


Abstract
Large neutral l-amino acids are substrates of system L amino acid transporters. The level of one of these, LAT1, is increased in many tumors. Aromatic l-amino acids may also be substrates of aromatic l-amino acid decarboxylase (AADC), the level of which is enhanced in endocrine tumors. Increased amino acid uptake and subsequent decarboxylation result in the intracellular accumulation of the amino acid and its decarboxylation product. 18F- and 11C-labeled neutral aromatic amino acids, such as l-3,4-dihydroxy-6-18F-fluorophenylalanine (18F-FDOPA) and 5-hydroxy-l-[β-11C]tryptophan, are thus successfully used in PET to image endocrine tumors. However, 5-hydroxy-l-[β-11C]tryptophan has a relatively short physical half-life (20 min). In this work, we evaluated the in vitro and in vivo characteristics of the 18F-labeled tryptophan analog 5-(2-18F-fluoroethoxy)-l-tryptophan (18F-l-FEHTP) as a PET probe for tumor imaging. Methods: 18F-l-FEHTP was synthesized by no-carrier-added 18F fluorination of 5-hydroxy-l-tryptophan. In vitro cell uptake and efflux of 18F-l-FEHTP and 18F-FDOPA were studied with NCI-H69 endocrine small cell lung cancer cells, PC-3 pseudoendocrine prostate cancer cells, and MDA-MB-231 exocrine breast cancer cells. Small-animal PET was performed with the respective xenograft-bearing mice. Tissues were analyzed for potential metabolites. Results: 18F-l-FEHTP specific activity and radiochemical purity were 50–150 GBq/μmol and greater than 95%, respectively. In vitro cell uptake of 18F-l-FEHTP was between 48% and 113% of added radioactivity per milligram of protein within 60 min at 37°C and was blocked by greater than 95% in all tested cell lines by the LAT1/2 inhibitor 2-amino-2-norboranecarboxylic acid. 18F-FDOPA uptake ranged from 26% to 53%/mg. PET studies revealed similar xenograft-to-reference tissue ratios for 18F-l-FEHTP and 18F-FDOPA at 30–45 min after injection. In contrast to the 18F-FDOPA PET results, pretreatment with the AADC inhibitor S-carbidopa did not affect the 18F-l-FEHTP PET results. No decarboxylation products of 18F-l-FEHTP were detected in the xenograft homogenates. Conclusion: 18F-l-FEHTP accumulates in endocrine and nonendocrine tumor models via LAT1 transport but is not decarboxylated by AADC. 18F-l-FEHTP may thus serve as a PET probe for tumor imaging and quantification of tumor LAT1 activity. These findings are of interest in view of the ongoing evaluation of LAT1 substrates and inhibitors for cancer therapy.
Most malignant lesions have an increased demand for amino acids. This property makes 18F- and 11C-labeled α-amino acids good candidates for tumor imaging by PET. Unlike 18F-FDG, the current gold standard for cancer imaging by PET, amino acids do not generally accumulate in the healthy brain or inflamed tissues. Amino acid–based PET probes thus provide good tumor-to-background ratios in the brain and allow differentiation between tumors and inflammation. Their accumulation in tumors is a consequence of increased amino acid uptake by cancer cells. In particular, the level of the heterodimer transport system for large neutral amino acids, LAT1/4F2hc (SLC7A5/SLC3A2; LAT1), is increased in many types of human tumors and may correlate with the malignancy of the lesion (1). The system L amino acid transporter (LAT) substrates l-[methyl-11C]methionine and O-(2-18F-fluoroethyl)-l-tyrosine (18F-FET) are used for brain tumor imaging (1,2).
The aromatic l-amino acids l-3,4-dihydroxyphenylalanine, l-tryptophan, and 5-hydroxy-l-tryptophan are substrates not only of LAT1 but also of aromatic l-amino acid decarboxylase (AADC), the level of which is enhanced in tumor cells with an endocrine character (3). Decarboxylation of those amino acids produces biogenic amines, which can subsequently be trapped in secretory vesicles. The combination of amino acid uptake and decarboxylation has been designated the APUD (amine precursor uptake and decarboxylation) concept (4). The PET probes l-3,4-dihydroxy-6-18F-fluorophenylalanine (18F-FDOPA) and 5-hydroxy-l-[β-11C]tryptophan (11C-5HTP) accumulate according to the APUD concept in endocrine tumors (1,5).
We aimed at synthesizing and evaluating an 18F-labeled alternative to 11C-5HTP for tumor imaging by PET because the longer physical half-life of 18F (110 min) has several advantages over the 20-min half-life of 11C (6). We hypothesized that 5-(2-18F-fluoroethoxy)-l-tryptophan (18F-l-FEHTP) (Fig. 1) is a substrate of LATs and possibly of AADC, similar to 11C-5HTP, and therefore would be a good PET probe for imaging endocrine tumors and possibly other tumors. During our evaluation of 18F-l-FEHTP, the synthesis of 18F-l-FEHTP and the first PET images and biodistribution results obtained with 18F-l-FEHTP in S180 fibrosarcoma–bearing mice were published by Li et al. (7).

Radiosynthesis scheme for generation of 18F-l-FEHTP. ACN = acetonitrile; TBAF = tetra-n-butylammonium fluoride; Ts = tosyl.
