


Abstract
Noninvasive monitoring of gene transfer will benefit basic research and patient care. Most gene-transfer imaging systems do not directly detect the gene of interest, and most do not exploit radiopharmaceuticals that have Food and Drug Administration approval for total-body use. 111In-Octreotide is used clinically to locate tumors overexpressing primarily somatostatin receptor type 2 (SSTR2). We report the in vitro and in vivo detection of SSTR2 chimeric gene transfer with this radiopharmaceutical. Methods: Full-length SSTR2A was ligated into a vector downstream of a 5′ Igκ leader sequence and the hemagglutinin A (HA) sequence. The vector plus insert was then introduced into HT1080 cells. Igκ and HA domain functions were confirmed by immunologic methods. Receptor binding was studied in transfected cells incubated with 111In-octreotide with and without somatostatin-28. Mice bearing tumors produced by transfected cells were injected with 111In-octreotide for biodistribution and imaging studies. Results: Cell-membrane localization by the amino-terminal Igκ domain was confirmed by immunofluorescence. The HA domain was identified by enzyme-linked immunosorbent assay, immunofluorescence, and Western blotting analysis with anti-HA antibodies. 111In-Octreotide detected the SSTR2 portion of the fusion protein in vitro (receptor-binding assay) and in vivo (biodistribution studies and gamma-camera imaging). In addition, in vitro studies using either the anti-HA antibody or 111In-octreotide correlated with biodistribution and imaging studies when cell clones expressing different levels of the fusion protein were tested. This approach may be feasible clinically because we were able to discern chimeric gene transfer in tumor-bearing animals with 111In-octreotide at doses similar to those already used in humans. Conclusion: With this method it may be possible to monitor transfer of a gene of interest directly and noninvasively.
Although there are many tools for monitoring gene transfer in vitro, few such tools are available for assessing gene transfer in vivo. Imaging agents are needed for studies of transgenic animals and for the future development of gene therapy, which will increasingly target specific tissues. Currently, the maintenance of long-term expression in target tissue is a challenge for the field of gene therapy. With the appropriate reporter, however, it should be possible to determine whether gene expression is achieved in the intended organ at a level necessary for therapeutic effect. With repeated visualization, the duration and level of expression in a target could assist in optimizing dosing schedules. Furthermore, following the level of expression in nontarget tissues may provide insight into toxicity.
Most in vivo methods for monitoring gene transfer capitalize on the sensitivity of gamma-camera, SPECT, or PET imaging for the detection of intravenously injected radiolabeled compounds localized to the products of transferred genes. For example, herpes simplex virus-1 thymidine-kinase gene transfer has been detected by gamma camera and PET in animal models using radiolabeled prodrugs that are entrapped in the cell after phosphorylation by the kinase (1–7). Transfer of the type-2 dopamine receptor has been detected by PET using a labeled antagonist. In this case, the antagonist binds to a receptor that is expressed on the cell surface (8,9). Transfer of the rat sodium-iodide symporter has also been detected by gamma camera using radioactive iodine entrapped in cells by the expressed protein (10). Iodine radioisotopes are used clinically, and this technique may also allow visualization by CT. However, the cellular consequences of the ectopic ion channel or iodine accrual outside the thyroid gland are unknown.
Several imaging systems will be needed for monitoring either single or multiple gene transfer in an individual patient. Although a few such systems have been designed, most do not use radiopharmaceuticals that are known to be safe in humans and that have Food and Drug Administration (FDA) approval (1–10). 111In-Octreotide is commercially available and is currently used clinically in humans primarily to detect tumors (e.g., rare neuroendocrine tumors) expressing somatostatin receptor type 2 (SSTR2) (11). There are 6 SSTRs: types 1, 2A and 2B, 3, 4, and 5. They belong to the 7–transmembrane domain family of receptors associated with G-proteins. Human type 2 has high affinity for octreotide, types 1 and 4 have low affinity (12,13), and types 3 and 5 have intermediate affinity (14–16). Types 2A and 2B are alternate splice variants; type 2A has a longer intracytoplasmic carboxy terminus than does type 2B, but otherwise, they are identical.
The 99mTc-labeled somatostatin derivative P829, as well as its 188Re-labeled analog, has been used to image nude mice with tumors made to express human SSTR2 by infection with an adenovirus vector encoding human SSTR2 (17). Two days after infection, tumors injected with vector plus SSTR2 were detected, whereas those injected with control virus were not. Imaging was performed 3 or 17 h after 99mTc- or 188Re-octreotide injection, respectively. The 188Re-labeled analog, which is not FDA approved, may be more useful for therapy because it decays by β-particle emission. Currently, the 99mTc-labeled molecule is FDA approved only for imaging the lungs, whereas 111In-octreotide is approved for imaging the total body.
Most imaging systems are developed to detect the transfer of a gene of interest or the reporter. A simple method to identify directly whether a gene of interest is being expressed in normal and tumor tissues would be to create a fusion protein that includes the gene and the reporter. We describe the successful use of 111In-octreotide, currently available in the clinic for imaging endocrine tumors, as a radiopharmaceutical for the in vitro and in vivo detection of a human SSTR2 chimeric gene product.
MATERIALS AND METHODS
Cloning
The polymerase chain reaction (PCR) was used to obtain SSTR2A from a phage containing the SSTR2A insert (American Type Culture Collection, Rockville, MD) using a forward primer (TCC CCG CGG CAT GGA CAT GGC GGA TGA) that contained a Sac II restriction site and a reverse primer (AAT CTG CAG CTG TCA GAT ACT GGT TTG GAG) that contained a Pst I restriction site, and a stop codon. The full-length SSTR2A insert was ligated into the pDisplay vector (Invitrogen, Carlsbad, CA), which was also cut with Sac II and Pst I. The insert was placed downstream of the membrane localization sequence (Igκ leader) and the sequence for the hemagglutinin A (HA) epitope tag. Next, Top 10 cells (Invitrogen) were transformed, using calcium chloride, with the vector only or the vector and the SSTR2A insert, and these were then selected for ampicillin resistance. The plasmid was purified using the Wizard Plus Midipreps DNA purification system (Promega, Madison, WI).
Cell Lines
HT1080 cells, a human fibroblast cell line, were grown in Dulbecco’s modified Eagle medium (DMEM) containing 1× glutamine, penicillin, and streptomycin (GPS) and 10% fetal bovine serum (FBS). For transfection, 1 μg DNA was added with lipofectin (Gibco BRL, Grand Island, NY) to 1 × 105 cells in 60-mm dishes according to the manufacturer’s instructions. After 5 h, the lipofectin–DNA solution was removed, and the cells were incubated in DMEM containing 1× GPS and 10% FBS for an additional 2 d. The cells were then diluted 1:20 and 1:5, and single colonies were isolated after G418 selection.
