


Abstract
Accumulation of hyperphosphorylated tau, a microtubule-associated protein, plays an important role in the progression of Alzheimer disease. Animal studies suggest that one strategy for treating Alzheimer disease and related tauopathies may be inhibition of O-GlcNAcase (OGA), which may subsequently decrease pathologic tau phosphorylation. Here, we report the pharmacokinetics of a novel PET radioligand, 18F-LSN3316612, which binds with high affinity and selectivity to OGA. Methods: PET imaging was performed on rhesus monkeys at baseline and after administration of either thiamet-G, a potent OGA inhibitor, or nonradioactive LSN3316612. The density of the enzyme was calculated as distribution volume using a 2-tissue-compartment model and serial concentrations of parent radioligand in arterial plasma. The radiation burden for future studies was based on whole-body imaging of monkeys. Oga∆Br, a mouse brain-specific knockout of Oga, was also scanned to assess the specificity of the radioligand for its target enzyme. Results: Uptake of radioactivity in monkey brain was high (∼5 SUV) and followed by slow washout. The highest uptake was in the amygdala, followed by striatum and hippocampus. Pretreatment with thiamet-G or nonradioactive LSN3316612 reduced brain uptake to a low and uniform concentration in all regions, corresponding to an approximately 90% decrease in distribution volume. Whole-body imaging of rhesus monkeys showed high uptake in kidney, spleen, liver, and testes. In Oga∆Br mice, brain uptake of 18F-LSN3316612 was reduced by 82% compared with control mice. Peripheral organs were unaffected in Oga∆Br mice, consistent with loss of OGA expression exclusively in the brain. The effective dose of 18F-LSN3316612 in humans was calculated to be 22 μSv/MBq, which is typical for 18F-labeled radioligands. Conclusion: These results show that 18F-LSN3316612 is an excellent radioligand for imaging and quantifying OGA in rhesus monkeys and mice. On the basis of these data, 18F-LSN3316612 merits evaluation in humans.
Neurofibrillary tangles, neuropil threads, and dystrophic neurites all contain hyperphosphorylated, insoluble tau protein and are a hallmark of Alzheimer disease and other tauopathies (1,2). The magnitude and location of pathologic tau correlate with the symptoms and severity of these diseases and suggest that reducing insoluble tau may have therapeutic benefits.
Studies in model systems have shown that hyperphosphorylation of tau partly depends on the extent of the protein’s O-GlcNAcylation, which is mediated by 2 enzymes (3). One enzyme, UDP-N-acetyl-d-glucosamine:polypeptidyl-transferase (OGT), adds β-N-acetylglucosamine (GlcNAc) to serine and threonine residues. Another enzyme, O-GlcNAcase (OGA) (4), removes the O-linked GlcNAc (O-GlcNAc). Although the mechanism by which glycosylation affects phosphorylation is poorly understood, the presence of this sugar moiety is associated with a decrease in pathologic tau accumulation. Thus, increased glycosylation by either activating OGT or inhibiting OGA should decrease tau accumulation (4,5). Indeed, studies have found that OGA inhibition markedly decreased the accumulation of hyperphosphorylated, aggregated tau in different tau-overexpression mice (4,6,7).
Because of the potential therapeutic benefit of OGA inhibitors in human tauopathies, several potent and selective OGA inhibitors have been developed (8). In parallel with these efforts, radiolabeled analogs have been sought to measure the target engagement and enzyme occupancy of therapeutic candidates with PET. To date, results for 1 PET radioligand for OGA, 18FMK8553, were reported in a meeting abstract (9). The ligand’s structure was not revealed, but it was reported to be suitable for imaging OGA and determining enzyme occupancy in both preclinical and clinical studies.
Our laboratory also developed a radioligand, 18F-LSN3316612, to measure OGA in the brain. LSN3316612 is selective and has high affinity (half-maximal inhibitory concentration, 1.9 nM) to inhibit human OGA in vitro (10). Here, we report PET imaging results using 18F-LSN3316612 in rhesus monkeys and mice (both Oga∆Br (11) and control mice) to assess selectivity of the radioligand for the target enzyme in vivo and to measure its associated radiation burden. The overall goal was to determine whether 18F-LSN3316612 would merit evaluation in human subjects.
MATERIALS AND METHODS
Radiochemistry
LSN3316612 (N-(5-(((2S,4S)-2-methyl-4-(6-fluoropyridin-2-yloxy) piperidin-1-yl)methyl)thiazol-2-yl)acetamide) and nitro precursor (N-(5-(((2S,4S)-2-methyl-4-(6-nitropyridin-2-yloxy)piperidin-1-yl)methyl)thiazol-2-yl)acetamide) were synthesized at Eli Lilly and Co. (Fig. 1). Kryptofix 2.2.2 was obtained from Sigma-Aldrich. C18 solid-phase extraction cartridges (Waters) were conditioned before use by elution with ethanol (10 mL) and then water (10 mL). Radiochemistry was performed in a lead-shielded hot-cell to minimize radiation exposure to personnel. All radioactivity measurements were performed with a calibrated Atomlab 300 ionization chamber (Biodex Medical Systems) and corrected for background and physical decay. No-carrier-added 18F fluoride ion was obtained through the 18O (p,n) 18F nuclear reaction by irradiating 18O water (95 atom %) for 90–120 min with a proton beam (17 MeV; 20 μA) generated from a PETrace cyclotron (GE Healthcare).

