


Abstract
Radiolabeled octreotide analogs are most successfully being applied today in clinical cancer imaging and treatment. Propagation of this paradigm to other radiopeptide families has been greatly hampered by the inherent poor metabolic stability of systemically administered peptide analogs. We hypothesized that the in vivo coadministration of specific enzyme inhibitors would improve peptide bioavailability and hence tumor uptake. Through single coinjection of the neutral endopeptidase inhibitor phosphoramidon (PA), we were able to provoke remarkable rises in the percentages of circulating intact somatostatin, gastrin, and bombesin radiopeptides in mouse models, resulting in a remarkable increase in uptake in tumor xenografts in mice. Methods: The peptide conjugates [DOTA-Ala1]SS14 (DOTA-Ala-Gly-c[Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys]-OH), PanSB1 (DOTA-PEG2-dTyr-Gln-Trp-Ala-Val-βAla-His-Phe-Nle-NH2), and DOTA-MG11 (DOTA-dGlu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) were labeled with 111In by 20 min of heating at an acidic pH. Metabolic stability was studied with high-performance liquid chromatography analysis of blood samples collected 5 min after the injection of the test radiopeptide alone or with PA into mice. Biodistribution was studied after injection of each 111In-labeled radiopeptide alone or after coinjection of PA in tumor-bearing severe combined immunodeficient (SCID) mice. Results: The amount of intact [111In-DOTA-Ala1]SS14 detected in the mouse circulation at 5 min after the injection of PA increased impressively—from less than 2% to 86%—whereas the uptake in AR4-2J xenografts rose from less than 1 percentage injected dose per gram of tissue (%ID/g) to 14 %ID/g at 4 h after injection. Likewise, the coadministration of PA resulted in a marked increase in the amount of circulating intact 111In-PanSB1—from 12% to 80%—at 5 min after injection, and radioligand uptake in human PC-3 xenografts in SCID mice escalated from less than 4 %ID/g to greater than 21 %ID/g at 4 h after injection. In a similar manner, the coadministration of PA resulted in an equally impressive increase in intact [111In-DOTA]MG11 levels in the mouse bloodstream—from less than 5% to 70%—at 5 min after injection, leading to a remarkable increase in radiotracer uptake—from 2 %ID/g to greater than 15 %ID/g—in both AR4-2J tumors and A431(CCKR+) tumors (i.e., tumors induced by A431 cells transfected to stably express the human cholecystokinin subtype 2 receptor) in mice at 4 h after injection. This effect was well visualized by SPECT/CT imaging of AR4-2J tumor–bearing mice at 4 h after injection. Conclusion: The results of this study clearly demonstrate that the coadministration of key enzyme inhibitors can effectively prolong the survival of radiolabeled peptides in the circulation, securing their safe transit to the target. This strategy clearly provoked an unprecedented increase in radiolabel accumulation in tumor xenografts in mice; this increase might translate into higher diagnostic sensitivity or improved therapeutic efficacy of radiopeptide drugs in cancer patients. Hence, our findings provide exciting new opportunities for the application of biodegradable (radio)peptide drugs of either natural or synthetic origin as well as for the rationale design of analogs that are stable in vivo.
The overexpression of peptide receptors on the cell membrane of cancer cells, as opposed to their lower level of expression in healthy surrounding tissues, has provided the unique opportunity to visualize and treat malignant tumors in cancer patients with the aid of radiolabeled peptide analogs, that is, radiopeptides (1–3). Radiopeptide drugs are carefully designed to carry the radionuclide of choice specifically to their receptor target located at primary and metastatic tumor sites. With a diagnostic radionuclide, γ photons detected by an external imaging device, such as a SPECT or PET camera, enable the highly specific visualization of tumor lesions. With a therapeutic radionuclide, particle radiation can induce tumor cell apoptosis and death (4–6). With the advent of [111In-diethylenetriaminepentaacetic acid (DTPA)]octreotide, this approach has been successfully implemented in clinics for the diagnosis of somatostatin receptor–positive neuroendocrine tumors (7). With the introduction of [177Lu-DOTA0,Tyr3]octreotate, this approach has been further pursued in phase 3 clinical studies of somatostatin receptor–targeted therapy of neuroendocrine tumors (8,9). Furthermore, other regulatory peptide receptors amply expressed in alternative cancer types have emerged as putative targets of newly developed radiopeptide ligands (3).
The success of this strategy relies largely on the safe delivery of the intact radiopeptide to its receptor target after intravenous injection into the living organism. Radiopeptide integrity and, hence, target delivery are challenged immediately on entry into the circulation by ubiquitous proteolytic enzymes present in the blood or anchored to the surface of blood cells and vasculature walls. Furthermore, during transit through major organs, such as the liver, lungs, kidneys, and gastrointestinal tract, radiopeptides are exposed to more enzymes residing in abundance in these compartments. The relationships among native peptide ligands, their cognate receptors, and endogenous enzymes orchestrating their close interactions have long been acknowledged both in normal conditions and in cancer. In the search for peptide analogs resistant to assailing enzymes, various structural modifications—such as key amino acid substitutions, reduction or methylation of biodegradable bonds, cyclization, multimerization, and other means—have been proposed (10,11). This search is costly and time-consuming and typically yields analogs with suboptimal pharmacologic characteristics.
