


Abstract
[S-methyl-11C](±)-7-methoxy-3-(4-(4-(methylthio)phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (11C-NR2B-SMe) and its enantiomers were synthesized as candidates for imaging the NR2B subunit within the N-methyl-d-aspartate receptor with PET. Methods: Brains were scanned with PET for 90 min after intravenous injection of one of the candidate radioligands into rats. To detect any NR2B-specific binding of radioligand in brain, various preblocking or displacing agents were evaluated for their impact on the PET brain imaging data. Radiometabolites from brain and other tissues were measured ex vivo and in vitro. Results: Each radioligand gave high early whole-brain uptake of radioactivity, followed by a brief fast decline and then a slow final decline. 11C-(S)-NR2B-SMe was studied extensively. Ex vivo measurements showed that radioactivity in rat brain at 30 min after radioligand injection was virtually unchanged radioligand. Only less lipophilic radiometabolites appeared in plasma. High-affinity NR2B ligands, Ro-25-6981, ifenprodil, and CO101244, showed increasing preblocking of whole-brain radioactivity retention with increasing dose (0.01–3.00 mg/kg, intravenously). Five σ1 antagonists (FTC146, BD1407, F3, F4, and NE100) and 4 σ1 agonists ((+)-pentazocine, (±)-PPCC, PRE-084, and (+)-SKF10047) were ineffective preblocking agents, except FTC146 and F4 at a high dose. Two potent σ1 receptor agonists, TC1 and SA4503, showed dose-dependent preblocking effects in the presence or absence of pharmacologic σ1 receptor blockade with FTC146. Conclusion: 11C-(S)-NR2B-SMe has adequate NR2B-specific PET signal in rat brain to warrant further evaluation in higher species. TC1 and SA4503 likely have off-target binding to NR2B in vivo.
N-methyl-d-aspartate (NMDA) receptors are ligand and voltage-gated ion channels that mediate influx of Ca2+, Na+, and K+ into the synapse (1). These receptors are expressed throughout the central nervous system and play key physiologic roles in synaptic plasticity, learning, and memory. NMDA receptors are also implicated in the pathophysiology of several central nervous system disorders (2–4) and more recently have been identified as a target for the treatment of disease-associated genomic variation (5). NMDA receptors exist as diverse subtypes as a result of variation in the assembly of 7 subunits (NR1, NR2 subunits [NR2A−NR2D], and NR3 [A or B]) into tetrameric receptor complexes. Unique structural features of the NMDA receptor subtypes account for the tuning of their physiologic roles and their distinct pharmacologic properties. NMDA receptors, especially those enriched with NR2B subunits, endow the prefrontal cortex not only with important functionality but also with a major vulnerability to environmental insults and to risk factors for psychiatric disorders (6).
NMDA receptors have distinct binding sites for l-glutamate, glycine, d-serine, polyamines, Mg2+, phencyclidine, and Zn2+. Ligands for NMDA are of 4 types, namely glutamate binding site ligands, glycine binding site ligands, channel blockers, and N-terminal domain binding ligands. Some NMDA ligands have been developed into drugs, such as memantine for the treatment of Alzheimer disease (7,8). Attempts to produce radioligands for imaging NMDA receptors in living subjects with PET is an active area of research that has nonetheless met with only limited success (9,10).
NR2B is the most studied subunit within the NMDA receptor complex. This subunit is expressed throughout the central nervous system, with the highest concentration in the forebrain and the dorsal horn of the spinal cord (11). The NR2B subunit is a therapeutic target for schizophrenia, stroke, and neurodegenerative diseases, especially neuropathic pain. Therapeutics targeting NR2B rather than the NMDA channel may have fewer side effects. The quantification of NMDA NR2B subunit receptors in vivo with PET could help to elucidate the contribution of this receptor to neuropsychiatric disorders and also assist in drug development (10). A promising PET radioligand for NR2B, named 11C-(R)-Me-NB1, has been reported very recently and shows modest displaceable signal for NR2B in rats in vivo (12,13).
Here, within a broader medicinal chemistry campaign to develop PET radioligands with high specific signal and low background noise, we discovered (±)-7-methoxy-3-(4-(4-(methylthio)phenyl)butyl)-2,3,4,5-tetrahydro-1H-benzo[d]azepin-1-ol (NR2B-SMe) (chemical structure is shown in Supplemental Fig. 1; supplemental materials are available at http://jnm.snmjournals.org). This compound shows high affinity for NR2B in the nanomolar range, moderate lipophilicity, and amenability to labeling with 11C (half-life, 20.4 min). We therefore prepared 11C-NR2B-SMe and its enantiomers from S-methyl propionyl precursors for evaluation as NR2B PET radioligands in rats.