Structure of 18F-LSN3316612.
18F-LSN3316612 was prepared in an automated TRACERlab FXF-N radiosynthesis apparatus (GE Healthcare). A solution composed of Kryptofix 2.2.2 (101 mg in 1.8 mL of MeCN) and K2CO3 (10.5 mg in 0.2 mL of H2O) was added to no-carrier-added 18F-fluoride ion in 18O-water from the cyclotron target and dried by 2 additions and evaporations of acetonitrile (2 mL each) under nitrogen at 65°C–88°C. Nitro precursor (1.6 ± 0.1 mg) in dimethyl sulfoxide (0.6 mL) was added to the dried 18F-fluoride complex and heated to 115°C for 20 min. The reaction mixture was then transferred into water (2 mL) and the 18F-LSN3316612 separated with high-performance liquid chromatography on a Luna C18 column (10 μm, 250 × 10 mm; Phenomenex) eluted with MeCN:0.01 M ammonium formate solution (30:70 v/v). The fraction containing 18F-LSN3316612 (Rt: 30–40 min, n = 6) was collected, diluted with water (70 mL), and passed through a preconditioned C18 solid-phase extraction cartridge. The cartridge was washed with water (5 mL) and then eluted with ethanol (1 mL) into a product collection vial. Saline (10 mL) was then passed through the C18 solid-phase extraction cartridge into the product collection vial to yield a fully formulated 18F-LSN3316612 solution. Finally, this solution was passed through a sterile filter (0.22-μm pore size; Millex-MP) into a sterile dose-vial (10 mL; Hospira) to yield a sterile 18F-LSN3316612 solution ready for injection. The identity of 18F-LSN3316612 was confirmed by its comobility with authentic LSN3316612 in high-performance liquid chromatography analysis on a Luna C18 column (10 μm, 250 × 4.6 mm; Phenomenex) eluted with MeCN:0.01 M ammonium formate solution (50:50 v/v). Liquid chromatography–mass spectrometry of radioligand carrier, calculated for C17H21FN4O2S m/z 364.1, found m/z 365.2 [M+H]+. The average molar activity of 18F-LSN3316612 at the time of injection was 48 ± 16 GBq/μmol at baseline as well as at the blocked and whole-body scans.
Animals
All animal experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals, eighth edition, and were approved by the National Institute of Mental Health Animal Care and Use Committee. PET imaging experiments were performed on rhesus monkeys (Macaca mulatta) and on Oga∆Br and control mice. Rhesus monkeys were anesthetized with ketamine (10 mg/kg intramuscularly) and then maintained with 1%−2% isoflurane and 98% O2. Mice were anesthetized with 1.5% isoflurane throughout the study.
Monkey Scan Acquisition
For the brain imaging studies, 4 rhesus monkeys (body weight, 10.8 ± 1.4 kg) underwent 6 PET scans: at baseline (n = 4) and after pretreatment with thiamet-G (10 mg/kg, n = 1) or with the nonradioactive parent compound (self-blocked, 1 mg/kg, n = 1). PET scans were acquired after injection of 18F-LSN3316612 (222 ± 19 MBq) using a microPET Focus 220 scanner (Siemens Medical Solutions) for 2–4 h, with frame duration ranging from 30 s to 10 min. Concurrent arterial blood sampling was performed in all scans to obtain metabolite-corrected input function for quantification. Electrocardiogram, body temperature, heart rate, and respiratory rate were monitored throughout the scans.
For whole-body imaging, 1 male (9.5 kg) and 1 female (8.6 kg) monkey underwent a whole-body scan at baseline. The male monkey was rescanned after pretreatment with thiamet-G (10 mg/kg intravenously). After 18F-LSN3316612 injection (230 ± 45 MBq), the animals were scanned for 2 h on the Siemens mCT scanner.
Measurement of 18F-LSN3316612 in Plasma
About 15 blood samples were drawn from the femoral artery during the 120-min PET scan every 15 s for the first 2 min followed by sampling at 3, 5, 10, 30, 60, 90, and 120 min (varying from 1.0 to 3.0 mL). The parent radioligand was separated from radiometabolites as previously described (12) except that an X-Terra C18 column (10 μm, 7.8 × 300 mm; Waters Corp.) and a mobile phase of MeOH:10 mM ammonium formate (1:3 v/v) were used.
Image Processing
PET images were reconstructed using Fourier rebinning plus 2-dimensional filtered backprojection with attenuation and scatter correction. Images were coregistered to a standardized monkey MRI template (13) using the FMRIB Software Library. Thirty-four predefined brain regions of interest from the template were applied to the coregistered PET image to obtain regional time–activity curves. Brain uptake was expressed as SUV, which normalizes for injected radioactivity and body weight.