Enzyme-Linked Immunosorbent Assay
Prospective colonies were assayed for expression by the enzyme-linked immunosorbent assay (ELISA). Cells were grown to confluence on 96-well plates, washed twice with phosphate-buffered saline (PBS), and fixed for 30 min with 2% formaldehyde in PBS or 10% formalin. They were then washed twice with PBS and blocked with 5% nonfat milk for 30 min. After another wash in PBS, the cells were exposed to 50 mU/mL horseradish peroxidase (HRP)–rat-anti-HA antibody conjugate (clone 3F10; Roche Applied Science, Indianapolis, IN) overnight at 4°C or 1 h at room temperature. Three washes with PBS for 5 min each followed. Positive cells exhibited a green-colored product after exposure to the HRP ELISA substrate (Bio-Rad Laboratories, Hercules, CA).
For quantitative ELISA, 30,000 cells were plated per well of a 96-well plate. The assay was then performed as above. The results were compared with a standard dilution curve of the HRP–rat-anti-HA antibody conjugate (1 U HRP enzymatic activity/μg antibody). A 0.98 correlation coefficient is routinely obtained between the color reaction product and serial dilution of the conjugated antibody.
Western Blot Analysis
Cells were grown to confluence in 6-well dishes, washed with PBS, and then exposed to Triton X-100–SDS lysis solution (0.1% sodium dodecyl sulfate [SDS], 1% Triton X-100, 0.1 mol/L Tris [pH 8], 0.14 mol/L sodium chloride, 0.025% sodium azide, 0.18% Complete protease inhibitor [Roche], and 1 mol/L iodoacetamide) for 1 h at 4°C. After a 30-min centrifugation of the suspension at 14,000g, the supernatant was collected and the protein concentration was determined using the Bradford method (Bio-Rad protein determination kit). Twenty micrograms of protein were loaded per lane on 7% SDS gels (18). The sample was then transferred to nitrocellulose using a semidry transfer apparatus (Fisher, Atlanta, GA) and Towbin buffer. The blots were dried and transfer was confirmed with Ponceau-S staining. Next, the membrane was washed in PBS, blocked with 5% nonfat milk for 1 h at room temperature, washed in PBS with 0.1% Tween, and exposed to 50 mU/mL HRP–rat-anti-HA antibody (clone 3F10) overnight at 4°C or 1 h at room temperature. After four 5-min washes with PBS, the membrane was covered with a chemiluminescent HRP substrate (Renaissance system; NEN Life Science Products, Boston, MA) for filming.
Immunofluorescence
Cells (10,000 per well) were plated in an 8-well silicone chamber slide system (Bellco Biological Glassware, Vineland, NJ) and incubated at 37°C overnight. The cells were then fixed with 10% formalin for 30 min, washed with PBS twice, blocked with 10% nonfat milk for 30 min, and exposed to diluted (1:250) mouse-anti-HA antibody (BabCo, Richmond, CA) for 1 h at room temperature. Five washes for 5 min each followed. Next, the cells were exposed to diluted (1:150) fluorescein isothiocyanate (FITC)–goat-anti-mouse antibody (Sigma, St. Louis, MO) for 30 min at room temperature. Again, 5 washes for 5 min each followed. The slides were then mounted with coverslips using Gel Mount (Biomeda, Foster City, CA) in preparation for evaluating immunofluorescence.
Receptor-Binding Studies
Cells (30,000 per well) were plated in a 96-well plate and incubated overnight. After a wash with PBS, they were exposed to 0.1 μmol/L 111In-octreotide or 111In-octreotide plus 1 μmol/L somatostatin-28 (Sigma) in binding buffer (20 mM Hepes [pH 7.4], 0.1% bacitracin, and 0.2% bovine serum albumin) for 1 h at room temperature. The cells were then washed with PBS 5 times and lysed with 0.1N NaOH, after which the radioactivity associated with the lysed cells was determined in a γ-counter. Controls included wells with cells exposed to binding buffer without radioactivity and wells without cells exposed to binding buffer with radioactivity.
Biodistribution and Imaging
In nude mice, the subcutaneous injection of 5 × 106 cells produced palpable tumors after 1 wk. One group of tumor-bearing mice was injected intravenously in the tail vein with 13 MBq (350 μCi) 111In-octreotide. Four hours and 24 h later, the animals were anesthetized (sodium pentobarbital, 40 mg/kg, intraperitoneally) and imaged using a gamma camera (Starcam 3200; General Electric Medical Systems, Milwaukee, WI) fitted with a medium-energy pinhole collimator. The mice were killed, each organ was dissected and weighed, and the associated radioactivity was determined in a γ-counter. In another set of experiments, tumor-bearing animals were injected intravenously with 0.37 MBq (10 μCi) 111In-octreotide and killed at 24 h. Next, the biodistribution of radioactivity in various organs and tissues was determined.
Statistical Methods
For in vivo assays, 1-tailed t tests were used to compare the uptake among tumors that differed in expression. Linear regression was used to analyze correlations between in vitro and in vivo studies. The analysis was performed using Lotus 123 ’97 software (IBM, Cambridge, MA) and Origin (OriginLab Corp., Northampton, MA) graphic software (version 6.1).
RESULTS
The SSTR2A receptor was cloned into the pDisplay vector containing a 5′ Igκ leader sequence for membrane localization of the expressed protein. This is followed by an HA sequence for antibody-based detection (19). The binding domain for octreotide is predicted to involve primarily the carboxy portion of the receptor between transmembrane domains III and VII (20–22). Amino-terminal fusion of the receptor is also less likely to interfere with receptor internalization, which is influenced by the carboxy-terminal intracellular tail (23,24). To prevent expression in addition to the carboxy-terminal tail of SSTR2A, a stop codon was introduced into the PCR back primer. After cloning the SSTR2 into pDisplay, the vector or the vector plus insert was introduced into HT1080 cells using lipofectin and G418 selection.
Expression
Using an antibody to the HA domain, protein expression was confirmed in whole cells by ELISA (normalized for cell number) and in cell lysates by Western blot analysis (normalized for protein). In a quantitative ELISA of clonal lines transfected with the same SSTR2A gene chimera (Fig. 1), clone 21 reacted more than (expressed more fusion protein than) clone 3, which reacted more than clone 11. In comparison, no reaction was seen in cells transfected with only vector. As seen on the Western blot in Figure 2, a distinct band (apparent molecular weight, 72,000 Da) is observed in all lanes representing cells transfected with SSTR2A chimeric gene but not in lanes representing cells transfected with only vector. The bands are progressively more intense in lanes marked clone 11, clone 3, and clone 21, implying that the greatest expression occurred in clone 21, followed by clone 3 and then clone 11. No expression was seen in cells transfected with only vector.