MATERIALS AND METHODS
General Methods
Animal Experiment Ethics and Approval
All animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals (https://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf) and were approved by the National Institute of Mental Health Animal Care and Use Committee.
Statistics
Subsequent numeric data are expressed as mean ± SD for n > 2, or as mean and range for n = 2.
Absolute Configuration and In Vitro Properties
The absolute configurations of (–)-NR2B-SMe and (+)-NR2B-SMe were determined by vibrational circular dichroism.
Radiosynthesis
Preparation of 11C-NR2B-SMe
(±)-methyl 3-((4-(4-(1-hydroxy-7-methoxy-1,2,4,5-tetrahydro-3H-benzo[d]azepin-3-yl)butyl)phenyl)thio)propanoate (NR2B-ester; 0.5 mg; 1.1 μmol) in dimethylformamide (0.40 mL) and tetra-n-butylammonium hydroxide in methanol (5 μL of 1 M stock solution in MeOH; 5 μmol) were loaded into a septum-sealed reaction vial (1-mL neck vial; Waters Corp.) of a Synthia apparatus (14). 11C-iodomethane (>37 GBq) was then swept with a stream of helium (15 mL/min) into the vial from a Microlab module and left at room temperature for 5 min. 11C-NR2B-SMe was separated out with high-performance liquid chromatography (HPLC) on a Luna C18 column (10 μm, 4.6 × 250 mm; Phenomenex) eluted at 6 mL/min for 20 min with trifluoroacetic acid (0.1% w/v) in MeCN water (35:65 v/v). Eluate was monitored for absorbance at 255 nm and radioactivity. The fraction containing 11C-NR2B-SMe (retention time, 10.0 min) was collected in ascorbic acid solution (10 mg/mL; 0.1 mL) and then rotary-evaporated to dryness (80°C, water bath). The residue was dissolved in sterile saline for injection (4 mL) containing ethanol (1 mL) and aqueous NaHCO3 solution (8.4% w/v; 40 μL). The solution (pH ∼5.5) was sterile-filtered into a sterile and pyrogen-free dose vial that had been preloaded with sterile saline for injection (5 mL).
This product was analyzed with HPLC on an X-Bridge C18 column (10 μm, 4.6 × 250 mm; Waters Corp.) eluted for 15 min with trifluoroacetic acid (0.1% w/v) in MeCN water (40:60 v/v) at 1.5 mL/min. Eluate was monitored for absorbance at 255 nm and for radioactivity. The retention time of 11C-NR2B-SMe was 9.5 min. The response of the analytic system had been calibrated for the mass of NR2B-SMe to allow molar activity to be calculated.
The identity of the radioligand was confirmed by its comobility in analytic HPLC with added reference compound and by liquid chromatography–mass spectrometry of residual carrier after decay of the formulated radioligand overnight. The formulated radioligand had greater than 98% radiochemical purity. The stability of formulated 11C-NR2B-SMe was assessed with radio-HPLC at 1 and 2 h after radiosynthesis.
Radiometabolites of 11C-NR2B-SMe in Rat Tissues Ex Vivo
Rats were anesthetized with 1.5% isoflurane in oxygen. Formulated 11C-NR2B-SMe (200 μL; 37 MBq) was then injected intravenously through the penile vein of each rat. Other experimental parameters related to this study are listed in Supplemental Table 1. Thirty minutes after injection of 11C-NR2B-SMe, a large anticoagulated (heparin) blood sample was drawn from each rat. The rats were then immediately sacrificed by decapitation, and their brains and myocardial tissues were excised.
For Brain and Myocardium
The radioactive tissue was weighed, placed in acetonitrile (1 mL), measured for radioactivity, and homogenized along with carrier NR2B-SMe (50 μg) using a hand-held tissue Tearor (model 985-370; BioSpec Products Inc.). Water (500 μL) was then added, and the tissue was further homogenized before measurement of radioactivity. The homogenate was centrifuged at 10,000g for 1 min. The supernatant was then analyzed with general HPLC method A (NR2B-SMe; retention time, ∼6.4 min). The precipitate was measured for radioactivity to determine the recovery of radioactivity in the supernatant that had been injected onto the HPLC column.