We propose instead the as-yet-unexplored application of enzyme inhibitors to optimize radiopeptide supply to tumor-associated receptor targets, with the aim of increasing diagnostic sensitivity and improving therapeutic efficacy against primary and disseminated disease. Through the coinjection of a single inhibitor of neutral endopeptidase (NEP; EC 3.4.24.11; neprilysin; CD10) (12) in experimental animals, we were able to provoke marked increases in the percentages of intact circulating radiopeptide analogs of the somatostatin, gastrin, and bombesin peptide families. Most importantly, we were able, for the first time, to our knowledge, to induce an impressive amplification of radiopeptide uptake in a variety of tumor xenografts in mice expressing the respective receptor targets.
Specifically, we intravenously injected phosphoramidon (PA) together with different radiopeptides in mice. PA is a potent and reversible competitive NEP inhibitor, and the structure of PA bound to the active site of human NEP has been elucidated (13). By virtue of its high level of water solubility, PA can be injected as a bolus together with a radiopeptide in doses ensuring the effective in vivo inhibition of NEP. It was first isolated from cultures of Streptomyces tanashiensis, but methods for its convenient synthesis recently became available (14,15).
Three types of 111In-labeled and DOTA-modified radioligands representing the somatostatin-14, bombesin, and gastrin peptide types were included in the present study. As a first example, we describe the effect of PA coinjection on the stability and localization of [111In-DOTA-Ala1]SS14 (111In-DOTA-Ala-Gly-c[Cys-Lys-Asn-Phe-Phe-Trp-Lys-Thr-Phe-Thr-Ser-Cys]-OH) in somatostatin subtype 2 receptor (sst2)–expressing AR4-2J tumors in mice (16). Our second example involves the bombesin analog 111In-PanSB1 ([111In-DOTA-PEG2-dTyr-Gln-Trp-Ala-Val-βAla-His-Phe-Nle-NH2), which is based on a previously reported sequence exhibiting a pan–bombesin receptor binding profile (17–19). The effect of PA on in vivo stability and tumor uptake was studied and assessed with human PC-3 xenografts expressing the gastrin-releasing peptide receptor (GRPR) in mice (20,21). Our third example encompasses the gastrin–cholecystokinin peptide family. The effect of PA coinjection on the in vivo stability of [111In-DOTA]MG11 (111In-DOTA-dGlu-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2) and on uptake in 2 tumor models in mice—a double A431-CCK2R(+) and A431-CCK2R(−) xenograft (i.e., induced by inoculation of A431 cells transfected to stably express the human cholecystokinin subtype 2 receptor [CCK2R] and of A431 cells devoid of CCK2R expression, respectively) and the CCK2R-expressing AR4-2J tumor model—was assessed (22–24). [111In-DOTA]MG11 was previously reported to have a favorably low level of renal accumulation; however, compared with full-chain gastrin analogs, [111In-DOTA]MG11 was associated with a lower level of metabolic stability and reduced tumor targeting (23,25).
The findings of the present study revealed a prominent role for NEP in the in vivo processing of N-terminal radiometallated peptides, consistent with its ubiquitous and abundant presence in the human body (12). The significance of NEP involvement in the catabolism of the radiopeptides tested in the present study and presumably further classes of radiopeptide ligands has not yet been adequately assessed and warrants further investigation. Therefore, we believe that the present preliminary study will stimulate further research to unravel the key role of major peptidases engaged in the catabolism of intravenously administered (radio)peptide drugs.
MATERIALS AND METHODS
Materials and Instrumentation
Chemicals were reagent grade. The peptides used in this study were synthesized or purchased from commercial sources. [DOTA-Ala1]SS14, [Tyr3]octreotate (H-dPhe-c[Cys-Tyr-dTrp-Lys-Thr-Cys]-Thr-OH), and demogastrin 2 [N4-Gly-dGlu-(Glu)5-Ala-Tyr-Gly-Trp-Met-Asp-Phe-NH2] were synthesized on solid support as previously described (16,24). PanSB1 and DOTA-MG11 were purchased from PiChem, and [Tyr4]bombesin (Pyr-Gln-Arg-Tyr-Gly-Asn-Gln-Trp-Ala-Val-Gly-His-Leu-Met-NH2) was obtained from Bachem AG. PA [N-(α-rhamnopyranosyloxyhydroxyphosphinyl)-l-leucyl-l-tryptophan × 2Na × 2H2O] was purchased from PeptaNova GmbH. For 111In labeling, 111InCl3 in 50 mM HCl at an activity concentration of 370 MBq/mL on the calibration date was purchased from Mallinckrodt Medical BV.
Radiochemical high-performance liquid chromatography (HPLC) analyses were performed on a Waters chromatograph with a model 600 solvent delivery system, coupled to both a model 996 photodiode-array UV detector (Waters) and a Gabi γ detector (Raytest RSM Analytische Instrumente GmbH); the Millennium Software (Waters) was used to control the HPLC system and process the data. Radioactivity in samples was measured in an automatic well-type γ counter (NaI(Tl) 3′′-crystal; Canberra Packard Auto-Gamma 5000 series instrument). SPECT/CT images were acquired with a 4-head multipinhole NanoSPECT/CT camera (Bioscan Inc.) at the Animal Imaging Facility at Erasmus MC.
