


Abstract
Astrocytes colocalize with fibrillar amyloid-β (Aβ) plaques in postmortem Alzheimer disease (AD) brain tissue. It is therefore of great interest to develop a PET tracer for visualizing astrocytes in vivo, enabling the study of the regional distribution of both astrocytes and fibrillar Aβ. A multitracer PET investigation was conducted for patients with mild cognitive impairment (MCI), patients with mild AD, and healthy controls using 11C-deuterium-L-deprenyl (11C-DED) to measure monoamine oxidase B located in astrocytes. Along with 11C-DED PET, 11C-Pittsburgh compound B (11C-PIB; fibrillar Aβ deposition), 18F-FDG (glucose metabolism), T1 MRI, cerebrospinal fluid, and neuropsychologic data were acquired from the patients. Methods: 11C-DED PET was performed in MCI patients (n = 8; mean age ± SD, 62.6 ± 7.5 y; mean Mini Mental State Examination, 27.5 ± 2.1), AD patients (n = 7; mean age, 65.1 ± 6.3 y; mean Mini Mental State Examination, 24.4 ± 5.7), and healthy age-matched controls (n = 14; mean age, 64.7 ± 3.6 y). A modified reference Patlak model, with cerebellar gray matter as a reference, was chosen for kinetic analysis of the 11C-DED data. 11C-DED data from 20 to 60 min were analyzed using a digital brain atlas. Mean regional 18F-FDG uptake and 11C-PIB retention were calculated for each patient, with cerebellar gray matter as a reference. Results: ANOVA analysis of the regional 11C-DED binding data revealed a significant group effect in the bilateral frontal and bilateral parietal cortices related to increased binding in the MCI patients. All patients, except 3 with MCI, showed high 11C-PIB retention. Increased 11C-DED binding in most cortical and subcortical regions was observed in MCI 11C-PIB+ patients relative to controls, MCI 11C-PIB (negative) patients, and AD patients. No regional correlations were found between the 3 PET tracers. Conclusion: Increased 11C-DED binding throughout the brain of the MCI 11C-PIB+ patients potentially suggests that astrocytosis is an early phenomenon in AD development.
The current predominant hypothesis for the cause of Alzheimer disease (AD) is related to dysfunction in the processing, turnover, deposition, and clearance of the amyloid-β (Aβ) protein (1). Astrocytes and microglia are fundamental in defending the brain against infection and injury. It has been proposed, because of histopathologic observations, that both forms of glial cell play an important role in neurodegenerative diseases such as AD and exacerbate disease effects when they are activated or reactive because of neuroinflammation (2,3).
PET with 11C-Pittsburgh compound B (11C-PIB) reliably measures fibrillar Aβ in the brain of AD patients in vivo (4). However, the relationship between fibrillar Aβ and neuroinflammation is less well understood. An increase in the binding of 11C-PK11195 was initially reported in the brains of AD patients (PK11195 binds to activated microglia) (5), but several later studies attempting to assess the relationship between fibrillar Aβ and activated microglia using both 11C-PIB and 11C-PK11195 have given conflicting results. One study suggests a regional relationship between fibrillar Aβ and activated microglia (6) and others that there is a limited or nonexistent relationship (7,8). To date, no in vivo PET studies have been published that investigate the relationship between fibrillar Aβ and astrocytes in AD.
L-deprenyl is an irreversible monoamine oxidase B (MAO-B) inhibitor. The enzyme MAO-B exists on the outer mitochondrial membrane, occurring predominantly in astrocytes (9,10). The PET tracer 11C-deuterium-L-deprenyl (11C-DED) has high affinity and specificity for MAO-B (11); a previous PET study with 11C-DED has shown that MAO-B increases in most brain regions in healthy older individuals (12). Thus far, studies using 11C-DED PET have been performed in chronic diseases such as epilepsy, amyotrophic lateral sclerosis, and Creutzfeldt–Jakob disease (13–16). These studies revealed changes in 11C-DED binding related to the epileptic lobe; increased 11C-DED binding in the white matter and pons (amyotrophic lateral sclerosis); and increased 11C-DED binding in the frontal, parietal, and occipital cortices (Creutzfeldt–Jakob disease). Currently, only 1 study has been published using 11C-DED PET in an AD population (17). This study used 11C-DED PET in a dose-finding paradigm to assess the occupancy of MAO-B in the brain of a novel reversible MAO-B inhibitor.