Compartmental Analysis
Total distribution volume (VT) for different regions was calculated using an unconstrained 2-tissue-compartment model with the metabolite-corrected input function (PMOD, version 3.6; PMOD Technologies Ltd.). Specific binding of the radioligand was estimated by comparing VT in baseline and blocked conditions.
PET Imaging of Oga∆Br and Control Mice
Two Oga∆Br and 2 control (OGA homozygous flox) mice (27 ± 1.5 g) were scanned after injection of 18F-LSN3316612 (12.6 ± 0.1 MBq) at baseline and after thiamet-G administration, in 2 different sessions. All mice except one were female. PET scans were acquired for 100 min using a microPET Focus 120 camera (Siemens Medical Solutions). Images were reconstructed using Fourier rebinning plus 2-dimensional ordered-subsets expectation maximization without scatter and attenuation correction.
Dosimetry Analysis
The following source organs were identified: brain, lungs, heart, liver, kidneys, urinary bladder, small intestine, testes, ovaries, and thyroid. Regions were drawn on the individual transaxial or coronal slices of whole-body PET images. For each organ, residence time was calculated from area under the time–activity curves uncorrected for decay as described by Kimura et al. (14). Absorbed radiation doses for individual organs and total body were obtained using OLINDA/EXM 1.1 and the model for a 70-kg adult.
RESULTS
Properties of 18F-LSN3316612
For brain and whole-body scans of monkeys, the average injected activity was 252 ± 28 MBq, the molar activity at the time of injection was 54 ± 13 GBq/μmol, and the mass dose was 0.45 ± 0.1 nmol/kg. These and subsequent data are expressed as mean ± SD. The distribution coefficient logD7.4 of 18F-LSN3316612 at room temperature in n-octanol was 3.04 ± 0.01 (n = 5). The apparent pKa of 18F-LSN3316612 was 6.8 ± 0.2 (n = 3), using cyclohexane as an extracting solvent.
Kinetic Analysis in Monkey Brain
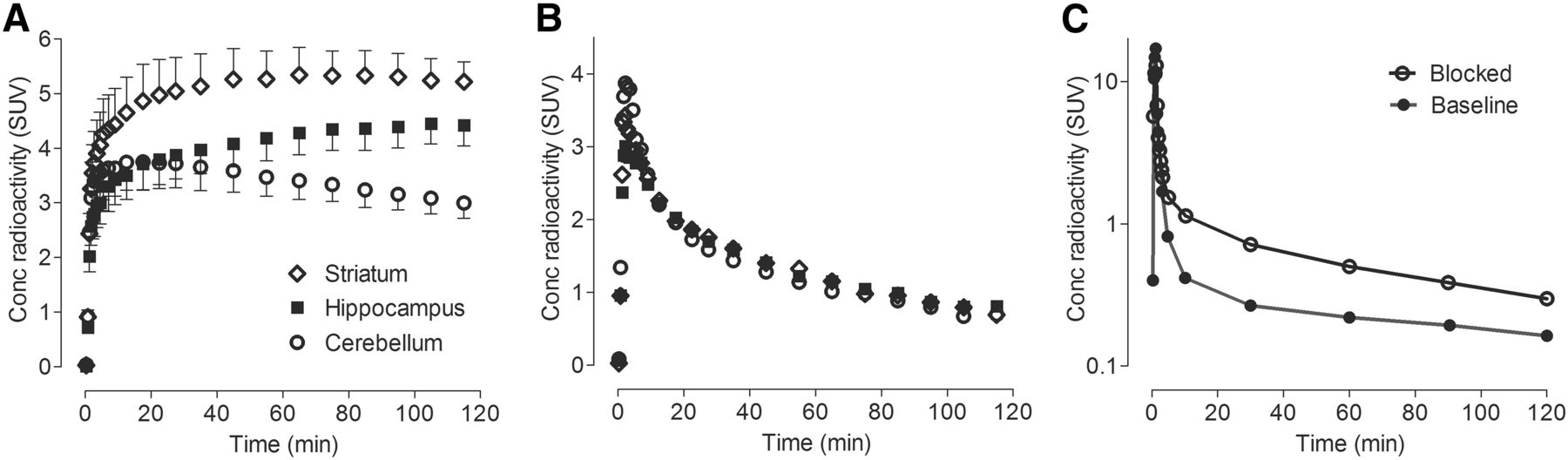
After 18F-LSN3316612 injection, radioactivity rapidly entered the brain within the first few minutes, peaked at about 30 min, and slowly washed out thereafter (Fig. 2A). The uptake of radioactivity was widespread, being highest (SUV, ∼5) in amygdala and striatum and lowest (SUV, ∼3.5) in cerebellum (Fig. 2A). Parent plasma concentrations for both baseline and self-blockade were similar, with moderate washout (Fig. 2C).

Concentration of radioactivity in brain at baseline (A) and after blockade (B) and concentration of parent radioligand in plasma (C). In baseline graph, symbols represent mean ± SD from 4 monkeys. The other 2 graphs show results from 1 animal. For blockade, thiamet-G (10 mg/kg intravenously) was injected 45 min before radioligand.