111In Labeling and Quality Control
Labeling with 111In was conducted by adding 10 nmol of peptide analog per 37–74 MBq of 111InCl3 in 0.1 M sodium acetate buffer and 10 mM sodium ascorbate. The typical end pH was 4.6. Labeling was complete after incubation in a boiling water bath for 20 min (26). Before HPLC quality control procedures, ethylenediaminetetraacetic acid in 0.1 M acetate buffer was added at a final concentration of 1 mM to the labeling reaction mixture as a free 111In3+ scavenger (27). For DOTA-MG11, Met (5 μL of a 0.1 M solution) was added to the labeling reaction mixture to prevent oxidation of the Met residue to the corresponding sulfoxide (28).
For radiochemical analyses, an XBridge Shield RP18 column (5 μm; 4.6 × 150 mm; Waters) coupled to the respective 2-cm guard column was used. Elution was accomplished with a linear gradient of a 0.1% trifluoroacetic acid–water solution and acetonitrile (starting at 10% acetonitrile, with a 1% increase per minute) at a flow rate of 1 mL/min.
Cell Cultures
Rat pancreatic tumor cell line AR4-2J endogenously expressing the sst2 and rat gastrin/CCK2R was kindly provided by Stephen Mather (St. Bartholomew’s Hospital, London, U.K.) and cultured as previously described (29,30). Human androgen–independent prostate adenocarcinoma PC-3 cells (LGC Promochem) endogenously expressing the human GRPR (20) were cultured in Dulbecco modified Eagle medium with GlutaMAX-I (Gibco BRL Life Technologies) and supplemented with 10% (v/v) heat-inactivated fetal bovine serum, penicillin at 100 U/mL, and streptomycin at 100 μg/mL. Human epidermoid carcinoma cell line A431 transfected to stably express the human CCK2R [A431-CCK2R(+)] or devoid of CCK2R expression [A431-CCK2R(−)] was a kind gift from Otto C. Boerman (Department of Nuclear Medicine, Radboud University Nijmegen Medical Centre, Nijmegen, The Netherlands) and Luigi Aloj (Istituto di Biostrutture e Bioimmagini, Consiglio Nazionale delle Ricerche, Naples, Italy). Transfected cells were cultured in Dulbecco modified Eagle medium with GlutaMAX-I and supplemented as described earlier but with G418 (Geneticin; Gibco BRL Life Technologies; 250 μg/mL) added to the medium (22). All culture reagents were obtained from Gibco BRL Life Technologies or from Biochrom KG Seromed. Cells were kept in a controlled humidified atmosphere containing 5% CO2 at 37°C. Cells were inspected for confluence, and 1:3 or 1:4 passages were performed (usually every 2–5 d, according to cell type) with a 0.05% trypsin–0.02% ethylenediaminetetraacetic acid (w/v) solution.
Metabolic Studies in Mice
A bolus of a test 111In-labeled radiopeptide solution (100 μL; 11–22 MBq; 3 nmol of total peptide in physiologic saline–ethanol [9:1, v/v]), together with vehicle (100 μL of physiologic saline–ethanol [9:1, v/v]) (control group) or PA (100 μL of vehicle containing 300 μg of PA) (PA-treated group), was injected into the tail veins of healthy Swiss albino mice (National Center for Scientific Research “Demokritos” Animal House). At 5 min after injection, blood (0.5–1 mL) was collected from the hearts of the animals and immediately transferred to prechilled polypropylene tubes containing ethylenediaminetetraacetic acid, and the tubes were placed on ice. For [111In-DOTA]MG11, the tubes also contained Met (final concentration of 1 mM) to prevent oxidation of the Met residues to the respective sulfoxides. Blood samples were centrifuged (10 min, 2,000g, 4°C; Hettich Universal 320R centrifuge), the plasma was collected and mixed with chilled acetonitrile in a 1:1 (v/v) ratio, and the samples were centrifuged again (10 min, 15,000g, 4°C). Supernatants were concentrated to a small volume under a gentle N2 flux at 40°C, diluted with physiologic saline (≈400 μL), and filtered through a Millex GV filter (0.22 μm).
Aliquots were analyzed by reversed-phase HPLC with a Symmetry Shield RP18 column (5 μm; 3.9 × 20 mm; Waters). Elution was accomplished in 50 min at a flow rate of 1 mL/min with the following gradient: 100% A and 0% B to 50% A and 50% B, where A was 0.1% trifluoroacetic acid in H2O and B was acetonitrile. The elution times of intact radiotracers were determined by coinjection of the respective 111In-labeled radiopeptide.
Biodistribution of 111In-Labeled Radioligands in Xenograft-Bearing severe combined immunodeficient (SCID) Mice
SCID mice that were 6 wk old on the day of arrival (National Center for Scientific Research “Demokritos” Animal House) were inoculated in their flanks with a bolus (≈150 μL) containing a suspension of 1 × 107–2 × 107 freshly harvested cells in sterile physiologic saline. The animals were kept under aseptic conditions until palpable tumors developed at the inoculation site. Palpable tumor development took 12 d, 3 wk, and 6 d for AR4-2J (350 ± 20 mg [mean ± SD]), PC-3 (150 ± 30 mg), and A431-CCKR(+) or A431-CCKR(−) tumors (260 ± 80 mg), respectively.