Activity of MAO-B increases in AD patients’ brains; the enzyme is overexpressed in reactive astrocytes, and significant numbers of astrocytes surround both fibrillar and diffuse Aβ plaques in postmortem AD brain tissue (18,19). A recent autoradiography study (20) suggested 11C-DED as a suitable in vivo PET ligand for assessing MAO-B in AD brains. This study demonstrated greater binding in the temporal lobe and white matter of AD brains compared with controls. The highest binding was observed in the earlier Braak stages (I–II). The observed pattern of 11C-DED binding in the AD brains colocalized with an increased number of astrocytes (glial fibrillary acid protein [GFAP] immunohistochemistry). In an autopsy-confirmed AD patient who had previously undergone 11C-PIB PET (21), reactive astrocytes were found throughout the brain, and there was a significant positive correlation between regional 11C-PIB retention and the total number of GFAP immunoreactive astrocytes, supporting the idea that astrocytosis occurs near fibrillar Aβ plaques. The same study, however, demonstrated that antemortem 11C-PIB retention did not correlate with postmortem binding of 3H-PK11195 (microglia) and 3H-L-deprenyl (MAO-B) in brain tissue.
Understanding the exact presence and time course of astrocytosis and its relationship to fibrillar Aβ deposition in those at risk of developing AD is of significant importance to develop a better understanding of the disease mechanism. The current investigation is the first, to our knowledge, to use 11C-DED, 11C-PIB, and 18F-FDG PET in a group of mild cognitive impairment (MCI) patients and mild AD patients. Along with the PET, structural T1 MRI and neuropsychologic data were acquired from the patients. Structural T1 MRI and 11C-DED PET were also acquired in a group of healthy age-matched controls.
The main objective of the current investigation was to determine whether there was a regional increase of MAO-B (potentially reflecting astrocytosis) in the brains of the MCI and AD patients, compared with the healthy controls, particularly in regions that are known to have high Aβ load (as measured by 11C-PIB PET), such as the temporal, parietal, and frontal cortices.
MATERIALS AND METHODS
Patients
Fourteen healthy age-matched controls, 8 MCI patients, and 7 AD patients were recruited (Table 1). The MCI and AD patients were referred to the Department of Geriatric Medicine, Karolinska University Hospital Huddinge, Stockholm, Sweden, for memory problems. They underwent comprehensive clinical examination, including neurologic and psychiatric examination, electroencephalogram, CT or MRI, cerebrospinal fluid (CSF) and blood analysis, and neuropsychologic testing. Diagnosis was made through a consensus committee that included doctors, clinical neuropsychologists, and specialist nurses. The AD patients fulfilled the diagnosis of probable AD according to the criteria of the National Institute of Neurologic and Communication Disorders, Alzheimer Disease and Related Disorders Association (NINCDS-ADRDA) (22). The MCI patients fulfilled the criteria for MCI established by Petersen (23).
Participant Information
The neuropsychologic testing covered global cognitive function, language, visuospatial ability, episodic memory, attention and cognitive speed, and executive function (Table 2). The patients’ neuropsychologic test scores were converted into z scores with respect to a reference group of healthy elderly controls from the Karolinska University Hospital, Huddinge (24).
Neuropsychologic Test Results (z Scores)
The healthy control 11C-DED PET data were originally acquired as part of a clinical drug trial and as such, no neuropsychologic, 18F-FDG, or 11C-PIB data were available from these participants. The healthy controls were nonsmokers; had normal blood pressure, heart function, normal MRI findings, no major or chronic illness within 1 mo of the 11C-DED PET scan, and no history of substance abuse, and they did not deviate from clinically normal in a physical examination.
All patients and their caregivers provided written informed consent to participate in the study, which was conducted according to the declaration of Helsinki and subsequent revisions. Ethical approval was obtained from the regional human ethics committee of Stockholm and the Faculty of Medicine and Radiation, Hazard Ethics Committee of Uppsala University Hospital, Sweden.
Imaging Methodology
Image Acquisition: PET Data
All PET investigations were performed at Uppsala PET center on ECAT EXACT HR+ scanners (Siemens/CTI). The orbitomeatal line was used to center the head of the participants. The PET data were acquired in 3-dimensional mode, yielding a 155-mm field of view. The 11C-DED acquisitions consisted of 19 time frames (4 × 30, 8 × 60, 4 × 300, and 3 × 600 s), with a total duration of 60 min. The 11C-PIB acquisitions consisted of 24 frames (4 × 30, 9 × 60, 3 × 180, and 8 × 300 s) over 60 min. The late 40- to 60-min 11C-PIB sum image was created and used for subsequent image analysis. For each 18F-FDG acquisition, 21 frames (4 × 30, 9 × 60, 3 × 180, and 5 × 300 s) were acquired over 45 min. A late 25- to 45-min 18F-FDG sum image was created and used for subsequent analysis. Patients fasted for 4 h preceding the 18F-FDG scan. The mean injected doses for each tracer were 211 ± 66 MBq for 11C-DED, 228 ± 70 MBq for 11C-PIB, and 229 ± 49 MBq for 18F-FDG. All but 2 PET scans were obtained within 14 d of each other; these exceptions were acquired within 20 d.
All emission data were reconstructed with filtered backprojection using a 4-mm Hanning filter, resulting in a transaxial spatial resolution of 5 mm in the field of view. The matrix included 128 × 128 pixels, and a zoom factor of 2.5 was used. All reconstructed frames were realigned to correct for patient motion during each PET scan.