For Plasma
Plasma was separated from blood cells, and a sample (50 μL) was measured for radioactivity. An aliquot (450 μL) was placed in acetonitrile (720 μL) along with carrier NR2B-SMe (5 μg) and mixed well. Water (100 μL) was added, mixed well, measured for radioactivity, and centrifuged at 10,000g for 1 min. The supernatant was analyzed with radio-HPLC and the precipitate measured for radioactivity, as described for brain.
The stability of 11C-NR2B-SMe in rat tissues ex vivo was determined by dividing the percentage of radioactivity present as radioligand in the tissue sample measured with HPLC by the fractional radiochemical purity of the radioligand (Table 1).
Ex Vivo and In Vitro Stabilities of 11C-NR2B-SMe in Rat Tissues
The SUV due to radioligand or radiometabolite only was calculated by multiplying the total SUV of the tissue by the fraction of the radioligand or radiometabolite measured with HPLC (Table 2).
Concentrations of 11C-NR2B-SMe in Rat Tissues and Tissue-to-Plasma Radioactivity Ratios Measured Ex Vivo at 30 Minutes After Intravenous Injection
PET Imaging in Rats
Selection of Agents for Preblocking and Displacement for PET Experiments in Rats
The aims of preblocking and displacement experiments were to assess radioligand target engagement and to determine radioligand selectivity in vivo. Many ligands do not show selectivity for binding to NR2B over σ1 receptors (15–19). In addition, σ1 receptors exist in close proximity on cell membranes and may have direct interactions with NMDA receptors, modulating the behavior of NMDA receptor complexes (20). NR2B-SMe showed high binding affinity to the NR2B receptor but also showed weak binding affinity to σ1 receptors in vitro (Table 3). To address NR2B-SMe selectivity in PET imaging, we chose various NR2B, σ1, and σ2 ligands for preblocking and displacement experiments in normal rats in vivo with PET. The studied NR2B ligands are summarized in Table 4, σ1 antagonists and ligands of undetermined σ1 intrinsic efficacy in Table 5, and putative σ1 agonists in Table 6.
Pharmacologic Parameters for 11C-NB1 and 11C-NR2B-SMe
In Vitro and In Vivo Pharmacologic Parameters for NR2B Ligands
In Vitro and In Vivo Pharmacologic Parameters for σ1 Receptor Ligands (Not Identified as Agonists)
In Vitro and In Vivo Pharmacologic Parameters for Putative σ1 Receptor Agonists
Estimation of ED50 Values In Vivo from Dose–Response Data

(A) Ro-25-6981 dose-dependently preblocks rat whole-brain radioactivity uptake from intravenous injection of 11C-(S)-NR2B-SMe. (B) AUC between 20 and 90 min from A for 11C-(S)-NR2B-SMe at different dose of preblocking agent Ro-25-6981, highly selective antagonist for NR2B receptor. Data are for n = 1.
After administering 11C-NR2B-SMe or one of its enantiomers to rats at baseline, the whole-brain radioactivity concentration (SUV) was seen to almost stabilize by 20 min (Fig. 1). The SUV unit normalizes radioactivity concentration for rat weight and injected dose. We used the SUVs of time–activity curves of the deployed radioligand between 20 and 90 min to calculate areas under the curve (AUCs). AUCs obtained from the same production of radioligand were scaled to the value (or mean value) for the baseline experiment to give relative values (dubbed relative AUC 20–90 min) (Fig. 1). These data were used to estimate the dose of blocking agent that was effective for a 50% reduction of the AUC 20–90 min at baseline, here termed ED50 and reported as moles of administered blocker per kilogram of body weight. Where data permitted, the dose–response curves were fitted with GraphPad Prism software (version 8.1.1) to estimate the ED50 values. A dummy value of 100 pmol per kilogram of body weight was used for zero concentration of the challenge agent in the construction of these dose–response curves.
RESULTS
NR2B-SMe and Its Chemical Properties
Absolute Configuration
(–)-NR2B-SMe and (+)-NR2B-SMe were found to have R and S absolute configuration, respectively, as determined by comparison of vibrational circular dichroism and infrared spectra measured with the calculated Boltzmann-averaged spectra of the calculated conformations (Supplemental Figs. 2 and 3; Supplemental Table 2), after separation of the racemic mixture by chiral HPLC columns (Supplemental Figs. 4 and 5).
Binding Affinities In Vitro and Other Physical Properties
The Ki value of NR2B-SMe in the in vitro assay in transiently transfected mouse fibroblast cells expressing NMDA was 2.2 nM (Table 3). The acid dissociation constant (pKa) was determined to be 5.04 (Supplemental Fig. 6), and the partition coefficient (logD7.4) was 3.41.