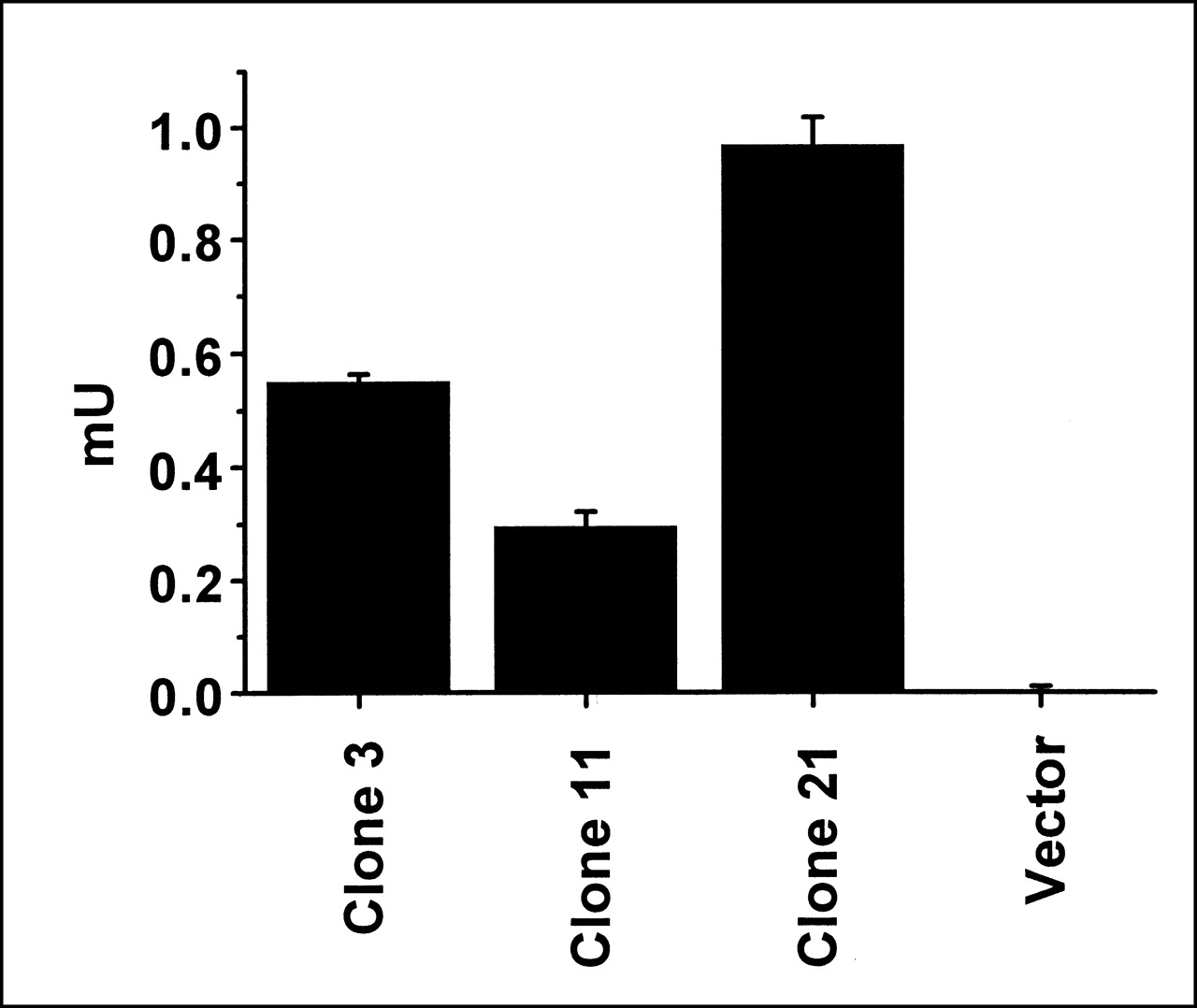
ELISA. Protein expression by cells (3 × 104 per well) transfected with SSTR2A gene chimera (clones 3, 11, and 21) or with only vector is compared by ELISA using an antibody to the HA tag. Error bars represent SD of duplicate samples.

Western blot. Twenty micrograms of protein were loaded per lane and detected with an antibody to the HA tag. Clones 3, 11, and 21 are transfected with SSTR2A gene chimera and vector is transfected with only vector.
Immunofluorescence
To determine intracellular localization of the expressed protein, HA-tag expression was determined by immunofluorescence. Figure 3 shows that the HA tag is detected in cells transfected with the SSTR2A gene chimera but not in cells transfected by only the vector or in untransfected cells (data not shown). The immunofluorescence is seen most clearly at the edges of the cell, following a cell-membrane distribution. In addition, immunofluorescence is excluded from the nucleus and is not diffusely distributed throughout the cytoplasm. Therefore, the expressed fusion protein localizes on the cell membrane. These data, the ELISA, and the Western blot analysis confirm proper function of the Igκ leader sequence and the HA domain.