On the day of the experiment, a bolus of a test 111In-labeled radiopeptide solution (100 μL; 37–74 kBq; 10 pmol of total peptide in vehicle), together with vehicle (100 μL) (control group), or PA (100 μL of vehicle containing 300 μg of PA) (PA-treated group), or PA plus a receptor blocker (100 μL of vehicle containing 300 μg of PA and excess receptor blocker) (in vivo receptor blockade group), was injected into the tail veins of mice. For [111In-DOTA]MG11, Met at a final concentration of 1 mM was also included. For in vivo receptor blockade, animals were coinjected with PA and 40 nmol of [Tyr3]octreotate (sst2 blockade), 40 nmol of [Tyr4]bombesin (GRPR blockade), or 40 nmol of demogastrin 2 (CCK2R blockade). Animals for each group were selected at random. Furthermore, 2 additional groups of AR4-2J tumor–bearing animals were injected with [111In-DTPA]octreotide together with either vehicle (100 μL) (control group) or PA (100 μL of vehicle containing 300 μg of PA) (PA-treated group).
Animals were euthanized in groups of 4 at 4 h after injection. Samples of blood and urine as well as organs of interest were collected and weighed, and radioactivity was measured in an automatic well-type γ counter. Data were calculated as the percentage injected dose per gram of tissue (%ID/g) with the aid of standard solutions and Microsoft Excel. Results are reported as the mean ± SD, as calculated with the PRISM 2.01 (GraphPad) software program.
Statistical analysis with the unpaired 2-tailed Student t test was performed to compare values for control, PA-treated, and in vivo receptor blockade animal groups at 4 h after injection. P values of less than 0.05 were considered statistically significant.
SPECT/CT Imaging of [111In-DOTA]MG11 in AR4-2J Tumor–Bearing Nude Mice
NMRI nu/nu mice (body weight, ≈30 g; Harlan CPB) were inoculated subcutaneously in the shoulder region with 106 AR4-2J cells (injection volume, 200 μL). When the tumors reached a diameter of approximately 1 cm, the animals were scanned. During imaging, the animals were anesthetized with isoflurane (Nicholas Pyramal Ltd.). Body temperature was controlled with a heated bed. After completion of the examinations, the animals were euthanized.
All animals received [111In-DOTA]MG11 (45 MBq; 0.5 nmol), together with vehicle or PA (300 μg), through the tail veins at 4 h before SPECT imaging (total injection volume, 200 μL). SPECT/CT images were acquired with a 4-head multipinhole SPECT/CT camera with energy peak settings at 171 and 245 keV (window width ± 10%) for 111In. Nine-pinhole apertures (diameter, 1.4 mm) were used on each camera head (detector, 230 × 215 mm). The matrix was 256 × 256. The animals were placed in the center of rotation; the radius of rotation was fixed. SPECT images were acquired with 20 projections, 60 s per projection, and a quality factor of 0.8. The images were reconstructed by use of the ordered-subset expectation maximization method with HiSPECT NG software (Scivis Wissenschaftliche Bildverarbeitung GmbH), 24 iterations, and a voxel size of 0.3 × 0.3 × 0.3 mm. CT images were acquired with the following settings: 180 projections, 45-kVp tube voltage, 500-ms exposure time, and a voxel size of 0.2 × 0.2 × 0.2 mm. The images were reconstructed by use of the exact-cone-beam filtered backprojection algorithm. SPECT images were resampled to the CT resolution and registered to CT images by use of the VivoQuant program (version 1.22; inviCRO LLC).
All animal experiments were performed in compliance with European and national regulations and after approval of protocols by national authorities.
RESULTS
111In-Labeled Radiotracers
The incorporation of 111In into the DOTA chelator coupled to the 3 peptide analogs was accomplished by heating at 90°C for 20 min in an acidic medium, in agreement with the results of a previous study (26). As demonstrated by reversed-phase HPLC of the labeling reaction mixtures, all three 111In-labeled radiotracers were eluted as a single radiochemical species (>95% purity).
Effect of PA on In Vivo Stability and AR4-2J Tumor Uptake of [111In-DOTA-Ala1]SS14 in Mice
The effect of PA coinjection on the stability and tumor localization of [111In-DOTA-Ala1]SS14 in mouse models is shown in Figure 1. Analysis of blood samples collected 5 min after [111In-DOTA-Ala1]SS14 injection into healthy mice revealed almost total degradation of the radiopeptide (<2% remaining intact analog) (Fig. 1A) (16,31). After the coinjection of PA, however, the amount of parent radiopeptide still present in the blood rose to 86% (Fig. 1B). Most importantly, this increase in the levels of circulating intact radiotracer translated into an impressive amplification of radiolabel levels in AR4-2J tumors at 4 h after injection into SCID mice. In particular, the uptake in sst2-expressing tumors rose from less than 1 %ID/g to 14 %ID/g (Fig. 1C and inset), whereas tumor uptake declined to less than 0.3 %ID/g after the coinjection of both PA and excess [Tyr3]octreotate; these results implied an sst2-mediated process. In contrast, at 4 h after the injection of metabolically stable [111In-DTPA]octreotide, the uptake in AR4-2J tumors (n = 7) was 5.5 ± 1.0 %ID/g in the control group and 5.6 ± 1.0 %ID/g in the PA-treated group.