Pharmacologic Screen
NR2B-SMe was found to be selective for binding to the NR2B subunit because a 10 μM concentration only weakly inhibited the binding of reference radioligands to numerous binding sites and receptors. At a 10 μm concentration, inhibition was greater than 10% for only a few binding sites and receptors. These were the calcium channel (10.6%), hERG channels (66.3%), guinea pig σ1 (83.7%), and PC12 cell σ2 (90.0%).
Experiments on HeLa Cells (21)
NR2B-SMe failed to compete with Cy3 dye for lysosome trapping in HeLa cells, in contrast with the positive control, loperamide (Supplemental Fig. 7).
Experiments with 11C-NR2B-SMe in Rats and Human Tissues
Stability of 11C-NR2B-SMe in Rat Whole Blood, Plasma, and Brain Ex Vivo and In Vitro
The radiochemical purity of 11C-NR2B-SMe was 98.4% throughout the experiment (duration, ∼3 h), after HPLC separation (Supplemental Fig. 8) and formulation (Supplemental Fig. 9).
At least 3 radiometabolites eluted before 11C-NR2B-SMe in the reversed-phase HPLC analyses of rat plasma ex vivo (Supplemental Fig. 10A). These were very minor in brain and myocardium ex vivo (Supplemental Figs. 10B and 10C). Unchanged radioligand at 30 min after injection accounted for 71.6% of radioactivity in rat plasma, 99.2% in brain, and 90% in myocardial tissue (Table 1). Brain and myocardium showed high ratios of radioligand concentration (SUV) to that in plasma (Table 2). Ex vivo radioactivity in plasma distributed 51% to blood cells (22). The radiometabolites in vitro are the same as those observed ex vivo (Supplemental Fig. 11).
Plasma Free Fraction of 11C-NR2B-SMe
The human plasma free fraction of 11C-NR2B-SMe was 0.82% ± 0.01% (n = 3).
Evaluation of 11C-NR2B-SMe and Its Enantiomers in Rats with PET
After intravenous injection of a bolus of racemic 11C-NR2B-SMe into rat, there was a rapid and high uptake of radioactivity in brain followed by a quick decline to a moderately high stable level (Fig. 2A; Supplemental Fig. 12A). Relatively high uptakes were seen in thalamus, striatum, and cortex (Fig. 2A). When rats were intravenously administered NR2B-SMe or the NR2B ligand ifenprodil, at 3.0 mg/kg at 10 min before 11C-NR2B-SMe, the peak whole-brain and brain regional radioactivity uptake declined to a common low level at 90 min, corresponding to about 10% of peak values in the baseline experiment (Fig. 2A). Accordingly, summed PET images before and after ifenprodil treatment were strikingly different (Fig. 2B). Administration of the NR2B ligand Ro-25-6981 at 10 min after radioligand resulted in a dose-dependently faster whole-brain radioactivity decline (Supplemental Fig. 12B).

PET imaging of rat brain with racemic 11C-NR2B-SMe. (A) PET time–activity curves for selected brain regions in rat after intravenous injection of 11C-NR2B-SMe showing that brain radioactivity accumulation is blocked by preadministration of ifenprodil at 3.0 mg/kg (intravenous). (B) Summed transaxial PET images (60–90 min) from 11C-NR2B-SMe at baseline and after preblocking with NR2B ligand ifenprodil. Data are for n = 1.
The separate homochiral radioligands gave slightly different time–activity curves in whole brain (Fig. 3). The R-enantiomer (Fig. 3A) showed a somewhat faster radioactivity decline from peak value than the S-enantiomer (Fig. 3B). Intravenous injection of the NR2B ligand Ro-25-691 at 10 min before either homochiral radioligand lowered whole-brain retention of radioactivity. The effect was greater at the higher dose of 0.05 mg/kg than the lower dose of 0.01 mg/kg. As seen for the racemic radioligand, Ro-25-6981 administered at 10 min after the R-enantiomer resulted in a dose-dependently faster whole-brain radioactivity decline (Supplemental Fig. 12C).

(A and B) Blocking of whole-brain radioactivity uptake in rat by dosing with NR2B ligand Ro-25-6981 before intravenous injection of 11C-(R)-NR2B-SMe (A) and 11C-(S)-NR2B-SMe (B). (C) Fitted dose–response curves derived from B for NR2B-SMe and from A in Figure 1 for NR2B ligand Ro-25-6981. Data are for n = 1.