Image Acquisition: MRI Data
All patients underwent a structural T1 magnetization-prepared rapid-acquisition gradient-echo sequence at 3 T (Siemens Trio scanner). Images were acquired with a matrix size of 192 × 256 × 256 and voxel sixe of 1.0 × 0.98 × 0.98 mm and were reconstructed to 1.0 × 1.0 × 1.0 mm isometric voxels, with an echo time of 3.42 ms, repetition time of 1,780 ms, inversion time of 900 ms, and flip angle of 9°. To exclude patients with non–AD-related brain abnormalities, T2 and diffusion-weighted images were also acquired.
Because the controls were recruited from another study, the MRI data were acquired at 1.5 T on a Philips Intera scanner. For examination of possible brain abnormalities, fluid-attenuated inversion recovery and T2 sequences were used. Furthermore, a structural T1 inversion recovery sequence was conducted to enable coregistration of PET and MR images. Parameters included a slice thickness of 4.5 mm, gap of 0.5 mm, matrix size of 256 × 256 × 30, echo time of 14 ms, and repetition time of 2,300 ms; therefore, these parameters provided structural MR images with a spatial resolution similar to that of the PET data.
Image Processing
After the 11C-DED PET data had been reconstructed, realigned, and modeled, the 11C-DED images were not processed further, to preserve the fidelity of the 11C-DED data. As a consequence, all data sampling and analysis were performed in native 11C-DED PET space. First, using SPM5 (Functional Imaging Laboratory, Wellcome Department of Imaging Neuroscience, University College London), we coregistered and resliced all available T1 MR images to their corresponding 11C-DED image. This process created a structural T1 reference image in 11C-DED PET space for each participant. Subsequently, each patient's 11C-PIB and 18F-FDG data were coregistered and resliced to their individual T1 reference image.
All T1 reference images were segmented into gray matter (GM) and white matter tissue classes using the unified segmentation algorithm of SPM5 (25). The resultant probabilistic GM map for each participant had a threshold of 0.5 applied to it, and a binary GM mask was created (0, no tissue, and 1, tissue with a ≥50% probability belonging to GM). The inverse nonlinear transformation parameter file from SPM's segmentation algorithm was used to warp a simplified digital probabilistic atlas (26), consisting of 24 cortical and subcortical regions, into each individual's native 11C-DED PET space. These atlases were multiplied by the corresponding binary GM mask, which generated a GM-specific digital atlas for each participant.
Raw coregistered and resliced 18F-FDG (Bq/mL) and 11C-PIB PET (Bq/mL) data for each patient were sampled using the same individual digital atlases previously created. A mean 18F-FDG uptake and 11C-PIB retention value was measured for each atlas region using this method. Regional GM ratio values were created for 18F-FDG and 11C-PIB and each atlas region by dividing by the respective mean cerebellar GM uptake.
11C-DED Modeling
PET data were analyzed using the individual brain atlases. It has previously been documented that 11C-DED is a highly flow-dependent tracer, and 11C-DED binding has a strong correlation with blood flow (11–13,27). In an attempt to avoid the early flow effects, regional parametric data were generated from dynamic 11C-DED data between 20 and 60 min. Because this was a clinical study, it was not considered feasible to obtain arterial blood samples as an input function for the 11C-DED modeling. As such, a modified reference-Patlak model was adopted for kinetic analysis (14).
The cerebellar GM from the individual atlases was used as the reference region in this model. This region had the lowest binding of all brain regions in an autoradiography study using 11C-DED that investigated controls and AD patients (20) and the lowest 11C-DED uptake of all investigated regions in the present study. However, because of net tracer accumulation in all brain regions, including the cerebellum, the reference Patlak method was modified by correcting k3 in the reference region (k3 = rate constant for irreversible binding to MAO-B) for irreversible binding with a fixed correction factor of 0.01 according to Johansson et al. (14). The correction factor was the minimum value of correction still leading to linearization in the model. This graphical reference Patlak model generated 2 useful measurements, an intercept (initial tracer distribution volume) value and a slope (kI = net 11C-DED binding to MAO-B) value, that were used in subsequent analyses.
RESULTS
Participants
Table 1 shows the demographic data for each group. Patients were subgrouped by their 11C-PIB PET scan status. A cutoff value of greater than 1.41 was used to determine regional 11C-PIB positivity (11C-PIB+; < 1.41 = 11C-PIB−), as previously described (28). No significant difference was observed in age and Mini Mental State Examination (MMSE) score between the groups. The 11C-PIB+ and 11C-PIB− MCI groups demonstrated comparable mean MMSE score values, whereas the MMSE score was lower for the AD group. The MCI 11C-PIB+ patients had significantly lower Aβ42 (Mann–Whitney U; P ≤ 0.05) and significantly higher τ-protein (Mann–Whitney U; P < 0.05) than did the MCI 11C-PIB− patients; there was no significant difference for hyperphosphorylated τ-protein (p-τ) CSF values. The AD patients had significantly lower Aβ42 (Mann–Whitney U; P ≤ 0.05), and they had increased τ (not significantly; Mann–Whitney U; P = 0.053); there was no significant difference for p-τ CSF values. There was no significant difference between the MCI 11C-PIB+ and AD patients for CSF data. The MCI 11C-PIB− patients had normal Aβ42, τ, p-τ values in CSF (Aβ42 < 450 pg/mL, τ > 400 pg/mL, and p-τ > 80 pg/mL are considered abnormal).