(A and B) HPLC analysis of blood collected 5 min after injection of [111In-DOTA-Ala1]SS14 into healthy mice without (A) or with (B) coinjection of PA. (C) Tissue distribution data at 4 h after injection of [111In-DOTA-Ala1]SS14 without PA, with PA, or with PA plus excess sst2 blocker coinjection into male SCID mice bearing AR4-2J xenografts; values for tumors are shown in inset. Ad = adrenal glands; Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = AR4-2J tumors.
Effect of PA on In Vivo Stability and Human PC-3 Xenograft Uptake of 111In-PanSB1 in Mice
Figure 2A shows the fast breakdown of 111In-PanSB1, within 5 min after entry into the bloodstream of healthy mice. Remarkably, the coadministration of PA resulted in an impressive increase in the levels of circulating 111In-PanSB1, from 12% to 80% (Fig. 2B). Consistent with this observation, the uptake of radioactivity in human GRPR–positive PC-3 xenografts in SCID mice escalated from less than 4 %ID/g to greater than 21 %ID/g at 4 h after coinjection of the radiopeptide with PA (Fig. 2C and inset). The uptake in human xenografts was reduced to less than 0.9 %ID/g by the coinjection of both PA and excess [Tyr4]bombesin, demonstrating GRPR specificity.

(A and B) HPLC analysis of blood collected 5 min after injection of 111In-PanSB1 into healthy mice without (A) or with (B) coinjection of PA. (C) Tissue distribution data at 4 h after injection of 111In-PanSB1 without PA, with PA, or with PA plus excess GRPR blocker coinjection into female SCID mice bearing human PC-3 xenografts; values for tumors are shown in inset. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = PC-3 tumors.
Effect of PA on In Vivo Stability and A431-CCK2R(+) Tumor Uptake of [111In-DOTA]MG11 in Mice
HPLC analysis of blood samples collected 5 min after injection of [111In-DOTA]MG11 revealed only 5% intact radiopeptide in mouse blood (Fig. 3A). Treatment with PA led to a notable increase in the levels of circulating [111In-DOTA]MG11 to 70% (Fig. 3B). The impact of this increase on the tumor uptake of [111In-DOTA]MG11 was studied in a double-tumor model in SCID mice (23). The coinjection of PA induced a striking increase in uptake, from 2 %ID/g to greater than 15 %ID/g, but only in A431-CCK2R(+) tumors and not in A431-CCK2R(−) tumors (Fig. 3C and inset). CCK2R-negative tumors had poor uptake (<0.5 %ID/g) independent of the coinjection of PA. An interesting feature of this experiment was the finding that renal radiopeptide uptake remained low and unaffected by treatment with PA.
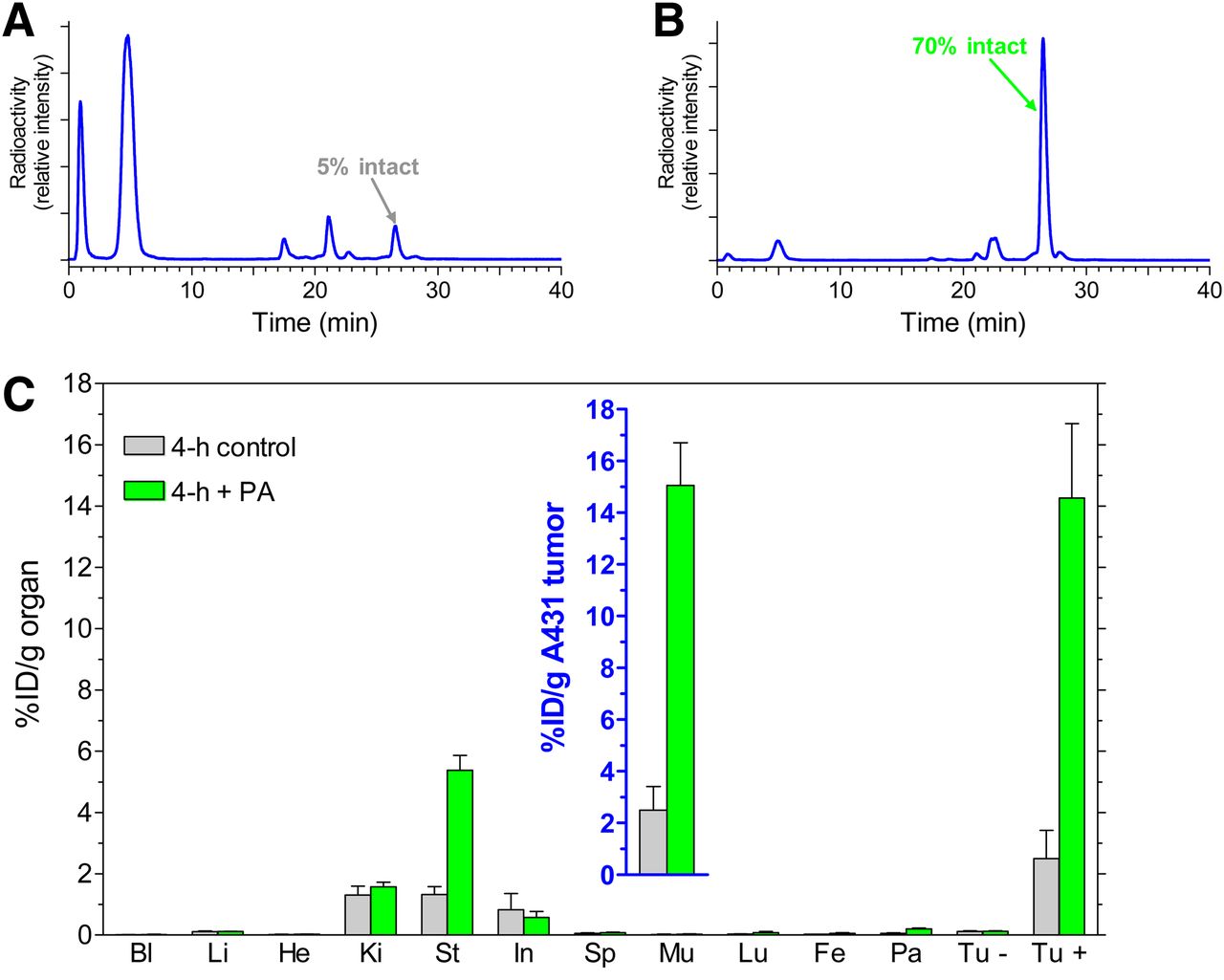