The abilities of NR2B ligands to preblock whole-brain radioactivity uptake before intravenous injection of 11C-(S)-NR2B-SMe into rats was studied in more detail. The time–activity curves for 11C-NR2B-SMe and each enantiomer showed dose-dependent preblocking by NR2B-SMe itself (Figs. 1 and 3C) and by the NR2B ligands ifenprodil (Supplemental Fig. 13A) and CO101244 (Supplemental Fig. 13C). Sigmoidal dose–response curves (relative AUC 20–90 min vs. dose) for NR2B-SMe (Fig. 3C), ifenprodil (Supplemental Fig. 13B), and CO101244 (Supplemental Fig. 13D), as well as for the NR2B ligand Ro-25-6981 (Fig. 3C), were readily fitted to provide ED50 values (nmol/kg) (Table 4). These ED50 values were in rank order of their NR2B binding potencies (1/Ki) in vitro (Table 4).
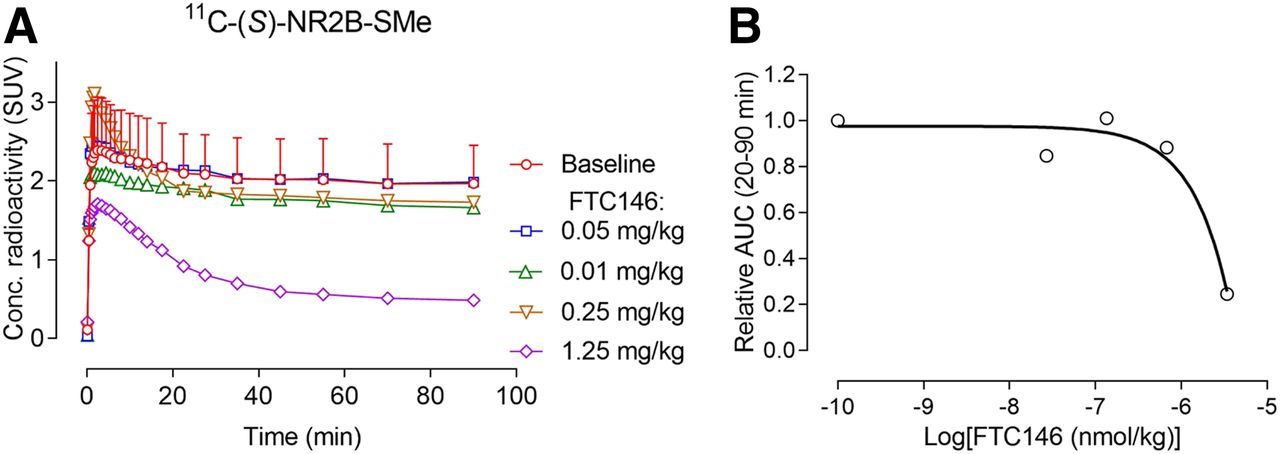
σ1 receptor antagonists, such as BD1047, NE100, and F4, showed minimal preblocking of radioactivity retention in whole rat brain before the administration of 11C-(S)-NR2B-SMe (Supplemental Fig. 14). No meaningful sigmoidal dose–response curves could be fitted for these data, showing that the ED50 values for these ligands were much greater than 1,000 nmol/kg, in contrast to their affinities for σ1 receptors in vitro, which are in the low nanomolar range (Table 5), or for the affinity of BD19147 at σ2 receptors, which is quite low at 47 nM (Table 5). The very high affinity σ1 receptor antagonist FTC146 caused preblocking only at a high dose of 1.25 mg/kg intravenously (Fig. 4A). The σ1 ligand F3 also showed a dose-dependent preblocking effect (Supplemental Fig. 14). Dose–response curves could be fitted for FTC146 (Fig. 4B) and F3 (Supplemental Fig. 14) and allowed ED50 values to be estimated. These ED50 values were found to be extremely high relative to their Ki values at σ1 receptors in vitro (Table 5).

(A) Low to moderate preadministered intravenous doses of highly potent σ1 receptor antagonist FTC146 do not reduce whole-brain radioactivity in rats after intravenous injection of 11C-(S)-NR2B-SMe. (B) Fitted dose–response curve for FTC146 derived from data in A. Baseline data are mean for n = 2. Error bar represents range. Other data are for n = 1.