Neuropsychology
Patients were divided on the basis of their 11C-PIB scan, as previously described. The AD patients were the most severely impaired on all neuropsychologic assessments, followed by the MCI 11C-PIB+ patients, and then by the less cognitively impaired MCI 11C-PIB− patients (neuropsychologic z scores below −1.645 are considered outside the reference range) (Table 2). There were no significant differences in neuropsychologic z scores among the 3 groups (Kruskal–Wallis; P = 0.05).
11C-PIB PET Data
All patients, except 3 with MCI, were 11C-PIB+. There was a clear significant regional difference between the MCI 11C-PIB− patients and 11C-PIB+ patients (MCI and AD) in all regions (Mann–Whitney U; P ≤ 0.05) except for the hippocampi when the 1.41 cutoff value was used. Figure 1 shows the regional values for the 11C-PIB data. Patients are stratified by 11C-PIB status (positive or negative) and by diagnosis. There were no significant differences in regional 11C-PIB retention ratio between the MCI 11C-PIB+ and the AD patients.

Regional cortical and subcortical 11C-PIB retention in MCI 11C-PIB−, MCI 11C-PIB+, and AD patients. Dashed reference cutoff line is set at 1.41, above which represents 11C-PIB positivity (28). Data are mean ± SD.
11C-DED PET Data
Intercept (initial tracer distribution volume) and slope (irreversibly bound tracer) values were calculated for each atlas region from the 11C-DED data. Initial examination of the 3 groups (healthy controls, MCI patients, and AD patients), independently of 11C-PIB status, using ANOVA on the regional 11C-DED slope data, revealed a significant group effect in the bilateral frontal cortex and bilateral parietal cortex; no subcortical regions demonstrated a significant group effect (Table 3 summarizes the F and P values for each atlas region). Post hoc testing (least significant difference P = 0.05) revealed that the group effect was related to the increased 11C-DED binding in the MCI patients relative to the controls.
Summary of F and P Values for Each 11C-DED Parameter
Performing ANOVA on the regional intercept data revealed a different pattern of significance between the groups to the slope data. There was a significant group effect in the left temporal, left insula, bilateral anterior cingulate, right parahippocampal cortex, right hippocampus, right caudate, and left putamen (Table 3). Post hoc testing (least significant difference P = 0.05) revealed that the group differences in these regions were largely due to the patients’ smaller intercept values.