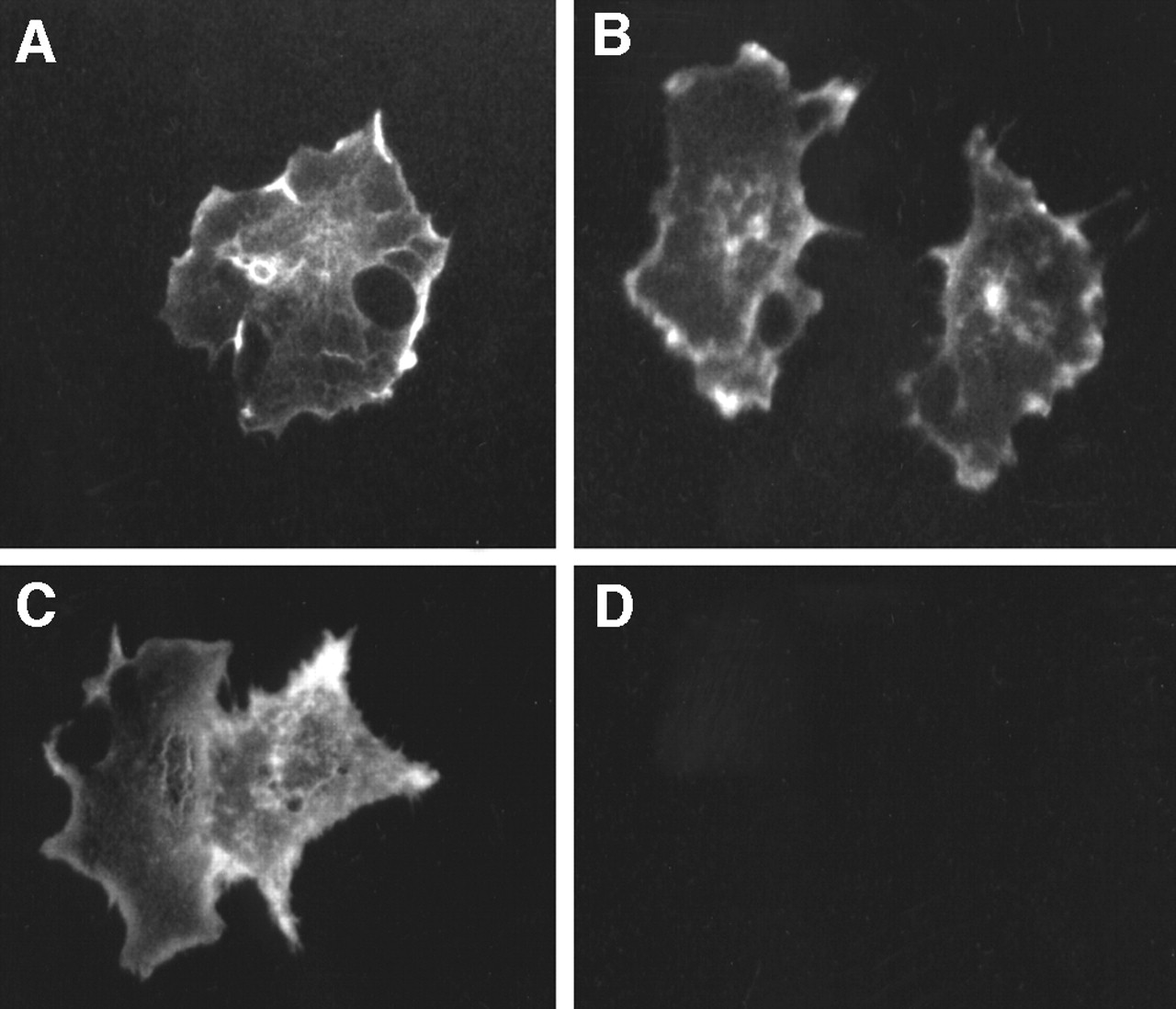
Immunofluorescence. Indirect immunofluorescence was assessed using antibody to HA tag and FITC-labeled secondary antibody. (A) Clone 3. (B) Clone 11. (C) Clone 21. (D) Vector-transfected cells.
Receptor Binding
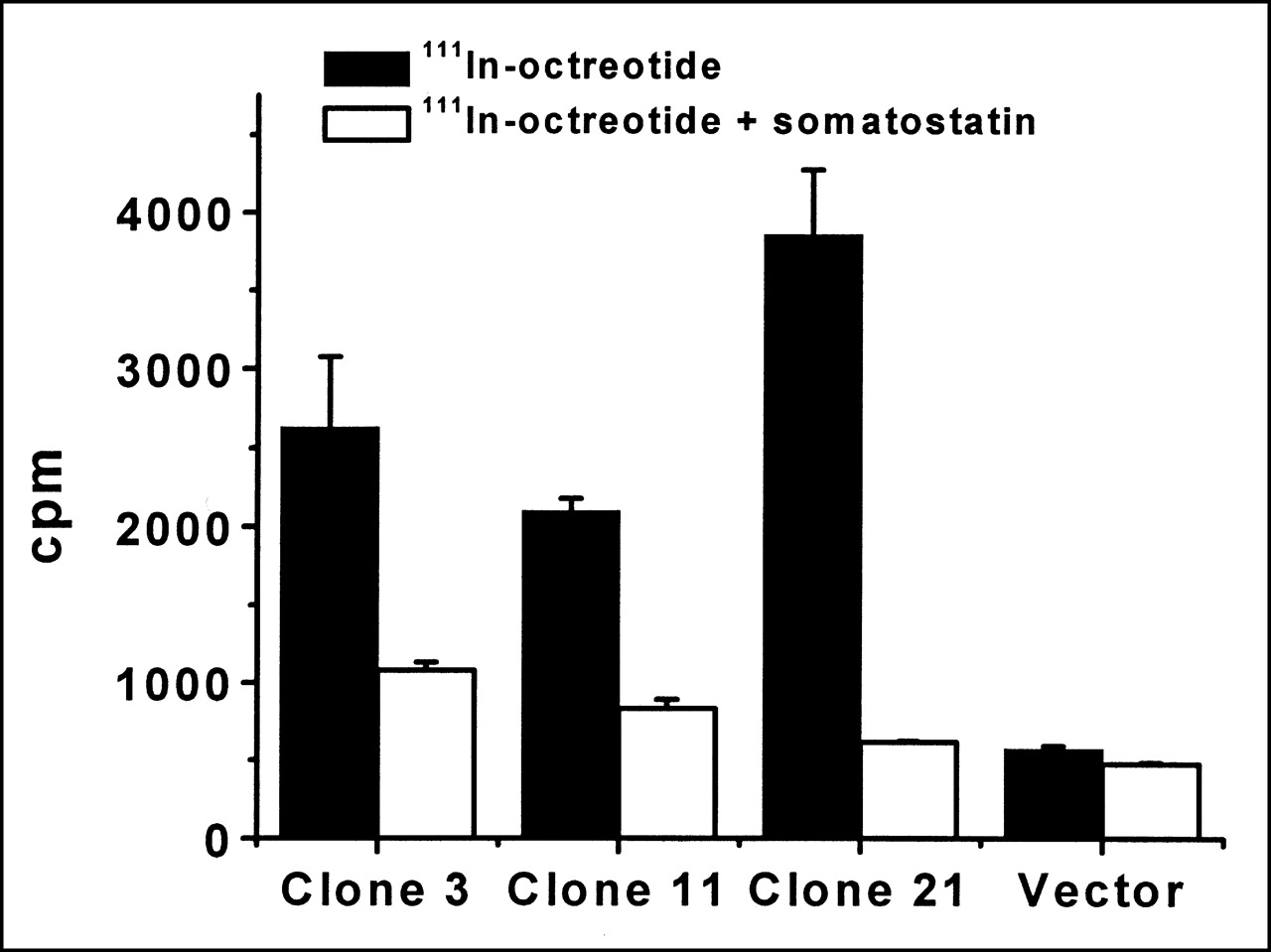
For confirming proper function of the SSTR2A portion of the fusion protein, receptor-binding assays were performed to show and compare the binding of octreotide to the various clones. Receptor saturation has been reported to occur when the octreotide concentration is 0.1 μmol/L (25,26). Binding of the different clones to 0.1 μmol/L 111In-octreotide was compared with or without 1 μmol/L somatostatin-28. Because all clones originate from the same transfection and are expressing the same-sized protein, differences in receptor binding should be attributed to protein expression. As seen in Figure 4, clones 21, 3, and 11 exhibit a greater number of counts per min without the addition of somatostatin-28 than with its addition. The specific binding to 111In-octreotide is greater for clone 21 than for 3, followed by clone 11. In the presence of somatostatin-28, lower counts are seen for all clones. This same low signal is also observed in cells transfected with vector or untransfected cells with or without the addition of somatostatin-28. The findings show that the SSTR2A domain of the fusion protein is functional. In addition, the receptor-binding data correlate with the Western blot analysis—that is, expression levels based on the HA tag corroborate receptor–ligand-binding data based on the SSTR2A that a greater amount of receptor fusion protein per cell is present in clone 21 than in clone 3, than in clone 11, all of which are transfected with the same fusion protein.