σ1 receptor agonists, such as (+)-pentazocine, (±)-PPCC, PRE-084, and (+)-SKF10047 (Supplemental Fig. 15), showed very weak preblocking effects on radioactivity retention in whole rat brain before administration of 11C-(S)-NR2B-SMe. These data could not be fitted to sigmoidal dose–response curves, and ED50 estimates were therefore extremely high (>1,000 nmol/kg) (Table 6). Exceptionally, the σ1 agonists TC1 (Fig. 5A) and SA4503 (Fig. 6A) showed dose-dependent preblocking effects that could be fitted to sigmoidal dose–response curves (Figs. 5C and 6C, respectively). The ED50 values for TC1 and SA4503 were estimated to be quite low at 45 and 29 nmol/kg, respectively (Table 6). However, the preblocking effects of these 2 σ1 agonists were unaltered when the potent σ1 receptor antagonist, FTC146, was used to pharmacologically block σ1 receptors (Figs. 5 and 6). The respective dose–response curves (Figs. 5B, 6B, 5C, and 6C) were congruent with those obtained in the absence of σ1 receptor blocking with FTC146.

(A) Intravenous preadministration of TC1 dose-dependently reduces rat whole-brain radioactivity uptake from intravenous injection of 11C-(S)-NR2B-SMe. (B) Intravenous preadministration of TC1 in rats that have been simultaneously pretreated with FTC146 (0.05 mg/kg, intravenously) at 10 min before 11C-(S)-NR2B-SMe dose-dependently reduces whole-brain radioactivity uptake. (C) Fitted dose–response curves from A and B data. Curves in A and B are highly congruent, as are curves in C, showing no effect of σ1 receptor blockade by FTC146. Baseline data are mean for n = 2. Error bar represents range. Other data are for n = 1.

(A) Preadministered intravenous doses of SA4503 reduced whole-brain radioactivity uptake in rats from intravenous injection of 11C-(S)-NR2B-SMe. (B) Simultaneous intravenous preadministration of SA4503 and FTC146 (0.05 mg/kg, intravenously) at 10 min before 11C-(S)-NR2B-SMe dose-dependently reduced whole-brain radioactivity uptake. (C) Fitted dose–response curves derived from A and B data. Curves in A and B are highly congruent, as are curves in C, showing no effect of σ1 receptor blockade by FTC146. Data are for n = 1.
DISCUSSION
We were able to resolve the enantiomers of NRB2B-SMe by chiral HPLC. Knowledge of the absolute configuration of NR2B-SMe could be valuable in future NR2B ligand design. We were able to assign the absolute configurations of NR2B-SMe enantiomers successfully with infrared and vibrational circular dichroism.
11C-NR2B-SMe and its enantiomers were readily prepared for intravenous injection into rats by treating NR2B ester with 11C-methyl iodide under basic conditions. The radiolabeling process is likely a reverse Michael addition of α,β-unsaturated acrylic acid or ester (Supplemental Fig. 1) (23). 11C-NR2B-SMe decomposed when concentrated immediately after HPLC isolation, but the decomposition was minimal after ascorbic acid was added during the removal of mobile phase. A constant radiochemical purity of greater than 98% was then achieved.
Initial PET experiments on rats used racemic 11C-NR2B-SMe. These experiments showed that 11C-NR2B-SMe readily entered brain, peaking at approximately 3 SUVs in whole brain at about 2.5 min (Fig. 2A). Peak radioactivity was followed by a slow decline in radioactivity. Radioactivity retention in brain regions such as thalamus, cortex, and cerebellum could be preblocked with NR2B-SMe itself and by the NR2B ligand ifenprodil (Fig. 2A). Thalamus and cortex are generally considered to be NR2B-rich regions. However, appreciable specific binding in cerebellum would not have been expected on the basis of previous in vitro autoradiography of postmortem rat brain with NR2-specific antibody (24). Our finding of specific binding in cerebellum that can be blocked by ifenprodil is consistent with findings of Krämer et al. (12), who observed high specific binding of their putative NR2B radioligand, 11C-Me-NB1, to rat cerebellum with both in vitro autoradiography and ex vivo biodistribution measurements. Yet, deeper investigations are needed to verify that specific binding in cerebellum is to NR2B. The radioactivity in brain could be partially displaced by the NR2B radioligand Ro-25-6981, suggesting reversibility of radioligand binding to NR2B (Supplemental Fig. 12).
At 30 min after intravenous administration, unchanged radioligand represented virtually all rat brain radioactivity (>99%), a finding that was highly favorable to pursuing further radioligand characterization. Parent 11C-NR2B-SMe represented close to 70% of radioactivity in plasma at 30 min after injection of 11C-NR2B-SMe, showing that peripheral metabolism in vivo was relatively slow (Table 1). 11C-NR2B-SMe showed a high accumulation in NR2B-rich heart (25), as well as in brain relative to blood (Table 2). 11C-NR2B-SMe also showed high metabolic stability in rat whole blood, plasma, and brain in vitro (Table 1).