To assess the amount of specific binding to OGA, animals were pretreated with either the nonradioactive parent ligand (LSN3316612, 1 mg/kg intravenously) or the prototypic inhibitor thiamet-G (10 mg/kg intravenously), both administered 45 min before the radioligand. Both compounds decreased brain uptake in all regions by about 90% (Figs. 2B and 3 and Supplemental Table 1; supplemental materials are available at http://jnm.snmjournals.org), indicating that about 90% of all radioligand uptake was specifically bound to the target enzyme OGA.

Parametric images of VT in rhesus monkey brain at baseline and after thiamet-G blockade. Uptake at baseline was especially high in striatum, hippocampus, frontal cortex, amygdala, and pituitary (arrows). Thiamet-G (10 mg/kg intravenously) decreased uptake by >90% in all regions. Skull shows negligible uptake of radioactivity. These parametric images quantified VT at voxel level using Logan plot.
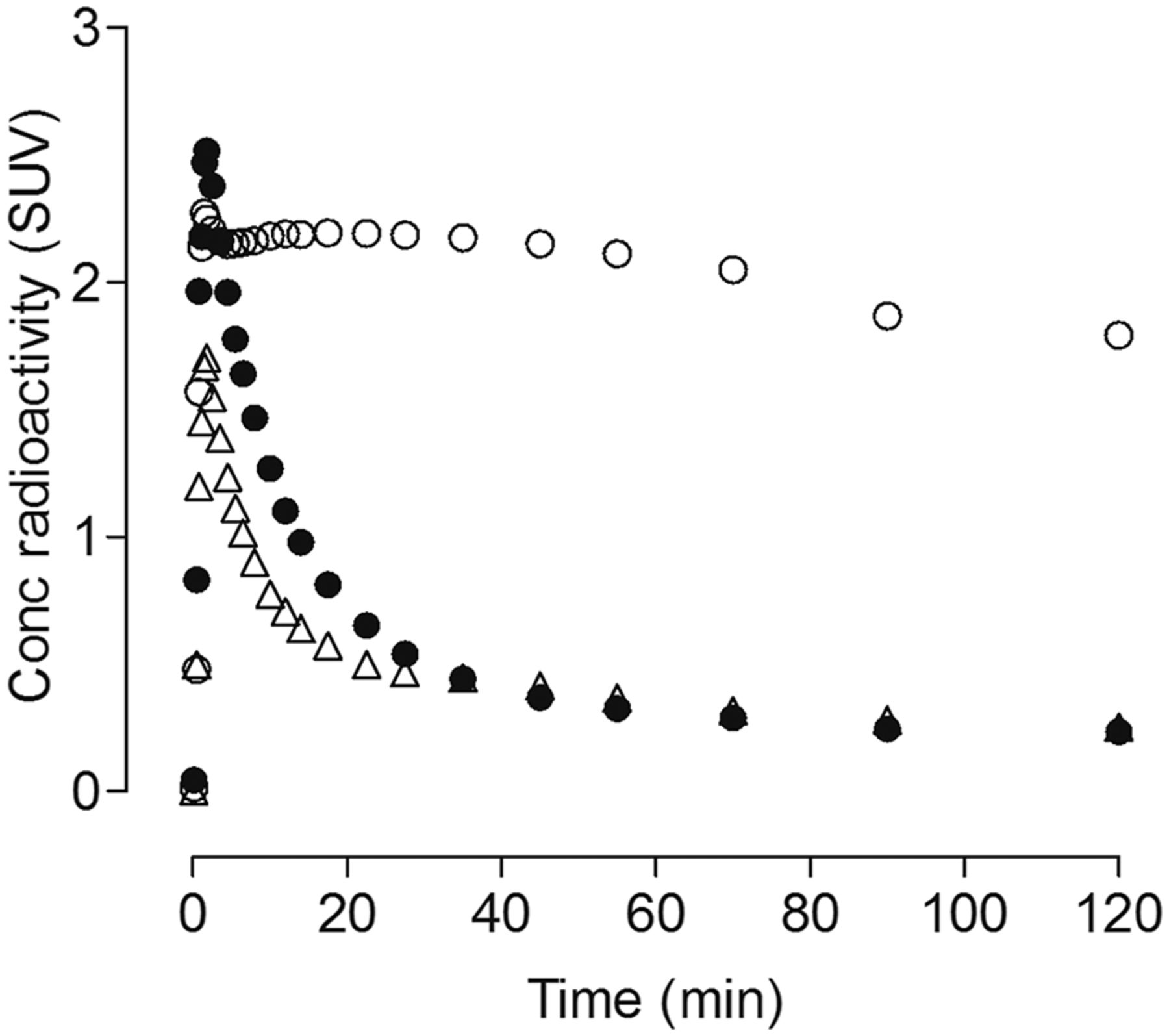
We quantified the total VT of OGA, which is proportional to the density of the enzyme, from time–activity curves of several brain regions and serial concentrations of parent radioligand in arterial plasma. In all baseline and blocked experiments, VT was reliably measured in all regions by an unconstrained 2-tissue-compartment model with good identifiability (i.e., SE < 10%). Results from the 2-tissue-compartment model are presented because blocking studies clearly showed the presence of both high-affinity specific binding and low-affinity nondisplaceable uptake; however, results from both the 1- and 2-tissue compartment models were essentially identical. The 3 brain regions with the highest VT (mL⋅cm−3) were amygdala (36.0), anterior cingulate (31.8), and striatum (30.6) (Supplemental Table 1). Cerebellum had the lowest VT (18.7). No reference tissue models were used because all brain regions showed displaceable/specific uptake. Even the corpus callosum, which is a pure white matter region, showed significant blockade by thiamet-G and nonradioactive LSN3316612 (Fig. 4).