Receptor binding. Cells (3 × 104 per well) were exposed to 0.1 μmol/L 111In-octreotide or 0.1 μmol/L 111In-octreotide plus 1 μmol/L somatostatin-28. Error bars represent SD of duplicate samples. cpm = counts per minute.
Imaging and Biodistribution
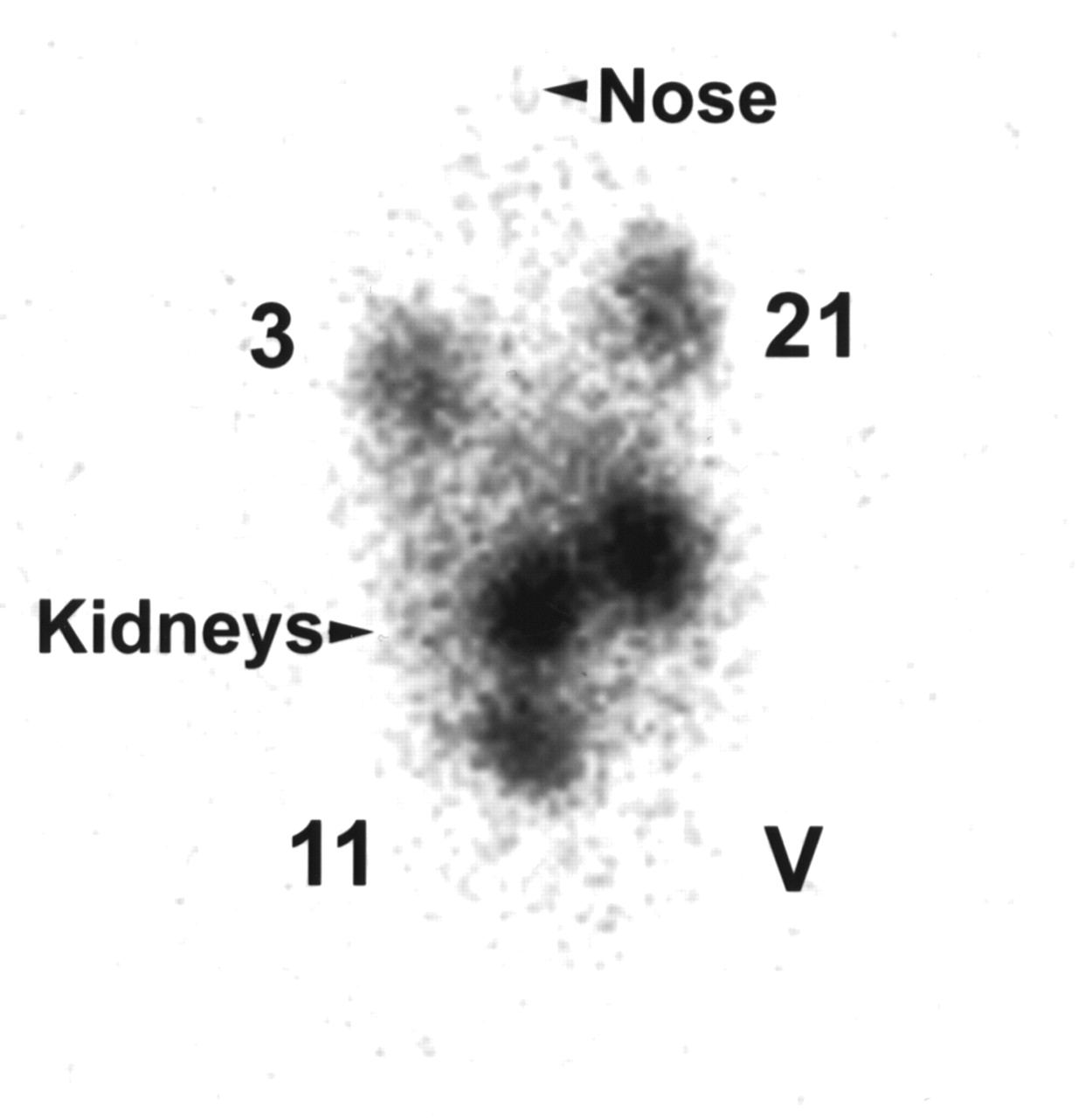
Nude mice injected with clone 21, 3, or 11 or vector-transfected cells were imaged 4 and 24 h after 111In-octreotide was administered via tail vein. Only background activity was apparent 4 h after injection (data not shown). Figure 5 shows that, 24 h later, tumors derived from vector-transfected cells are not visible (Fig. 5, lower right) and those from clone 11 (Fig. 5, lower left) are difficult to see. In comparison, tumors derived from clone 21 (Fig. 5, upper right) are easily visible and are more conspicuous than those from clone 3 (Fig. 5, upper left). These visual findings were confirmed by measuring activity in regions of interest.

Image of nude mouse. Mice bearing subcutaneous tumors (in both shoulders and thighs) were injected intravenously with 111In-octreotide (13 MBq) and imaged 24 h later: 3, tumor derived from clone 3; 21, tumor derived from clone 21; 11, tumor derived from clone 11; and V, tumor derived from vector-transfected cells.
The mice were killed to evaluate the biodistribution of radiopharmaceutical. As seen in Figure 6, more radioactivity per gram is present in tumors derived from clone 21 than in those from clone 3, followed by clone 11 and then only vector-transfected cells. There is a statistically significant difference between tumors originating from vector or SSTR2A–chimeric gene-transfected cells and between clone 11 and clone 3 or clone 21 (P < 0.05). Although the difference between tumors arising from clones 3 and 21 is not statistically significant, the trend is apparent. These data correlate with the Western blot, receptor-binding, and imaging data.