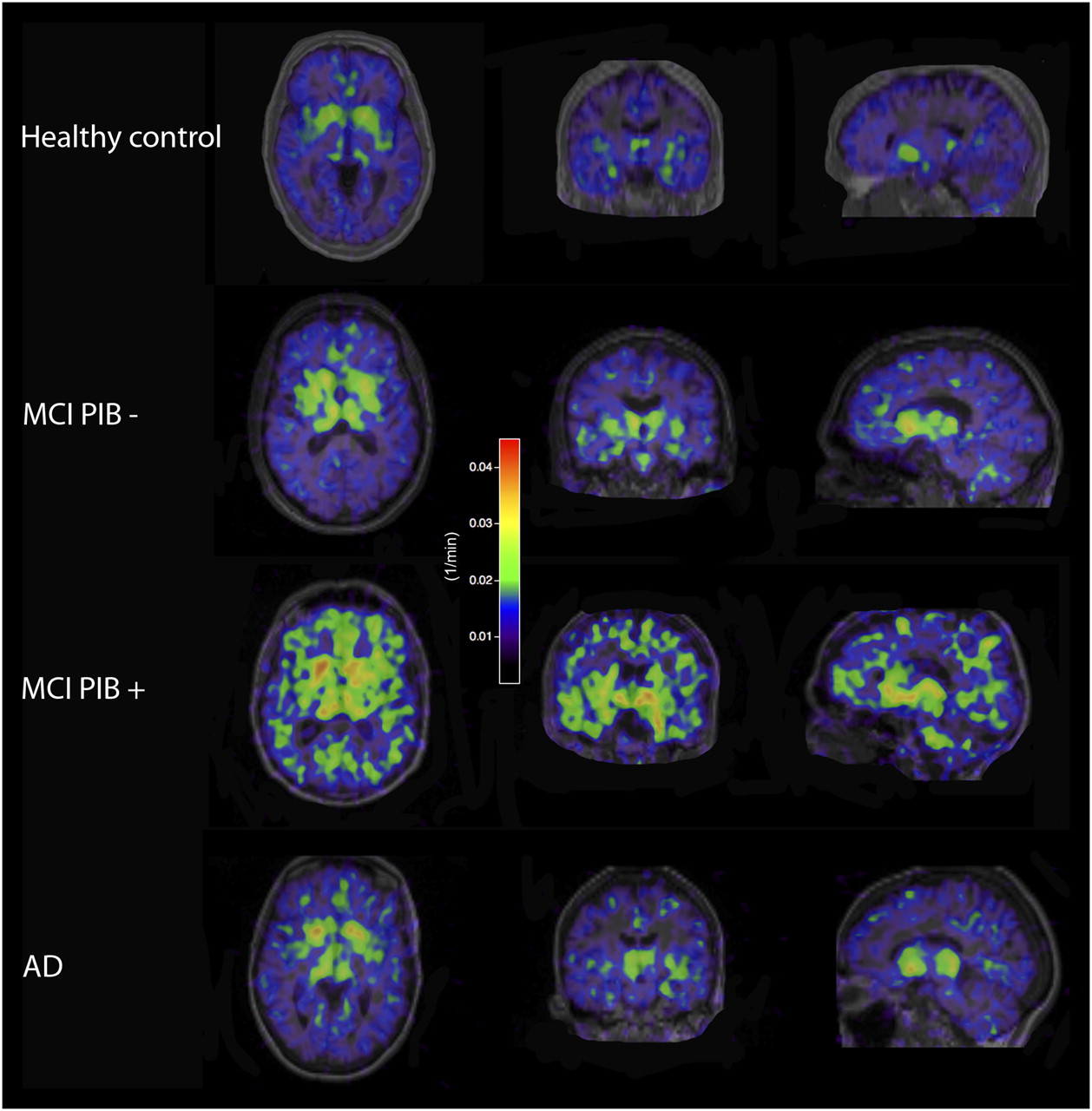
Because no 11C-PIB data were available for the healthy controls, only the MCI and AD patients were divided into 11C-PIB+ and 11C-PIB− subgroups (Fig. 2 displays representative 11C-DED scans following the subgrouping of the MCI and AD patients). After subdivision, Kruskal–Wallis tests were performed on the 11C-DED data. Significant effects of 11C-DED slope values (P ≤ 0.05) were found in the right occipital cortex and right hippocampus. These differences were due to the MCI 11C-PIB+ patients having the highest 11C-DED slope values in the right occipital cortex and the AD patients having the lowest values in the right hippocampus. No significant differences were found for 11C-DED intercept. Figure 3 displays the regional 11C-DED slope data for all subjects. The MCI 11C-PIB+ patients showed the greatest 11C-DED binding in all regions.

Representative parametric images of 11C-DED binding (slope).

Regional cortical (A) and subcortical (B) binding (slope) of 11C-DED in healthy controls and MCI 11C-PIB−, MCI 11C-PIB+, and AD patients. Data are mean + SD.
18F-FDG PET Data
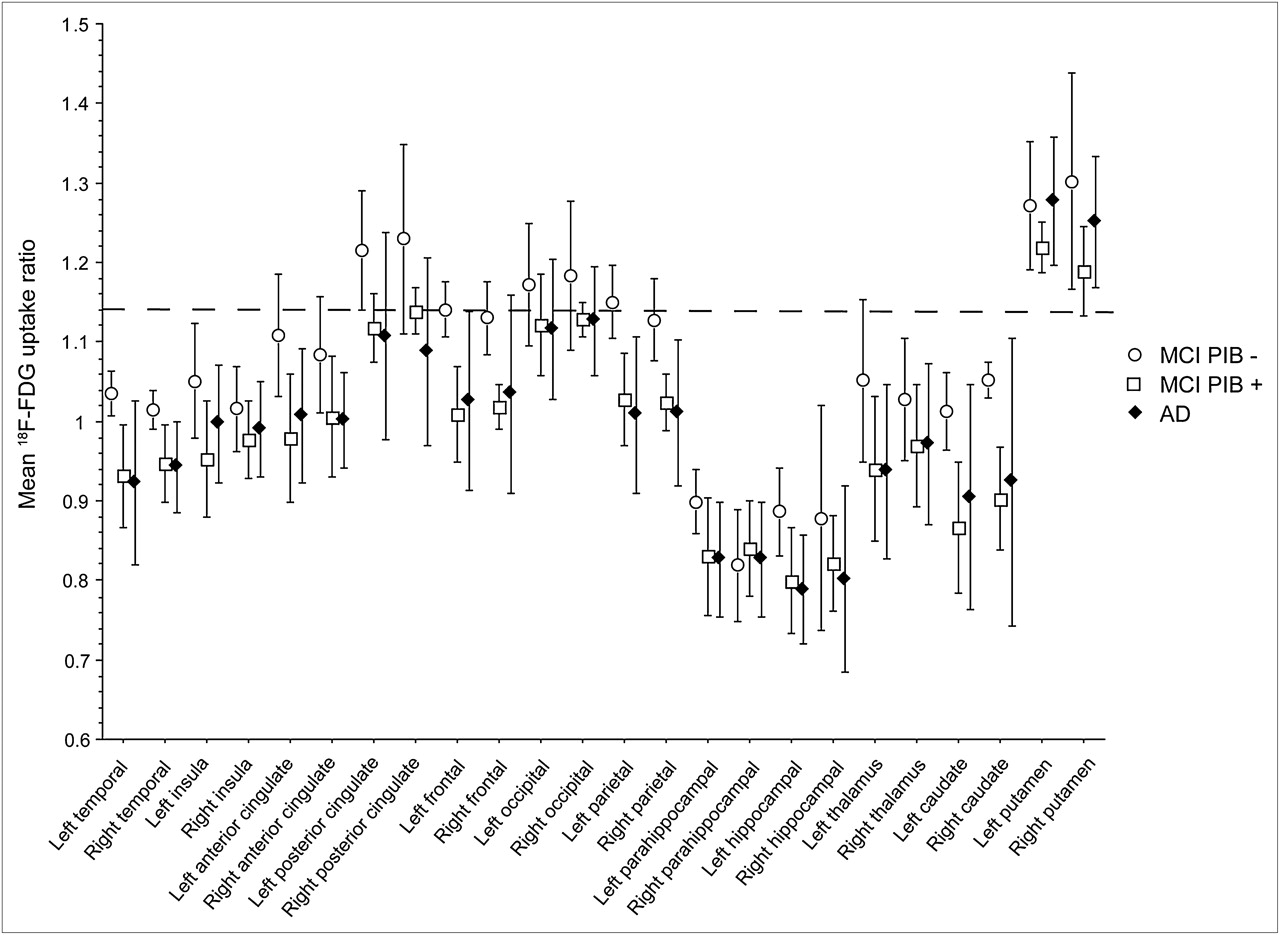
There was no difference in regional 18F-FDG uptake ratios between the MCI and AD patients and no difference between the MCI 11C-PIB+ and AD patients (Mann–Whitney U). However, the MCI 11C-PIB− patients had significantly greater 18F-FDG uptake ratios than did the 11C-PIB+ patients (MCI and AD) (Mann–Whitney U; P ≤ 0.05) in the bilateral temporal, left frontal, bilateral parietal, right caudate, and left hippocampus. Figure 4 shows the regional values for the 18F-FDG uptake data expressed in ratio to the cerebellum; the dashed cutoff line is at 1.138, which defines abnormality in cortical regions (29). Patients are stratified by 11C-PIB status (positive and negative) and diagnosis.

Regional cortical and subcortical 18F-FDG uptake in the MCI 11C-PIB-, 11C-MCI PIB+, and AD patients. Dashed reference cutoff line is set at 1.138, below which represents abnormal in neocortex (29). Data are mean ± SD.
Interrelationship of 11C-DED, 11C-PIB, and 18F-FDG PET Data