Uptake of radioactivity in corpus callosum (white matter region) of rhesus macaque brain. Uptake at baseline (○) was significantly blocked by nonradioactive LSN3316612 (●), 1 mg/kg intravenously, administered 45 min before radioligand.
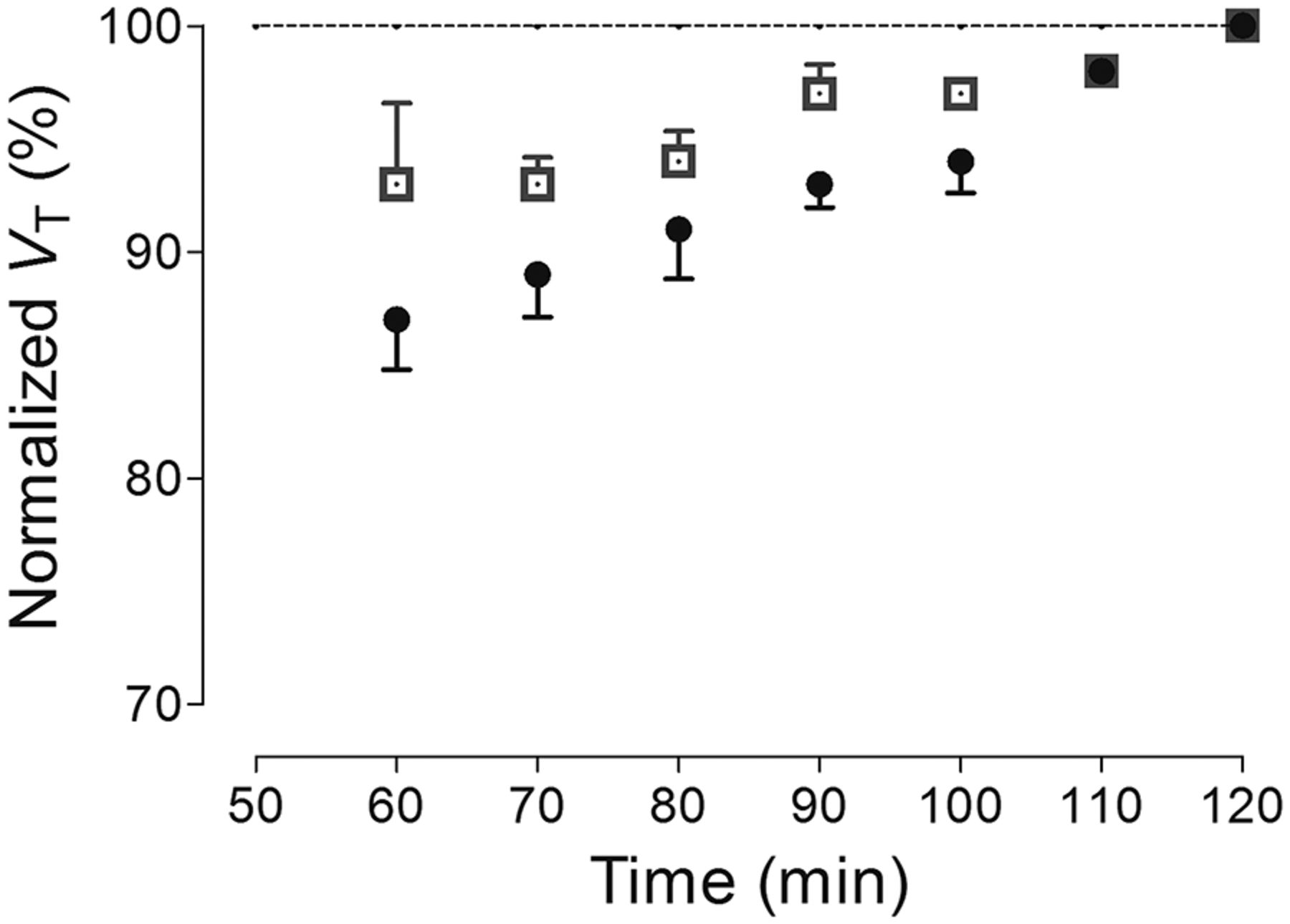
To indirectly assess whether radiometabolites accumulated in the brain during the scan, the stability of VT was calculated over time by truncating the period of brain data used for compartmental modeling. Despite the relatively slower-than-normal washout of 18F-LSN3316612, VT asymptotically reached the terminal value calculated from the complete 120 min of scanning, and the VT values for all brain regions were within 10% of their terminal values by 90 min (Fig. 5). Nevertheless, the VT values did increase slightly during the last 30 min, suggesting that either equilibrium was slow to be achieved or that a small amount of radiometabolites accumulated in brain. Consistent with the notion of minimal accumulation of radiometabolites in brain, most 18F-LSN3316612 radiometabolites were much less lipophilic than the parent radioligand and, therefore, less likely to enter brain. Even at the end of the scan (120 min), 31% of the radioactivity in plasma represented parent radioligand, and about 69% represented radiometabolites that were much less lipophilic than LSN3316612, as indicated by their earlier elution on reverse-phase high-performance liquid chromatography (Supplemental Fig. 1, peaks A–C). We have measured the free fraction of radioligand in all studies. They range from 5%–16%. However, the free fraction was similar within the same monkey between baseline and blocked scans.