(A and B) HPLC analysis of blood collected 5 min after injection of [111In-DOTA]MG11 into healthy mice without (A) or with (B) coinjection of PA. (C) Tissue distribution data at 4 h after injection of [111In-DOTA]MG11 without PA or with PA coinjection into male SCID mice bearing A431-CCK2R(+) and A431-CCK2R(−) xenografts; values for tumors are shown in inset. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu − = A431-CCK2R(−) tumors; Tu + = A431-CCK2R(+) tumors.
Effect of PA on AR4-2J Tumor Uptake of [111In-DOTA]MG11 in Mice: Visualization with SPECT/CT
Treatment with PA resulted in a profound increase in [111In-DOTA]MG11 uptake in AR4-2J tumors (endogenously expressing the rat CCK2R), from less than 2 %ID/g to greater than 16 %ID/g, at 4 h after injection. After the coinjection of PA and excess demogastrin 2 (24), tumor uptake declined to 0.3 %ID/g, indicating CCK2R-mediated accumulation (Fig. 4A and inset). These findings were reproduced by the excellent tumor visualization on images acquired by SPECT/CT small-animal imaging of AR4-2J tumor–bearing mice, certifying the positive impact of this approach on tumor visualization (Figs. 4B and 4C).
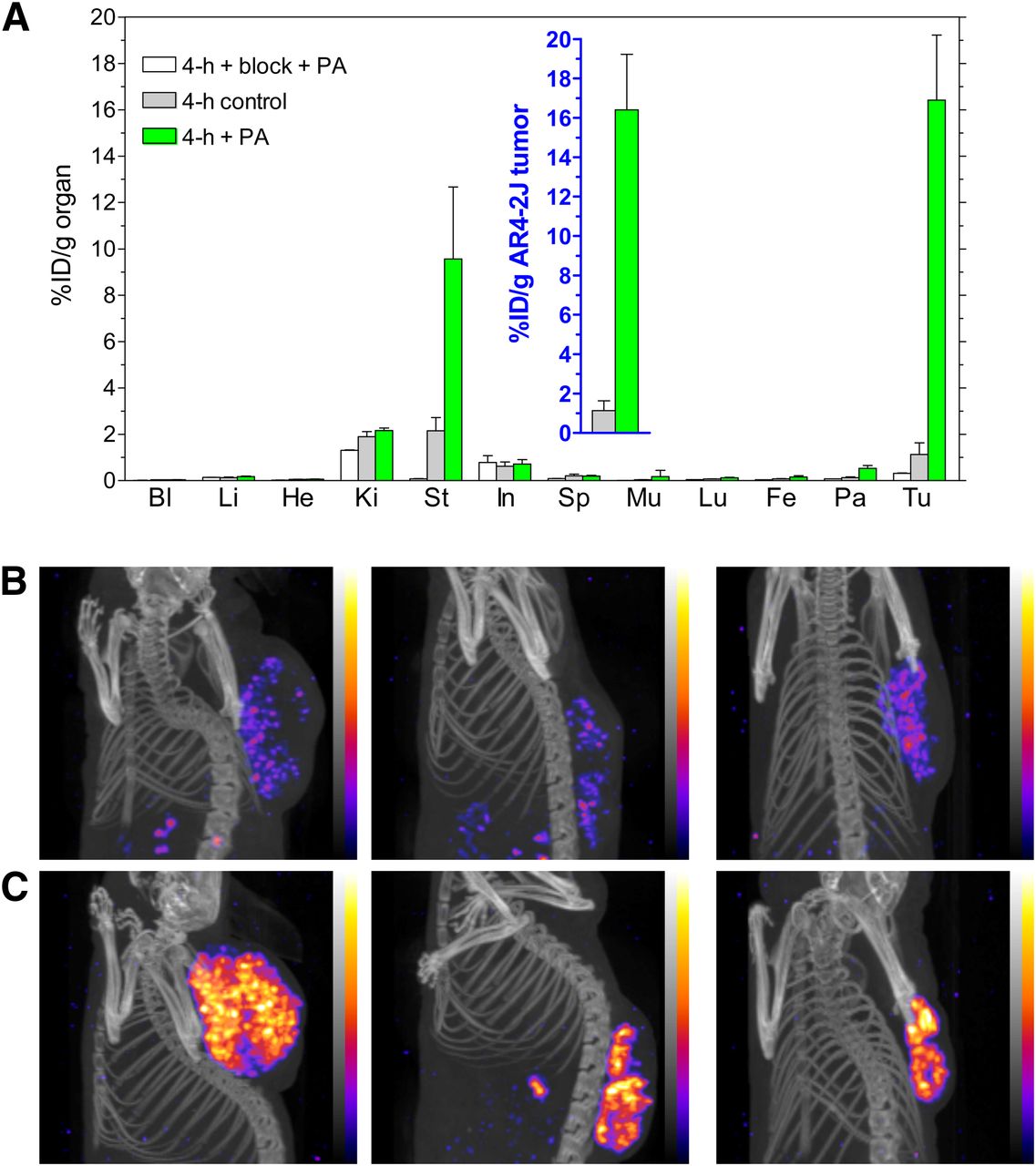
(A) Biodistribution of [111In-DOTA]MG11 at 4 h after injection without PA, with PA, or with PA plus excess CCK2R blocker coinjection into AR4-2J tumor–bearing mice; values for tumors are shown in inset. Bl = blood; Fe = femur; He = heart; In = intestines; Ki = kidneys; Li = liver; Lu = lungs; Mu = muscle; Pa = pancreas; Sp = spleen; St = stomach; Tu = AR4-2J tumors. (B and C) SPECT/CT at 4 h after injection of [111In-DOTA]MG11 without (B) or with (C) coinjection of PA (n = 3 per group). Note the highly improved tumor visualization in C.
DISCUSSION
The last 2 decades have witnessed spectacular progress in experimental and clinical oncology. Recent advances include the documentation of peptide receptor expression on the surface of cancer cells at a density exceeding that in healthy surrounding tissues. Peptide receptors belong to the superfamily of serpentine G protein–coupled receptors (GPCRs), and their role in cancer is becoming increasingly recognized (1–3). Nuclear medicine researchers have taken advantage of recent advances and have implemented new molecular tools based on radiolabeled peptide ligands to localize and combat primary and metastatic disease. Radiopeptides have been suitably designed to carry radionuclides of choice, attractive either for SPECT (γ emitters) or PET (positron emitters) imaging or for radionuclide therapy (α, β, or Auger electron emitters) to lesion-associated GPCR targets with a high specificity (4,6).