To determine the interrelationship between the different PET tracers, 1-tailed Pearson correlations were performed. To control for the large number of pairwise comparisons being calculated (12 bilateral regions and 4 different PET parameters, 11C-PIB retention, 18F-FDG uptake, 11C-DED slope, and 11C-DED intercept; 96 regions in total), a Bonferroni-corrected P value of 0.05 was used (yielding a significance level of 0.05/96). No pairwise correlations exceeded this P value. Some limited regional correlations between the tracers were revealed but at much lower levels of significance (P = 0.05 uncorrected; Supplemental Table 1 displays these correlations [supplemental materials are available online only at http://jnm.snmjournals.org]).
DISCUSSION
The main objective of the investigation was to determine whether there was a regional increase of MAO-B in the brains of MCI and AD patients. Significantly increased 11C-DED binding was found in the frontal and parietal cortices of the MCI group only. A significant group effect of 11C-DED intercept values was also found; this effect was related to the healthy controls having the greatest values (healthy controls > MCI > AD) in several cortical and subcortical regions (Table 3). The combined MCI group had increased 11C-DED binding in the frontal and parietal cortices, and it is well established by many previous studies (4,30–32) that there are high levels of fibrillar Aβ in these cortical regions in MCI and AD patients.
The increase of 11C-DED binding to MAO-B was more evident in the MCI 11C-PIB+ patients when the MCI group was split into 11C-PIB+ and 11C-PIB− subgroups. The increased 11C-DED binding in these patients might be related to an increased number of astrocytes reacting to greater quantities of Aβ. Activity of MAO-B increases in AD patients’ brains, and the enzyme is overexpressed in reactive astrocytes that colocalize with Aβ plaques (18–20). The fact that the MCI PIB+ patients demonstrated the highest 11C-DED binding (slope) of the 3 groups possibly suggests that reactive astrocytosis could be an early dynamic phenomenon in the time course of AD; this finding is supported by greater 11C-DED binding in the earliest Braak stages (I–II) (20) of AD postmortem brain tissue. The idea that astrocytosis is an early phenomenon that occurs along with early amyloid deposition in the time course of AD has been previously hypothesized (3,33).