Time stability curves of total VT for 2 regions: striatum (□), which had high uptake, and cerebellum (●), which had lowest uptake. VT was calculated using unconstrained 2-tissue-compartment model with increasingly truncated acquisition times. All values of VT were normalized as percentage of terminal value attained from 120 min of imaging. Data represent mean ± SD of 4 monkeys.
Brain Uptake in Oga∆Br Mice
To further explore the specificity of 18F-LSN3316612 for its target, control and Oga∆Br mice were imaged. Uptake in control mice peaked at an SUV of about 2 within 1 to 2 min and had negligible washout during the 120 min of scanning (Fig. 6A). Oga∆Br mice also had an early peak, indicating that the radioligand was delivered to brain but then washed out very rapidly thereafter. During the last 80 min of scanning (i.e., 40−120 min), brain uptake of the radioligand in Oga∆Br mice was reduced to 18% of that in control mice (Fig. 6A). The lack of retention of radioligand in Oga∆Br mice confirmed that 18F-LSN3316612 binds selectively to its target in mouse brain (Supplemental Fig. 4). As confirmation of this genetic selectivity, blockade by thiamet-G reduced uptake in control mice to levels seen in Oga∆Br animals (Fig. 6).

Uptake of radioactivity in whole brain from 2 controls (○) and 2 Oga∆Br (●) mice. Uptake in control mice preblocked with thiamet-G (△) (10 mg/kg intravenously) was similar to that in Oga∆Br mice.
Prior studies found that loss of OGA in Oga∆Br animals is exclusive to the brain and that OGA is still expressed in peripheral organs (11). Indeed, whole-body PET imaging demonstrated that Oga∆Br mice had minimal radioligand uptake in the brain but that uptake in peripheral organs such as the heart was analogous to that observed in control mice (Supplemental Fig. 2).
To determine whether radiometabolites enter the brain, 3 control mice were euthanized 30 min after 18F-LSN3316612 injection (3.5 ± 0.6 MBq), and the composition of extracted radioactivity was measured with radio–high-performance liquid chromatography. Parent radioligand represented 99.7% ± 0.1% of radioactivity in control mouse brain and only 42.8% ± 9.0% of radioactivity in plasma.
Whole-Body Biodistribution and Dosimetry
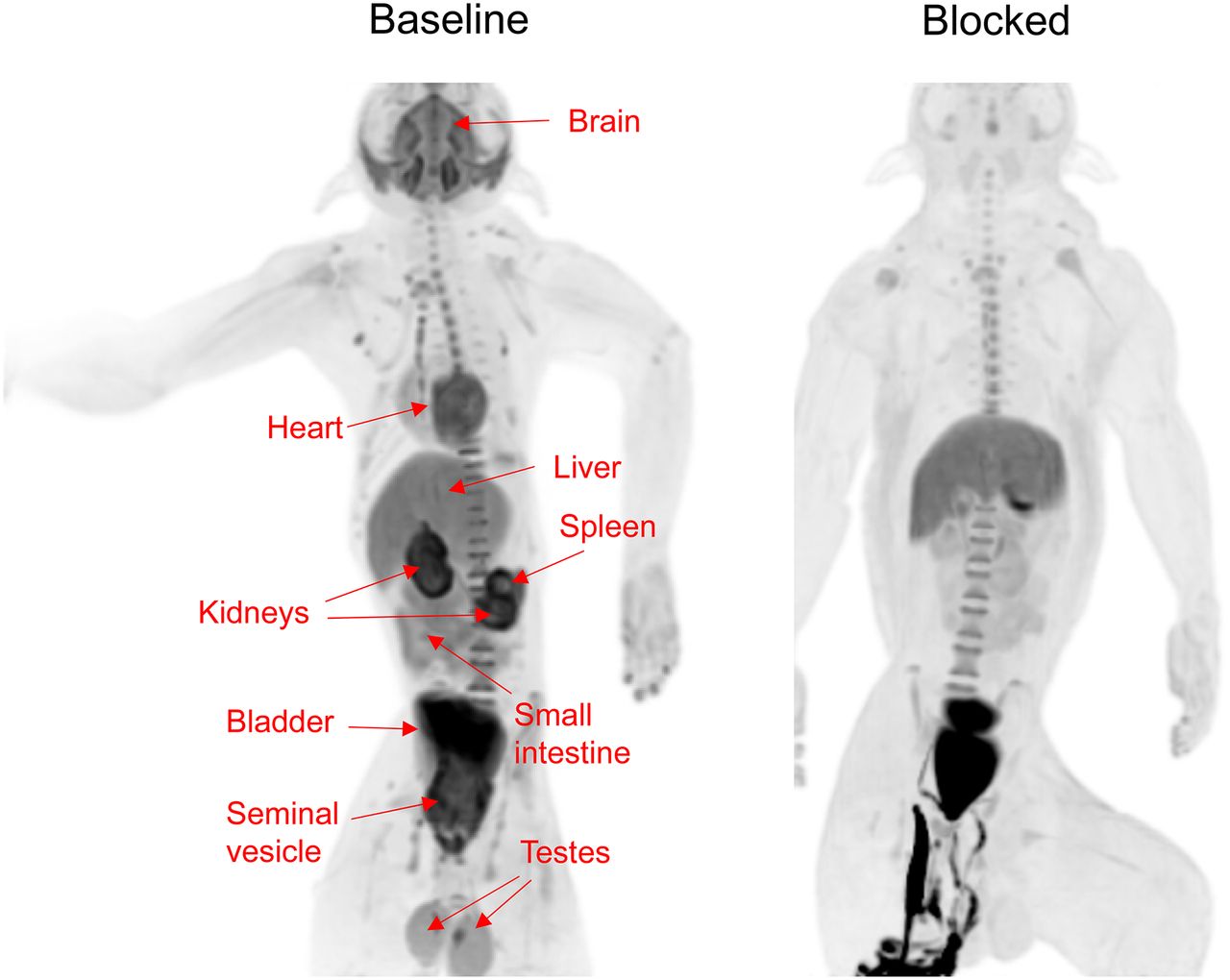