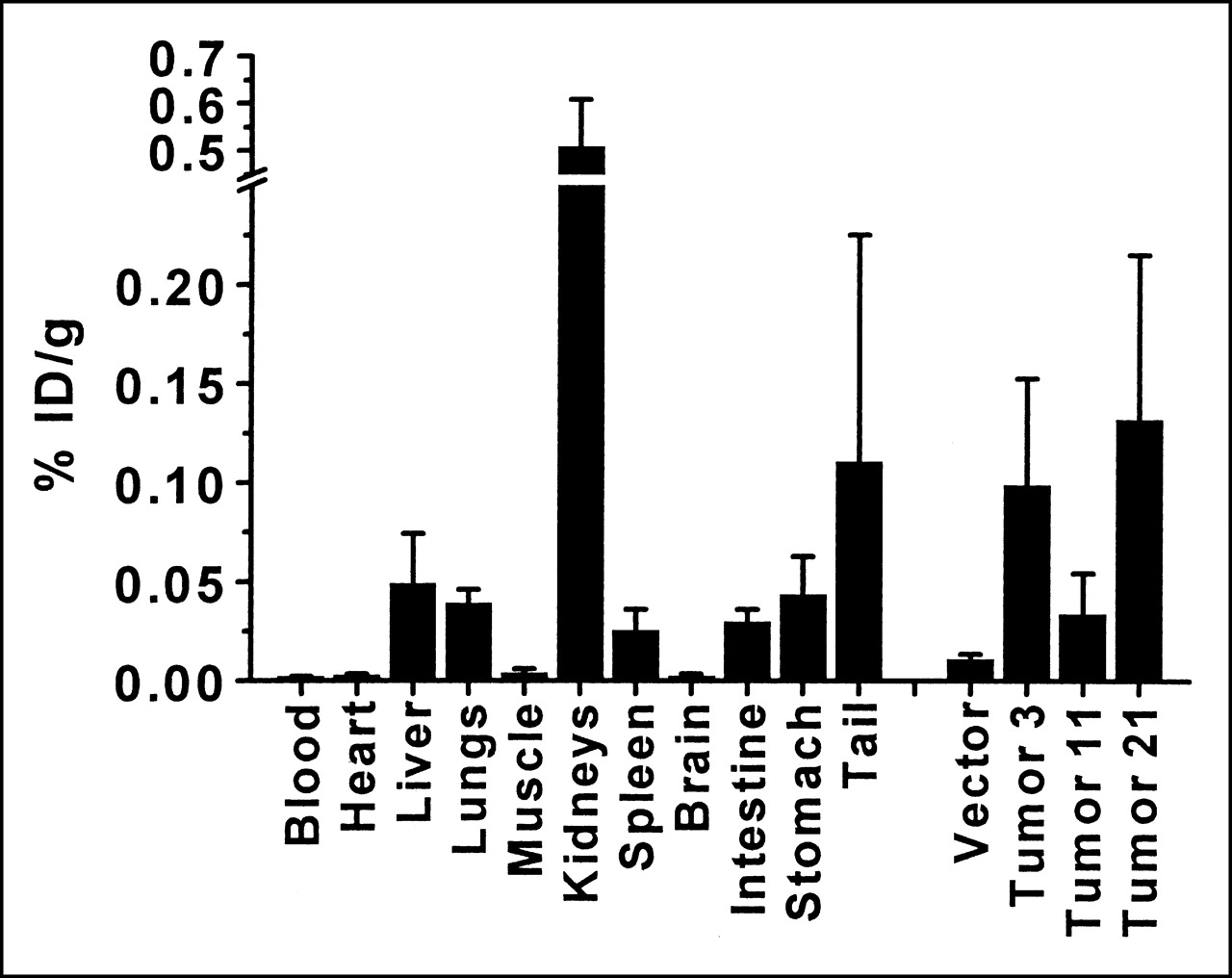
Biodistribution after intravenous administration of 13 MBq 111In-octreotide. Mice bearing subcutaneous tumors (in both shoulders and thighs) were injected intravenously with 111In-octreotide and killed 24 h later. Organs and tumors were weighed and radioactivity was counted. Error bars represent SD (n = 4 mice). % ID/g = percentage injected dose per gram.
The above experiments used large doses of 111In-octreotide (13 MBq [350 μCi]) to produce sufficient counts for imaging mice. Biodistribution experiments were also performed after the injection of 0.37 MBq (10 μCi) of radiopharmaceutical, a dose similar on a per kilogram basis to those used in human tumor-imaging studies (222 MBq [∼6 mCi]) (Fig. 7). To further test the robustness of the method, cells with the least fusion protein expression were used. Figure 7 indicates that, as with larger doses of radiopharmaceutical, organ uptake in the mice mimics clinical findings in humans. Radioactivity per gram is greatest in the kidneys, followed by the liver, gastrointestinal tract, lungs, and spleen. In live animals, air filling the lungs decreases their background activity. In comparison, blood and muscle uptake per gram is quite low. The greater percentage injected dose per gram seen with the lower dose (Fig. 7) likely reflects the greater excretion that occurs when larger doses of the radiotracer are delivered. Figure 7 shows that tumors derived from clone 11 have statistically greater uptake of radiotracer per gram than tumors derived from cells transfected with only vector (P < 0.05). The data imply that gene transfer into human cells can be detected with doses of 111In-octreotide similar to those currently used clinically.