Subdividing the MCI patients also revealed a clear difference between them. The 11C-PIB− MCI patients (n = 3) had normal CSF values for Aβ42, τ, and p-τ; these patients were less cognitively impaired and had significantly more preserved 18F-FDG uptake throughout the neocortex. Crucially, the bilateral posterior cingulate in the 11C-PIB− patients was above the cutoff line (18F-FDG uptake ratio > 1.138), suggestive of a non-AD syndrome. The MCI 11C-PIB+ patients were on a continuum between the MCI 11C-PIB− patients and the AD patients for CSF measures. The MCI 11C-PIB+ group was more cognitively impaired than the MCI 11C-PIB− group but not as severely as the AD patients. With respect to 18F-FDG uptake, the MCI 11C-PIB+ group essentially already had an AD-like pattern throughout the brain. It is highly likely that the MCI 11C-PIB+ patients are actually prodromal AD; however, at the time of investigation they were not sufficiently impaired cognitively to meet the NINCDS-ADRDA criteria for AD.
There were no significant relationships when the regional data from the 3 PET tracers (11C-DED, 11C-PIB, and 18F-FDG) were correlated. In agreement with the poor association between the in vivo PET tracers in this investigation, a lack of significant correlation was found between in vivo 11C-PIB retention and postmortem 3H-PIB binding, binding of 3H-PK11195 (microglia), and 3H-L-deprenyl binding (MAO-B) in AD brain tissue from 1 patient (21). Furthermore, regional glucose metabolism (18F-FDG PET) in vivo did not correlate with binding of 3H-PK11195 or 3H-L-deprenyl binding at autopsy. This postmortem study revealed a significant positive correlation between regional in vivo 11C-PIB retention and the total number of GFAP immunoreactive cells. In vivo 11C-PIB might correlate with postmortem GFAP immunohistochemistry but not 3H-L-deprenyl because of the fact that GFAP is not specific to reactive astrocytes (34). Measurement of astrocytes postmortem in an AD brain using GFAP immunohistochemistry is likely to be measure of the total number of astrocytes after the early neuroinflammatory phase has finished. This idea is supported by the 11C-DED data from the MCI 11C-PIB+ and AD patients in the current investigation, in which the 11C-PIB+ patients had the greatest 11C-DED binding and the AD patients had the least.
Regional atrophy is unlikely to explain the results of the current investigation because a GM-specific atlas was adopted. These individual atlases sampled only voxels having greater than 50% probability of belonging to the GM, limiting partial-volume effects on the current data. Also, one would expect the pattern of atrophy to be as follows: AD > MCI > healthy controls, whereas the 11C-DED binding data demonstrated a pattern of MCI > AD ≥ healthy controls. Another methodologic factor to consider is the effect of blood flow on the 11C-DED PET data. 11C-DED is a highly flow-dependant tracer (12,13,27), and it has long been known that cerebral blood flow (CBF) is reduced in AD when measured with PET, SPECT, and arterial spin labeling (35–38). In brain regions that have reduced CBF and high MAO-B, the critical factor is 11C-DED delivery and not 11C-DED binding per se. If MAO-B activity is increased in MCI and AD because of the presence of greater astrocytosis, then the possibility remains that the 11C-DED binding in these individuals is actually underestimated because of their reduced CBF.
One of the limitations of the current investigation is that no arterial blood samples were obtained. Acquiring arterial blood samples would have allowed the 11C-DED data to be modeled more accurately and would have permitted the dynamic interaction between tracer delivery (blood flow or intercept) and tracer binding (slope) to be investigated in more detail. The current data, even without an arterial input function, indicate that the contribution of CBF and vascular integrity needs further investigation to determine the relative contribution of cerebral amyloid angiopathy to vascular integrity. In pure AD cases, the contribution of cerebral amyloid angiopathy is thought to be minimal (39). However, this idea still requires further investigation because cerebral amyloid angiopathy will not only limit in vivo 11C-DED delivery but also generate a proportion of the in vivo 11C-PIB signal.
Investigation of amyloid in the vasculature will need to be assessed along with the absolute quantification of the different types of amyloid deposits. Postmortem investigations (21,39) have shown that in vivo 11C-PIB retention is strongly correlated with the 11C-PIB binding in postmortem tissue throughout the brain. It is also known that of the 2 main types of Aβ deposits (neuritic and diffuse), reactive astrocytes and activated microglia are associated only with the neuritic type (19). If 11C-PIB in vivo labels all fibrillar amyloid but only neuritic plaques are surrounded by reactive astrocytes, then this might partially explain the absence of correlation between the 11C-DED and 11C-PIB tracers. This suggestion reinforces the need for more specific amyloid and astrocyte PET tracers.
Along with addressing the absolute quantity and type of amyloid deposits relative to reactive astrocytes, several factors remain unanswered with respect to 11C-DED. It is highly specific for the MAO-B enzyme; however, MAO-B is found in astrocytes and within the serotonergic neurons (9,10). Therefore, it is unknown how much of the 11C-DED signal is truly related to astrocytosis only. The regional location and relative quantification of MAO-B in other cell types (serotonergic neurons and nonreactive astrocytes) not related to astrocytosis still needs further examination.