Because 11C-NR2B-SMe was strongly retained in rat brain at baseline, we investigated whether trapping in lysosomes might be responsible. Lysosomes are membrane-bound organelles found in nearly all animal cells (26). With a pH ranging from 4.5 to 5.0, the interior of the lysosomes is acidic whereas cytosol is slightly basic (pH 7.2). Protonated weak bases may accumulate within lysosomes, in some cases reaching 100- to 1,000-fold higher concentrations than the extracellular concentration, a phenomenon known as lysosomotropism (27) or proton pump effect (28). The accumulation of compounds in lysosomes may be estimated from compound pKa (29). NR2B-SMe is monobasic. The optimal pKa range for lysosomal trapping of a monobasic compound is between 6 and 10. Therefore, we measured the pKa of 11C-NR2B-SMe. We also investigated the uptake of NR2B-SMe into HeLa cell lysosomes in vitro.
The apparent pKa of 11C-NR2B-SMe was found to be 5.04 ± 0.01 (n = 3) (Supplemental Fig. 6). Therefore, 11C-NR2B-SMe will be protonated to only a very low extent (0.63%) at the physiologic pH of cytosol (pH 7.2) but will be protonated to a greater extent (76%–50%) in lysosomes (pH 4.5–5.0). The pKa of 11C-NR2B-SMe, however, suggested a low risk for lysosomal trapping.
Conclusive evidence against lysosomal trapping of 11C-NR2B-SMe came from experiments on HeLa cells, in which NR2B-SMe competes with the cyanine dye Cy3 for cell uptake. The greenish yellow fluorescence of the dye did not change on addition of NR2B-SMe, just as with addition of dimethylsulfoxide only, whereas the dye was excluded from the cells with loperamide (50 μM), a known ligand for lysosome trapping (30).
The lipophilicity of a PET radioligand, as indexed by logD at pH 7.4, is a key property that influences many aspects of PET radioligand behavior in vivo (31), including brain entry, metabolism, and protein binding. We found the logD7.4 of 11C-NR2B-SMe to be 3.41, which is close to that predicted by computation (2.98) and in the range for many successful PET radioligands for central nervous system targets.
The stability of 11C-NR2B-SMe in human brain homogenate and human plasma and the plasma free fraction in vitro were of interest with respect to possible eventual radioligand application in human subjects. 11C-NR2B-SMe was virtually unchanged when exposed to these tissues for 30 min at room temperature in vitro, suggesting that radiometabolites would not be generated in human brain in vivo.
The plasma free fraction of a PET radioligand can be an important parameter for the use of a PET radioligand for quantifying a protein target in human subjects. The human plasma free fraction for 11C-NR2B-SMe in human plasma was relatively low, but measurable with good precision (0.82% ± 0.01%, n = 3).
Generally, PET radioligands should not be used as racemates because their enantiomers may show differences in binding affinities for their targets and possibly other differences in, for example, kinetics and metabolism. Given the encouraging PET results and encouraging in vitro measures with racemic 11C-NR2B-SMe, we proceeded to evaluate the enantiomers of 11C-NR2B-SMe. Each homochiral radioligand showed brain uptake similar to that of the racemic radioligand in rats. The S-enantiomer showed a slower decline in whole-brain radioactivity concentration from peak value than did the R-enantiomer. The NR2B ligand Ro-25-6981 was effective in blocking the brain retention of radioactivity from both enantiomers, suggesting they both show specific binding to NR2B (Fig. 3). As also seen for the racemic radioligand, radioactivity in brain from the R-enantiomer could be displaced with Ro-25-6981 in a dose-dependent manner, thereby confirming the reversibility of specific binding (Supplemental Fig. 12). Because 11C-(S)-NR2B-SMe showed the stronger retention in rat brain, subsequent experiments were performed with this radioligand. They were primarily directed at exploring the specificity of the PET signal.
In total, we used 3 NR2B ligands (Ro-25-6981, CO-101244, and ifenprodil) and NR2B-SMe itself as preblocking agents at various doses for PET experiments on 11C-(S)-NR2B-SMe in rats. Each ligand gave data that could be fitted to a dose–response curve for the estimation of ED50 values in vivo. The ED50 values were in rank order of the Ki values in vitro (Table 6). These results are therefore consistent with 11C-NR2B-SMe selectively occupying the NR2B binding site in rat brain in vivo.