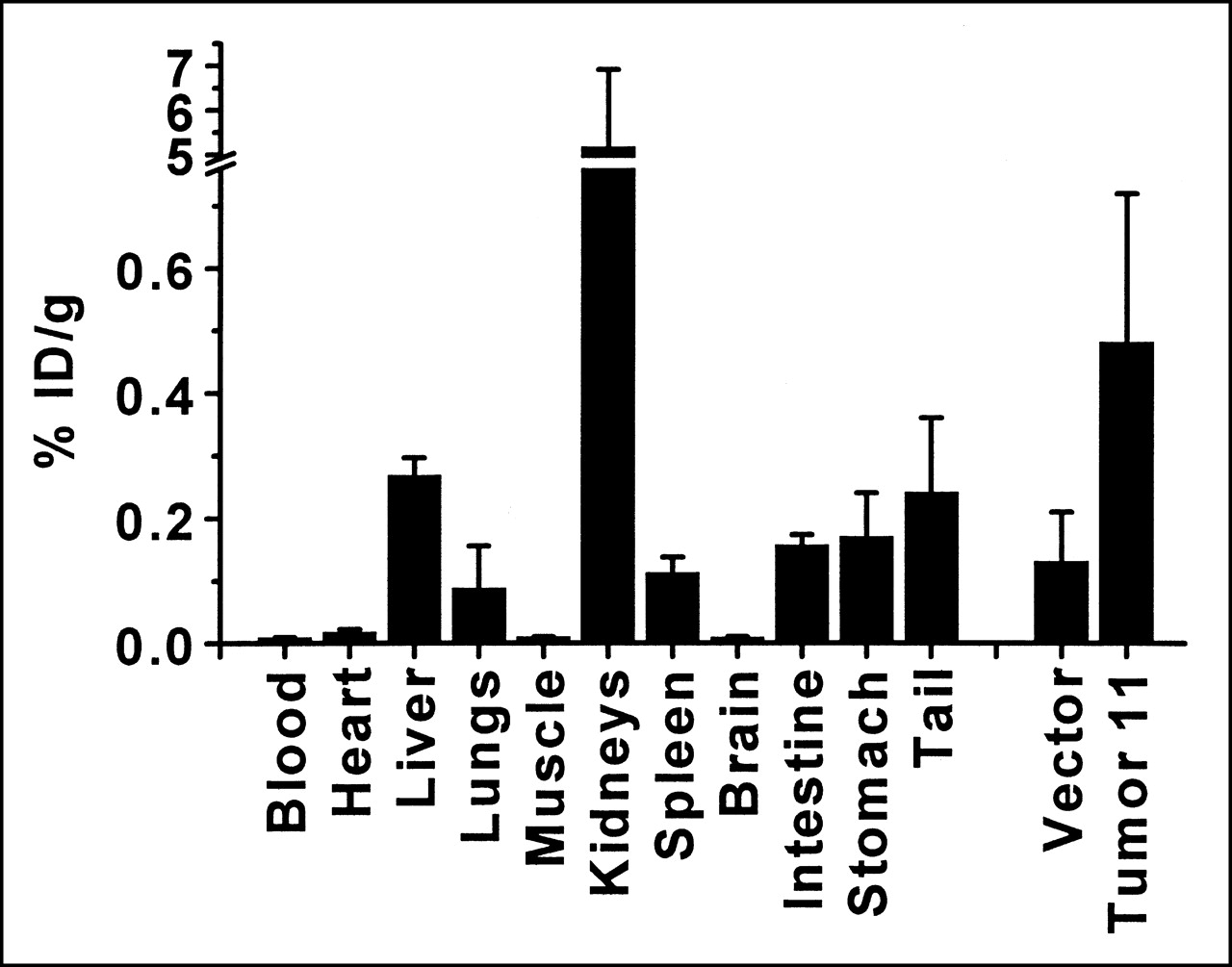
Biodistribution after intravenous administration of 0.37 MBq 111In-octreotide. Mice bearing subcutaneous tumors (in both shoulders and thighs) were injected intravenously with 111In-octreotide and killed 24 h later. Organs and tumors were weighed and radioactivity was counted. Error bars represent SD (n = 3 mice bearing duplicate tumors). % ID/g = percentage injected dose per gram.
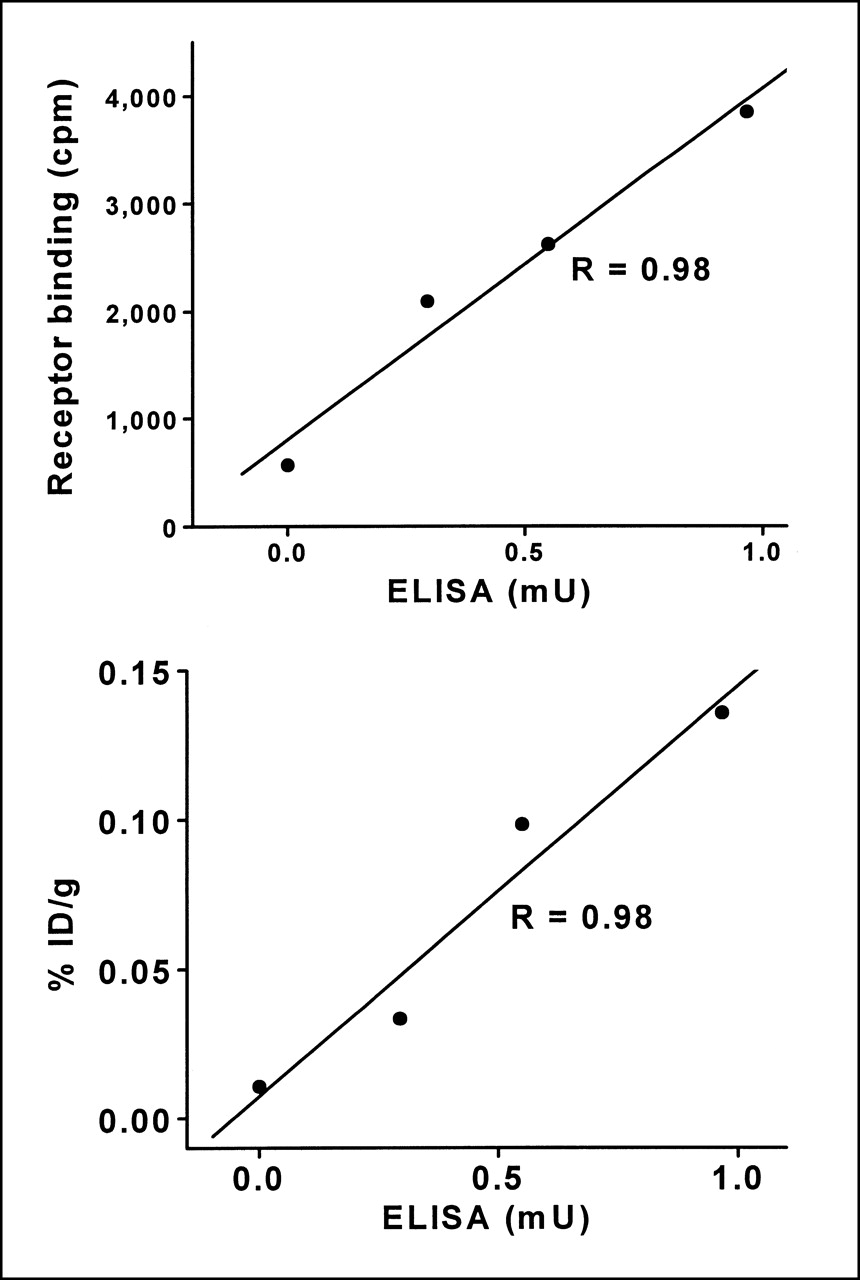
The association of the in vitro and in vivo findings was further analyzed by regression analysis. Chimeric gene expression as measured by ELISA correlates with the receptor-binding data (Fig. 8, top) (correlation coefficient, 0.98). Further, the ELISA also shows strong correlation with radioligand uptake by tumors in the biodistribution experiments (Fig. 8, bottom) (correlation coefficient, 0.98). Thus, for the gene chimera, protein expression monitored through the HA domain correlates with ligand binding monitored through the SSTR2A domain, in vitro and in vivo.