Of particular relevance is the current successful clinical application of radiolabeled somatostatin analogs in the diagnosis, staging, and therapy of neuroendocrine tumors overexpressing the sst2. The success of this strategy has been largely dependent on the high level of metabolic stability of the radiopeptides used. In contrast to the rapidly biodegradable native somatostatin-14 (31), sst2-directed radiopeptide drugs (e.g., [111In-DTPA]octreotide) are based on cyclic octapeptide analogs, such as octreotide, that are robust in vivo (7–9). The prevalence of neuroendocrine tumors is rather low, and other putative GPCR targets for radiopeptide ligands are abundantly expressed in frequently occurring human cancers (3). For example, GRPRs are highly expressed in human prostate, breast, and lung cancers and have been proposed as targets for bombesin-like radioligands (32,33). The impact of radiopeptides on clinical oncology could be tremendously increased if the success of [111In-DTPA]octreotide could be translated to another radiopeptide family.
However, success in this respect remains limited, partly because of the poor metabolic stability of radiopeptide drugs. To overcome this problem, several structural modifications have been pursued, but metabolic stability typically improves only at the cost of receptor affinity or the pharmacokinetic profile after radiopeptide injection into a living organism (10,11). On the other hand, assays widely used to assess radiopeptide metabolic stability usually involve in vitro incubation in mouse or human serum. As a result, the action of enzymes anchored to vasculature walls or blood cells or residing in major organs (lungs, kidneys, liver, and intestines) exposed to circulating drugs is totally disregarded, and metabolic stability is erroneously overestimated (34).
In the present study, we explored an exciting new approach to enhancing tumor targeting; in this approach, an enzyme inhibitor and a radiopeptide are intravenously coadministered in vivo. The inhibitor–radiopeptide pair are advantageously selected so that the radiopeptide is protected by the inhibitor immediately on entry into the circulation and until it reaches its tumor-associated receptor target. Therefore, only transient inhibition has been pursued and has been found adequate to safeguard the transit of the radiopeptide to the target. It is reasonable to assume that the increased tumor uptake induced by this strategy will eventually translate into higher diagnostic sensitivity and improved therapeutic efficacy of radiopeptide drugs in cancer patients.
