C-labeled counterpart, which has a half-life of 20 min. Therefore, it is desirable to label these diarylsulfides with 18F.
Among the fluorinated diarylsulfides that have been developed, N,N-dimethyl-2-(2-amino-4-fluorophenylthio)benzylamine (4-F-ADAM) and N,N-dimethyl-2-(2-amino-4-fluoromethylphenylthio)benzylamine (AFM) (Fig. 1) show high affinity and specificity toward SERT (Ki = 4.8, 5,358, and 137 nmol/L toward SERT, DAT, and NET, respectively, for 4-F-ADAM and 1.8, 2,055, and 96 nmol/L toward SERT, DAT, and NET, respectively, for AFM expressed in cloned human transporters) (24). The fluorine atom of 4-F-ADAM that is directly attached to one of the phenyl rings may provide its in vivo stability. Therefore, we have labeled 4-F-ADAM with 18F and evaluated it as a PET SERT radioligand.
The Ki values of 4-F-ADAM for SERT, DAT, and NET—0.081, 2,267, and 117 nmol/L, respectively—were significantly different from those (4.8, 5,358, and 137 nmol/L, respectively) reported by Huang et al. (24). Although our absolute Ki values were different from those reported by Huang et al., both studies showed a similar trend of specificity toward monoamine transporters. This discrepancy is probably due to the difference in cell lines used for these studies. For this reason, it is important to compare different tracers under identical conditions, as illustrated in a recent report (37). For comparison, the in vitro bindings of (+)McN5652 (a selective SERT agent), AFM, ADAM, and DASB to monoamine transporters were also included in Table 1. Compared with (+)McN5652, 4-F-ADAM displayed a greater selectivity for SERT.
The log P of 4-18F-ADAM was 2.73, which was higher than the log P values of AFM and DASB (2.44 and 2.38, respectively) but lower than those of DAPA and ADAM (3.06 and 3.31, respectively) (37). It is generally assumed that the higher the lipophilicity of a radioligand is, the higher the nonspecific binding to plasma proteins and transporter or receptor will be. Therefore, a desirable radioligand will have high affinity to the target transporter or receptor but have relatively low lipophilicity. Although there is no gold standard log P value for an ideal SERT radioligand, in view of the reported values (37), a log P of ∼3.0 is probably desirable for a useful SERT imaging agent.
The uptake of 4-18F-ADAM in rat brain was high and rapid, indicating an efficient passage of the tracer through the intact blood–brain barrier. The high lipophilicity of this tracer was reflected in its log P value (2.73) between n-octanol and phosphate buffer, pH 7.4. The slow washout of 4-18F-ADAM from rat brain suggests its high affinity to the binding sites of the brain. The radioactivity in the femur increased with time, indicating that in vivo defluorination of 4-18F-ADAM may occur in rats.
Regional brain uptake and retention of 4-18F-ADAM in rats was high in regions rich in SERT (hypothalamus, hippocampus, and striatum). The ratio of specific to nonspecific binding increased with time, reaching a peak at 2 h after injection. The ratios of hypothalamus to cerebellum were 5.57 and 12.49 at 1 and 2 h, respectively, after injection. In comparison, the ratios of hypothalamus to cerebellum for 18F-ACF at the same time points were 3.53 and 3.23, respectively (30). The ratio of hypothalamus to cerebellum was about 6 for 18F-AFM at 1 h after injection (32). The specific uptake in other brain regions containing a high concentration of SERT (hippocampus and striatum) also had high target-to-nontarget ratios, suggesting that the distribution of 4-18F-ADAM followed the distribution of SERT in the brain.
Blocking experiments showed that the uptake of 4-18F-ADAM in rat brain regions rich in SERT was significantly inhibited by (+)McN5652 but was less significantly inhibited by either methylphenidate or nisoxetine, suggesting that in vivo uptake of 4-18F-ADAM in rat brain was specific to SERT binding sites.
Metabolic studies showed that greater than 96% of the radioactivity in the rat brain was parent 4-18F-ADAM at 1 h after injection. If there are no differences in the metabolism of 4-18F-ADAM between rat and primate brain, then we may assume only a minor contribution from radioactive metabolites during PET brain studies with this radioligand.
The PET study of 4-18F-ADAM in a baboon showed that the ratios of radioactivity in the midbrain to those in the cerebellum at 1, 2, and 3 h after injection were 2.1, 3.2, and 4.2, respectively (Fig. 4). In comparison, the midbrain-to-cerebellum ratios in a baboon or cynomolgus monkey were 1.73, 3.53, 1.6–2.1, and 1.4–1.6 for 11C-labeled (+)McN5652 (38), DASB (37), MADAM (18), and N,N-dimethyl-2-(2-amino-4-ethylphenylthio)benzylamine (39), respectively, at 75–95 min after injection. The corresponding ratio for 123I-ADAM (16) in a baboon was 2.41 at 180 min after injection. However, a direct comparison of these radiotracers under the identical conditions will make these data more meaningful. The ratio of the radioactivity in the striatum to that in the cerebellum was also high (1.8, 2.4, and 2.7 at 1, 2, and 3 h, respectively, after injection), whereas its ratio was lower in the cortex (1.1, 1.2, and 1.4 at 1, 2, and 3 h, respectively, after injection) (Fig. 4). The radioactivity in the skull did not increase with time, suggesting that defluorination of 4-18F-ADAM may not occur in the baboon. A recent PET study of 11C-4-F-ADAM (11C-AFA) in a baboon showed that uptake of 11C-4-F-ADAM in SERT-rich regions, such as the thalamus, midbrain, and striatum, was blocked by pretreatment of the baboon with citalopram (4 mg/kg) (40). These results suggest that 4-18F-ADAM has specific binding in baboon brain regions with a high concentration of SERT and that 4-18F-ADAM is suitable for studying SERT in the brain using PET.
CONCLUSION
Taken together, the results showed that 4-18F-ADAM has high affinity and selectivity to SERT, has high uptake and specific binding to rat brain regions (hypothalamus, hippocampus, and striatum) rich in SERT, is metabolically stable in rat brain, and localizes in the SERT-rich midbrain of the baboon. The ratios of the radioactivity in the midbrain to that in the cerebellum at 2 and 3 h after injection were 3.2 and 4.2, respectively. To date, 4-18F-ADAM is one of a few 18F-labeled radioligands that are suitable for studying SERT in the living brain using PET. The high target-to-nontarget ratio can provide better images in human applications. Direct comparison with 18F-AFM and other potent 18F-labeled SERT imaging agents in baboons under the identical conditions and further characterization of this new radioligand in humans are warranted.
Acknowledgments
We are grateful to Harry. J. White, who produced 18F-fluoride for this work.
Footnotes
Received Dec. 30, 2002; revision accepted Aug. 14, 2003.
For correspondence or reprints contact: Chyng-Yann Shiue, PhD, Department of Radiology, University of Pennsylvania, 3400 Spruce St., Philadelphia, PA 19104.
E-mail: shiue{at}rad.upenn.edu




{kind=link}
{kind=link}
{kind=link}
{kind=link}