To estimate the radiation burden of 18F-LSN3316612 and to explore the peripheral distribution of the target enzyme, whole-body imaging was performed on 2 rhesus monkeys (1 male, 1 female). In addition, the male monkey was rescanned after blockade by thiamet-G (10 mg/kg intravenously). In the baseline scans, high uptake was seen in brain, heart, testes, kidneys, liver, small intestine, and urinary bladder (Fig. 7). Some of the uptake (e.g., urinary bladder) clearly represented excretion of the parent radioligand or of a radiometabolite. In addition to brain, the blocking drug thiamet-G decreased uptake in heart, kidneys, small intestine, spleen, testes, and seminal vesicle, suggesting that the radioligand was binding to the target enzyme in these organs (Fig. 7).

Whole-body distribution of radioactivity in male monkey after injection of 18F- LSN3316612 at baseline and with thiamet-G blockade (10 mg/kg, intravenously). Images were averaged for entire 120-min scan and presented as maximal-intensity projection, which enhances contrast and allows easier visualization of organs. Clear blocking effect was seen in brain, heart, kidneys, small intestine, spleen, testes, and seminal vesicle. In blocked scan, radioactivity below pelvis was due to urine excreted into animal’s diaper.
Organ doses were converted into human equivalents by correcting for the relative weight of the organ to the body in the 2 species (15). The organs with the 3 highest doses (μSv/MBq) were kidneys (46.5), testes (41.1), and spleen (38.0) (Supplemental Table 2). The dose to the ovaries was estimated to be 14.4 μSv/MBq. The estimated effective dose in humans was calculated to be 22 μSv/MBq, which is similar to other 18F-based radioligands; a previously published average effective dose for 54 radioligands was found to be 20.6 ± 6.8 μSv/MBq (16).
DISCUSSION
The present PET imaging findings in rhesus macaques and transgenic mice suggest that 18F-LSN3316612 is an excellent radioligand for imaging its target enzyme, OGA. Brain uptake was moderately high (SUVpeak, ∼5), and approximately 90% was specifically bound to OGA, on the basis of blocking studies with thiamet-G and the nonradioactive parent ligand. Serial measurements of radioactivity in brain and parent radioligand in arterial plasma indicated that the regional density of OGA was well identified by compartmental modeling as VT. After radioligand injection, 80 min of imaging were required to identify VT values that were relatively stable over time. Transgenic Oga∆Br mice had negligible binding of radioligand in brain, further demonstrating the specificity of 18F-LSN3316612 for its target. Whole-body imaging in monkeys showed high uptake in organs known to have the target enzyme (e.g., kidney and small intestine) and also showed that the radioligand has a radiation burden (effective dose, 22 μSv/MBq) similar to that of other 18F-labeled radioligands.