Investigating these factors might permit increased understanding of the dynamic relationship between perfusion, amyloid deposition, and total MAO-B occupancy in the brains of MCI and AD patients.
CONCLUSION
This investigation shows that increased 11C-DED binding to MAO-B occurs early in the progression of AD, particularly in MCI 11C-PIB+ patients’ brains. This finding provides evidence that astrocytosis could be an early phenomenon in AD evolution that decreases with advanced disease severity. This suggestion requires further investigation, with parallel in vivo neuroimaging and follow-up postmortem tissue analysis. The present data further highlight the need to establish specific biomarkers to precisely explain the biologic complexity of prodromal AD.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We are grateful to Kerstin Heurling (PET modeling), Jan Axelsson (MATLAB expertise), and Gunnar Blomquist (PET modeling), whose help completing the data analysis was invaluable. This study was supported by the Knut and Alice Wallenberg Foundation, the Swedish Research Council (project 05817), Swedish Brain Power, the regional agreement on medical training and clinical research (ALF) between Stockholm County Council and the Karolinska Institutet, the Strategic Research Program in Neuroscience at Karolinska Institutet, the Foundation for Old Servants, the Gun and Bertil Stohnes Foundation, Karolinska Institutet foundations, the Swedish Brain Foundation, and the Alzheimer Foundation in Sweden. No other potential conflict of interest relevant to this article was reported.
Footnotes
↵* Contributed equally to this work.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication December 25, 2010.
- Accepted for publication August 24, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Cell Adhesion Molecule Protocadherin-{gamma}C5 Ameliorates A{beta} Plaque Pathogenesis by Modulating Astrocyte Function in Alzheimer's Disease
- Effects of Pesticide Exposure on Neuroinflammation and Microglial Gene Expression: Relevance to Mechanisms of Alzheimers Disease Risk
- Assessment of Neurovascular Uncoupling: APOE Status is a Key Driver of Early Metabolic and Vascular Dysfunction
- Cerebrovascular disease drives Alzheimer plasma biomarker concentrations in adults with Down syndrome
- Relationship between reactive astrocytes, by [18F]SMBT-1 imaging, with amyloid-beta, tau, glucose metabolism, and microgliosis in mouse models of Alzheimers disease
- "FDA-approved carbonic anhydrase inhibitors reduce Amyloid {beta} pathology and improve cognition, by ameliorating cerebrovascular health and glial fitness"
- Assessing Reactive Astrogliosis with 18F-SMBT-1 Across the Alzheimer Disease Spectrum
- First-in-Humans Evaluation of 18F-SMBT-1, a Novel 18F-Labeled Monoamine Oxidase-B PET Tracer for Imaging Reactive Astrogliosis
- Visualization of reactive astrocytes in living brain of Alzheimers disease patient
- High-contrast in-vivo imaging of tau pathologies in Alzheimers and non-Alzheimers disease tauopathies
- Development of an In Vivo Method to Estimate Effective Drug Doses and Quantify Fatty Acid Amide Hydrolase in Rodent Brain using Positron Emission Tomography Tracer [11C]DFMC
- Cortical microstructural correlates of astrocytosis in autosomal-dominant Alzheimer disease
- Endothelin type B receptor promotes cofilin rod formation and dendritic loss in neurons by inducing oxidative stress and cofilin activation
- Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer's disease model
- Correlations of 18F-THK5351 PET with Postmortem Burden of Tau and Astrogliosis in Alzheimer Disease
- Sembragiline: A Novel, Selective Monoamine Oxidase Type B Inhibitor for the Treatment of Alzheimers Disease
- Calcineurin/NFAT Signaling in Activated Astrocytes Drives Network Hyperexcitability in A{beta}-Bearing Mice
- Comparison of Early-Phase 11C-Deuterium-L-Deprenyl and 11C-Pittsburgh Compound B PET for Assessing Brain Perfusion in Alzheimer Disease
- In Vivo and In Vitro Characterization of a Novel MAO-B Inhibitor Radioligand, 18F-Labeled Deuterated Fluorodeprenyl
- Imaging of neuroinflammation in dementia: a review
- In Vivo Detection of Age- and Disease-Related Increases in Neuroinflammation by 18F-GE180 TSPO MicroPET Imaging in Wild-Type and Alzheimer's Transgenic Mice
- Enhancing Astrocytic Lysosome Biogenesis Facilitates A{beta} Clearance and Attenuates Amyloid Plaque Pathogenesis
- Targeting Astrocytes Ameliorates Neurologic Changes in a Mouse Model of Alzheimer's Disease
- Early Stage Drug Treatment That Normalizes Proinflammatory Cytokine Production Attenuates Synaptic Dysfunction in a Mouse Model That Exhibits Age-Dependent Progression of Alzheimer's Disease-Related Pathology