We demonstrated the feasibility of this approach with a series of radiopeptides based on the somatostatin, bombesin, and gastrin peptide families. Through the coinjection of a single NEP inhibitor, PA (12,13), and a test radiopeptide, we were able, for the first time, to our knowledge, to induce a marked increase in tumor uptake in mouse models and to relate this result to a notable improvement in the level of the circulating intact drug. Of great significance is the fact that PA had no measurable effect on the tumor uptake of metabolically stable [111In-DTPA]octreotide in the same animal sst2-positive model used for [111In-DOTA-Ala1]SS14. This finding strongly supports our basic assumption that the observed increase in tumor uptake was a direct result of enhanced radiopeptide bioavailability induced through NEP inhibition by PA. The profound impact of the administration of a single NEP inhibitor on the bioavailability and in vivo tumor targeting of the radiopeptides tested in the present study is indeed astonishing in view of the large number of proteases present in the human body. The human degradome features at least 569 proteases and their homologs, comprising the 5 classes of metalloproteinases, serine proteases, cysteine proteases, threonine proteases, and aspartic proteases. Even more proteases are represented in the mouse and rat degradomes, which encompass 644 and 629 members, respectively (35,36). Therefore, although NEP seems to play a key role in initiating the breakdown of the radiopeptides tested in the present study and perhaps other radiopeptides, we continue our studies to reveal other potential candidates.
The proposed strategy may have significant consequences for biodegradable radiopeptides, even those based on native sequences (16,31). The latter have been evolutionarily optimized for best interacting with their cognate receptors. It is expected that this approach will rapidly broaden the application of more radiopeptide analogs to an increasing number of cancer classes. In addition, this approach provides a versatile means for identifying enzymes actually responsible for the in vivo degradation of a particular radiopeptide drug. This information may be essential for the rationale design of new, stabilized radiopeptide analogs but may also have consequences for other peptide drugs, such as peptide-conjugated cytotoxic drugs, optical imaging peptide probes, or peptidic ligands selected by phage display technologies (37).
CONCLUSION
The results of the present study emphasize the significance of radiopeptide in vivo stability as a key element of successful tumor targeting for cancer visualization and therapy in living subjects. Unexpectedly, our findings indicate that the action of a single peptidase (i.e., NEP) is responsible for the rapid in vivo breakdown of intravenously administered radiopeptides from at least the somatostatin, bombesin, and gastrin peptide families.
Most importantly, our findings provide the exciting opportunities to “protect” biodegradable radiopeptides and to “serve” in enhancing their supply and accumulation at tumor sites through the mere coinjection of a protease inhibitor, such as PA. Such amplification of radiolabel levels on tumor lesions is expected to strongly boost diagnostic sensitivity and therapeutic efficacy. In addition, there is a rich reservoir of information about existing peptidase inhibitors, a few of which have successfully been tested in human studies or are even approved drugs, highlighting the potential of this strategy for translation into clinical practice.
The proposed novel concept of radiopeptide drug–enzyme inhibitor coinjection to optimize tumor uptake is particularly valid for molecular imaging and therapy procedures. Because nuclear medicine protocols involve single or a limited number of systemic drug administrations, they require short-term enzyme inhibition only. Protection is essentially needed for the period between intravenous radiopharmaceutical injection and delivery to the tumor-associated receptor target—a period that is rather brief for small and rapidly localizing radiopeptide ligands. For such purposes, water-soluble agents “escorting” the radiopeptide over the hydrophilic body compartments and inducing fast and transient enzyme inhibition represent an elegant route for optimizing tumor targeting.
DISCLOSURE
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734. No potential conflict of interest relevant to this article was reported.
Acknowledgments
We thank Aikaterini Tatsi, Panteleimon J. Marsouvanidis, and Aikaterini Kaloudi for their assistance in parts of the metabolic and biodistribution experiments as well as Saskia Berndsen, Stuart Koelewijn, Jan de Swart, and Harald Groen for expert support during the SPECT/CT study.
Footnotes
Published online Nov. 28, 2013.
- © 2014 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication July 15, 2013.
- Accepted for publication September 25, 2013.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Targeting of the Cholecystokinin-2 Receptor with the Minigastrin Analog 177Lu-DOTA-PP-F11N: Does the Use of Protease Inhibitors Further Improve In Vivo Distribution?
- International Union of Basic and Clinical Pharmacology. CV. Somatostatin Receptors: Structure, Function, Ligands, and New Nomenclature
- Clinical History of the Theranostic Radionuclide Approach to Neuroendocrine Tumors and Other Types of Cancer: Historical Review Based on an Interview of Eric P. Krenning by Rachel Levine
- Evaluation of a Fluorescent and Radiolabeled Hybrid Somatostatin Analog In Vitro and in Mice Bearing H69 Neuroendocrine Xenografts
- Radiopeptides for Imaging and Therapy: A Radiant Future
- In Vitro and In Vivo Application of Radiolabeled Gastrin-Releasing Peptide Receptor Ligands in Breast Cancer
- Comparative Studies of Three 68Ga-Labeled [Des-Arg10]Kallidin Derivatives for Imaging Bradykinin B1 Receptor Expression with PET
- In Vivo Radioimaging of Bradykinin Receptor B1, a Widely Overexpressed Molecule in Human Cancer