Central and Peripheral Distribution of Radioligand Binding in Monkey
Brain uptake of 18F-LSN3316612 was widespread and had a relatively narrow 2-fold range, with the highest VT (mL⋅cm−3) being in amygdala (36.0) and the lowest in cerebellum (18.7). One implication of this distribution is that OGA inhibitors may have pharmacologic effects in all regions of the brain. The relative distribution of OGA in primate brain has not been reported; thus, we cannot say whether the regional distribution of radioligand uptake reflects that of the protein itself. Because the relative density of the messenger RNA transcript can sometimes be used as a surrogate for protein density, we correlated the regional uptake of 18F-LSN3316612 in monkey brain with that of MGEA5, an OGA gene transcript, as identified via the Allen Brain Atlas (http://human.brain-map.org/). However, brain uptake of 18F-LSN3316612 did not correlate with messenger RNA transcript density (Supplemental Fig. 3), suggesting regional differences in the relative efficiency of translating messenger RNA into OGA protein.
With regard to the distribution of OGA in peripheral organs, we found high densities of specific/displaceable binding in kidney, testes, small intestine, liver, heart, and spleen. The Human Protein Atlas (http://proteinatlas.org/) reports moderate to high expression of OGA in all these organs with the exception of spleen, which is reported as low. In contrast, our PET imaging studies showed that spleen had especially high and displaceable radioligand uptake. The cause of this apparent discrepancy is unknown but could reflect a species difference between the Human Protein Atlas and nonhuman primate imaging.
In addition, uptake in the testes was especially high and even allowed visualization of the seminal vesicle (Fig. 7). This uptake presumably reflected specific binding to the target enzyme, as it was blocked by nonradioactive LSN3316612. Given the radiation sensitivity of the rapidly dividing cells in the testes, we extrapolated the radiation expected in a human man. The resulting organ dose was high (41.1 μSv/MBq) but will probably not be dose-limiting under most radiation guidelines.
Potential Uses of the Radioligand
Like other PET radioligands, 18F-LSN3316612 could potentially be useful for 2 general purposes: to facilitate therapeutic drug development and to explore pathophysiology. Drug development for central nervous system disorders, in contrast to most disorders associated with peripheral organs, is complicated by the blood–brain barrier, which can block entry to the target organ. To deal with this potential problem, most central nervous system drugs developed over the last 10–20 y have required demonstrable measures indicating that the drug enters brain and engages a reasonable percentage of its target. The present study indicates that 18F-LSN3316612 can clearly be used for this purpose, at least for drugs that bind at the same site as the radioligand.
The results further suggest that 18F-LSN3316612 may be useful for exploring the status of OGA in patients with tauopathy, for example to assess whether the density of this enzyme is altered in brain areas with tau deposits in ways that may reflect either a cause or an effect of those deposits. O-GlcNAc cycling has been strongly implicated, such as cardiac hypertrophy and heart failure (3), B cell homeostasis and antibody responses in the spleen (3,17), and diabetic nephropathy (3). Several peripheral organs showed specific (i.e., displaceable) binding to OGA (Fig. 7) and might be studied with this radioligand. However, unlike the brain, which had no radiometabolites, peripheral organs may accumulate radiometabolites, which must be accounted or corrected for.
Limitations of the Radioligand
Although 18F-LSN3316612 has exemplary properties for imaging and quantifying its target enzyme in monkey brain, its extension to humans may have limitations. The current study suggests that the most worrisome characteristic of 18F-LSN3316612 is that VT measurements did not stabilize; instead, they increased by 5%–10% in the last 40 min of a 2-h scan. Such a relatively small increase in humans would be acceptable, but greater increases may be problematic. Such instability over time could reflect that equilibrium binding has not been achieved (e.g., because of high density of the enzyme or high affinity of the radioligand) or that radiometabolites accumulate in brain. Thus, we recommend that time stability of VT be carefully assessed in any future human studies over a potentially longer scanning period. It should be noted, however, that we found no radiometabolites in control mouse brain 30 min after injection. If metabolism of the radioligand is similar between species, radiometabolites did not contaminate the PET signal from monkey brain; instead, the radioligand was slow to equilibrate.
CONCLUSION
18F-LSN3316612 is an excellent radioligand for imaging and quantifying its target enzyme in monkey brain, demonstrating good uptake, a high percentage of specific binding, selectivity for the target, and amenability to quantitation with compartmental modeling. The results suggest that 18F-LSN3316612 is suitable for extension in human subjects. This would assist therapeutic development of OGA inhibitors, which may reduce tau protein depositions that could provide a potential treatment for several neurodegenerative disorders.
DISCLOSURE
This work was supported by the Intramural Research Programs of NIDDK and NIMH (ZIAMH002793; ZIAMH002795) and a research grant from Eli Lilly & Co. Vanessa Barth, Nancy Kant, Lawrence Slieker, Sergey Shcherbinin, Hugh Nuthall, and Cynthia Jesudason were employees of Eli Lilly and Co. at the time the study was conducted. No other potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank the dedicated staff of the Molecular Imaging Branch of the NIMH, the PET Department of NIH’s Clinical Center, and the NIMH veterinary staff for help in successfully completing the studies and Ioline Henter for invaluable editorial assistance.
Footnotes
Published online Sep. 13, 2018.
- © 2019 by the Society of Nuclear Medicine and Molecular Imaging.
REFERENCES
- Received for publication April 18, 2018.
- Accepted for publication July 9, 2018.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}