Correlation of ELISA with other measurements. (Top) Receptor-binding assay. cpm = counts per minute. (Bottom) Radiotracer uptake by tumors (as determined in Fig. 6). % ID/g = percentage injected dose per gram. In both cases, the correlation coefficient is 0.98.
DISCUSSION
The capacity to noninvasively monitor gene transfer will benefit the future development of gene therapy; in particular, this technique can be used for analysis of efficacy and toxicity and for dose scheduling. It should also be an aid in the basic sciences (e.g., in studies involving transgenic mice). Clinical agents known to be safe in humans are desirable for rapidly implementing imaging techniques. 111In-Octreotide, FDA approved for whole-body imaging, is primarily used for the diagnosis of rare neuroendocrine tumors. In this study, we have shown that this radiopharmaceutical can be used to image noninvasively the transfer of a gene chimera and the presence of the subsequent fusion-protein product. We further illustrate a direct correlation between the imaging of the fusion protein and the expression of different functional domains.
The data show that transfer of the SSTR2A gene chimera can be detected in vitro and in vivo. In addition, the different domains of the expressed fusion protein retain function. At its amino terminus, the expressed protein contains a 5′ Igκ leader sequence for membrane localization in addition to an HA tag for detection by an anti-HA antibody. The SSTR2A portion binds octreotide.
Although SSTR–carboxy-terminal fusion proteins have been studied in vitro (24), amino-terminal fusion was chosen because the binding domain for octreotide is predicted to be distal, primarily involving SSTR2 amino acids between transmembrane domains III and VII (20–22). Because the membrane localization signal at the SSTR2A amino terminus is disrupted by the fusion, a new membrane localization signal is created by the Igκ leader sequence. In addition, amino-terminal fusion is also less likely to interfere with receptor internalization, which involves the carboxy-terminal intracellular domain (23,24). The HA domain of the heterologous protein was exploited to confirm expression. The ELISA allowed us to quickly screen small numbers of cells, saving the time usually needed to expand large numbers of resistant colonies. Moreover, the initial screening was performed without using radioactivity. Again capitalizing on the HA domain, immunofluorescence localized the fusion protein to the cell membrane, verifying a functional Igκ membrane-localization domain.
Protein expression was also detected by Western blot analysis. The fusion protein was not found in lysates made with RIPA buffer (1% Triton X-100, 1% bovine hemoglobin, 1 mmol/L iodoacetamide, Aprotinin (0.2 U/mL), 1 mmol/L phenylmethylsulfonylfluoride, 0.1 mol/L Tris chloride [pH 8.0], 0.14 mol/L NaCl, 0.025% NaN3, 1% sodium deoxycholate, and 0.1% SDS) (data not shown), possibly because of disruption of the epitope tag by deoxycholate. However, it was readily identified in Triton X-100–SDS lysates. The expressed protein has an apparent molecular weight of 72,000 Da, which is less than that previously reported for SSTR2A and greater than that predicted for the protein-only components. Previous studies have shown that, because of glycosylation, the molecular weight of wild-type SSTR2 is greater than that predicted by its protein components (27). The Igκ leader sequence and HA domain likely alter the normal glycosylation of the receptor component. Because somatostatin-28 inhibited binding of 111In-octreotide to cells expressing the fusion protein, the data also imply that such receptor modifications do not prevent somatostatin-28 binding. The increasing expression of clones 11, 3, and 21 seen by ELISA correlates with receptor-binding data (Fig. 8, top). Thus, findings with an antibody against the HA domain correlate with 111In-octreotide binding to the SSTR2A portion of the receptor. This in vitro correlation is also noted in the in vivo biodistribution and imaging data. This significantly enhances the SSTR as a reporter of gene transfer, because the HA domain enables cell biology assays, such as ELISA, Western blotting, immunoprecipitation, and immunofluorescence. It may also be exploited for immunohistochemistry of tissue sections.
For the in vivo experiments, subcutaneous tumors were grown from clonal populations of cells that were selected after plasmid transfection. Others have injected virus intratumorally into previously established tumors in mice (17). With this approach, however, only cells along the needle tract express the receptor and, as such, the uptake per gram of tissue does not accurately reflect tumor uptake because many uninfected cells will be included in the tumor sample—hence, falsely increasing weight. Expression outside the tumor, which decreases uptake measurements, is also possible. In our approach, each cell in the implanted tumor expresses the protein of interest at essentially the same level, obviating such issues.
The adenovirus-infected cells were imaged by 99mTc- or 188Re-labeled somatostatin analog P829. The 188Re-labeled analog, which may be more useful for therapy, is currently not FDA approved; the 99mTc-labeled variant is approved only for imaging the lungs. 111In-Octreotide, which was used in these studies and by others in previous studies (28), has been approved by the FDA for whole-body imaging. Because of its longer half-life (2.8 d vs. 6 h for 99mTc), 111In decay does not significantly influence interpretation of images after the delay of 24 h between injection and obtaining the final patient scan. In patients, the images at 4 and 24 h are often compared to detect movement of radiotracer, signifying gut activity. The images at 24 h always have the highest signal-to-background information because of washout of nonspecific interstitial activity. In humans, 111In-octreotide is excreted primarily through the kidneys, with a second route of excretion through the liver to the intestine. Activity is also found in the spleen. This biodistribution is mimicked in the mouse, where radioactivity per gram is primarily found in the kidneys, followed by the liver and the spleen. Finally, 111In can be detected by a standard gamma camera. These are favorable characteristics for imaging gene transfer compared with those of possible PET agents, which are also expensive and less readily available.
CONCLUSION
Our findings show that the three portions of the expressed fusion protein are functional and that the in vitro expression data correlate with the in vivo imaging and biodistribution data. In addition, the chimeric gene transfer can be discerned using 111In-octreotide. Thus, the approach described may enable the noninvasive detection of gene transfer, including chimeric gene transfer, in humans.
Footnotes
Received Jul. 5, 2001; revision accepted Dec. 3, 2001.
For correspondence or reprints contact: Vikas Kundra, MD, PhD, Department of Radiology, Division of Diagnostic Imaging, Box 57, M.D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030-4095.
Email: vkundra{at}di.mdacc.tmc.edu




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}