σ1 receptors are transmembrane proteins expressed in many different tissues. They are particularly concentrated in certain regions of the central nervous system (32) and may function as a chaperone to NMDA receptors (33,34). Five antagonists of the σ1 receptor (FTC146, BD1047, NE100, F3, and F4) had very limited preblocking effects on the PET radioligand 11C-NR2B-SMe (Fig. 4; Supplemental Fig. 14; Table 5), further suggesting that 11C-NR2B-SMe does not occupy σ1 receptors. Four of 6 tested σ1 receptor agonists had only weak preblocking effects (i.e., high ED50 values) (Supplemental Fig. 14; Table 6). However, 2 putative selective σ1 agonists, TC1 and SA4503, had strong effects on the brain uptake of 11C-NR2B-SMe (Figs. 5 and 6; Table 6). We considered that these 2 ligands might have off-target binding to NR2B receptors. Therefore, we designed preblocking PET experiments with TC1 or SA4503 and 11C-(S)-NR2B-SMe in which σ1 receptors would be pharmacologically blocked with the high-affinity antagonist FTC146 (KD = 2.5 pM). For these experiments, we selected an FTC146 dose of 0.05 mg/kg (intravenous), which we estimated would achieve an exceptionally high peak brain concentration of 134 nM, which should assuredly occupy all σ1 receptors, even if the brain free fraction was exceptionally low. This estimate was based on published data (18F-FTC146 peak brain uptake, ∼1 SUV in mice) (35,36). The effects of TC1 and SA4503 on brain uptake of 11C-(S)-NR2B-SMe were the same as in the absence of FTC146 (Figs. 5 and 6, respectively). Therefore, preblocking of σ1 receptors did not prevent interactions of TC1 and SA4503 with the NR2B receptor. This strongly suggests that these σ1 agonists have some affinity for NR2B receptors. Also, these data further suggest that 11C-(S)-NR2B-SMe does not have appreciable off-target binding to σ1 receptors in rat brain in vivo.
CONCLUSION
Racemic 11C-NR2B-SMe binds selectively and with high affinity to the NR2B subunit. Absolute configurations were successfully assigned to the enantiomers of NR2B-SMe. 11C-NR2B-SMe and its enantiomers were readily prepared as prospective radioligands for PET imaging of NR2B subunits within NMDA receptors. In normal rats, these radioligands show high initial brain radioactivity uptake, followed by slow washout. The radioactivity in brain is predominantly unchanged radioligand and is not trapped in lysosomes. 11C-(S)-NR2B-SMe shows a slower decline in brain concentration than its antipode. Brain uptake of this radioligand is reversible and can be preblocked up to 90% with a variety of NR2B ligands in a dose-dependent way. Brain uptake of 11C-NR2B-SMe cannot be preblocked by σ1 receptor antagonists, consistent with the radioligand occupying the NR2B binding site, not the σ1 receptor. However, brain uptake of 11C-(S)-NR2B-SMe can be preblocked by 2 of 6 σ1 receptor agonists, consistent with a direct interaction of the 2 agonists with NR2B. Altogether, this study suggests that 11C-(S)-NR2B-SMe has adequate NR2B-specific PET signal in rat brain to warrant yet deeper investigation of its specific binding across brain regions, including cerebellum, and further evaluation as a radioligand in higher species.
DISCLOSURE
This study was funded by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health: projects ZIAMH002795 and ZIAMH002793. No other potential conflict of interest relevant to this article was reported.
KEY POINTS
QUESTION: Can we image the brain NR2B receptor using PET?
PERTINENT FINDINGS: PET imaging using normal rats has shown that 11C-NR2B-SMe in both racemic and chiral forms has specific signal that can be preblocked and displaced using ligands specifically targeting NR2B receptors but not using ligands specifically targeting σ1 receptors.
IMPLICATIONS FOR PATIENT CARE: 11C-NR2B-SMe has potential as a PET radioligand to measure the distribution of NR2B receptors in human brain.
Acknowledgments
We thank Dr. Mary Herman (CDBD, National Institute of Mental Health) for postmortem human brain tissues. We also thank the NIH PET Department for 11C production, PMOD Technologies for providing the image analysis software, and PDSP for performing assays: the PDSP is directed by Bryan L. Roth, PhD, with project officer Jamie Driscoll (National Institute of Mental Health) at the University of North Carolina Chapel Hill (contract NO1MH32004).
Footnotes
Published online Jan. 10, 2020.
- © 2020 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication August 14, 2019.
- Accepted for publication December 19, 2019.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}