In this work, we describe the synthesis of 18F-l-FEHTP and compare its biochemical and pharmacologic characteristics in cell cultures and in small-animal PET with those of 18F-FDOPA. For the in vitro and in vivo evaluations, we chose an endocrine tumor model, the small cell lung cancer cell line NCI-H69 with high AADC activity (8); the so-called “pseudoendocrine” prostate cancer cell line PC-3, which has been shown to express AADC at the messenger RNA level (9); and exocrine breast cancer cell line MDA-MB-231, which displays no AADC activity. LAT1 expression has been shown to occur in all 3 cell lines (10–12).
MATERIALS AND METHODS
Radiosynthesis
18F-l-FEHTP was prepared with a 2-step radiolabeling approach similar to that described recently (7) and as depicted in Figure 1. The disodium salt of 5-hydroxy-l-tryptophan was prepared by reacting 5-hydroxy-l-tryptophan with 2 equivalent sodium methoxide (0.5N) in dry methanol at room temperature. No-carrier-added 18F-fluoride was produced via the 18O(p,n)18F nuclear reaction by irradiation of enriched 18O-water in a cyclotron (Cyclone 18/9; IBA). 18F-fluoride was immobilized on an anion-exchange cartridge (QMA Light; Waters) preconditioned with K2CO3 (5 mL, 0.5 M) and then 5–10 mL of water. The activity was eluted with tetrabutylammonium hydroxide (0.6 mL, 0.18 mM) and dried under vacuum with a stream of nitrogen at 110°C. Azeotropic drying was repeated 3 times with 1 mL of acetonitrile each time. To the dried 18F-fluoride complex (typically 20 GBq), ethylene glycol ditosylate (5–6 mg in 0.9 mL of CH3CN) was added, and the mixture was heated at 105°C for 6 min. The reaction mixture was cooled, diluted with 35% ethanol in water (10 mL), and passed through a LiChrolute EN cartridge (Merck). The 18F-labeled product was eluted with dimethyl sulfoxide (DMSO; 0.8–0.9 mL) to the second reactor, which had been preloaded with 5-hydroxy-l-tryptophan disodium salt (5–10 mg) in a mixture of DMSO (0.20 mL) and water (0.15 mL). The reaction mixture was heated at 120°C for 9 min. After cooling, the reaction mixture was diluted with water (1.8–2.0 mL) and purified by semipreparative high-performance liquid chromatography (HPLC) (Supplemental Material) (supplemental materials are available online only at http://jnm.snmjournals.org). The 18F-l-FEHTP product was collected at about 14 min and neutralized with sodium hydrogen carbonate (8.4%). The solution was passed through a sterile filter and used for in vitro and in vivo studies.
18F-dl-FEHTP was produced by the same procedure but with the racemic 5-hydroxy-tryptophan disodium salt.
18F-FDOPA was obtained at the University Hospital Zurich from routine production for clinical use (13). The specific activity was 9–120 GBq/mmol at the end of synthesis.
In Vitro Cell Studies
PC-3, NCI-H69, and MDA-MB-231 cells were purchased from the German Collection of Microorganisms and Cell Cultures, Cell Lines Service, and the American Type Culture Collection, respectively. Cells were grown to subconfluence in 48-well plates (Costar; Corning) or 5-mL plastic tubes (for NCI-H69). Cells were washed and incubated for 1 h at 37°C with Earle balanced salt solution containing Ca2+ and Mg2+ (EBSS; Invitrogen). The irreversible monoamine oxidase A and B inhibitors clorgyline (Sigma) and pargyline (Acros Organics), respectively, and—if indicated—the LAT inhibitor 2-amino-2-norboranecarboxylic acid (BCH) were added at final concentrations of 0.1, 0.1, and 10 mM, respectively. At time zero, about 20 kBq of 18F-l-FEHTP or 18F-FDOPA were added, and the cells were incubated at 37°C or on ice. The final tracer concentrations were 0.3–4 nM 18F-l-FEHTP and 0.3–4 μM 18F-FDOPA, that is, less than typical Michaelis-Menten constants KM values for LAT1 and AADC (14). Total uptake was less than 15% of the added tracer after 1 h. At various time points, the cells were washed twice with ice-cold EBSS, and adherent cells were detached with trypsin–ethylenediaminetetraacetic acid (Invitrogen). All cells were transferred to Eppendorf tubes, and the radioactivity was quantified in a γ-counter (Wizard 1480; PerkinElmer). To inhibit AADC, we added 80 μM S-carbidopa (Santa Cruz Biotechnology) in DMSO–100 mM phosphate-buffered saline 30 min before tracer addition. The final DMSO concentration was 0.3%. Cell viability was 80%–90% after 60 min for all conditions, as tested with trypan blue staining.
For efflux assays, cells were preincubated for 1 h as described earlier, and then the cultures were washed twice with ice-cold EBSS and incubated with EBSS or EBSS containing 0.8 mM l-leucine (time zero). At various time points, the cells were washed twice with ice-cold EBSS, detached as described earlier, and analyzed in the γ-counter. Protein was quantified with the DC Protein Assay (Bio-Rad) after cell lysis with 2% sodium dodecyl sulfate. Bovine serum albumin was used for calibration.
In Vivo PET Experiments
Animal care and experiments were conducted in accordance with Swiss Animal Welfare legislation and were approved by the Veterinary Office of Canton Zurich, Zurich, Switzerland. Five-week-old female NMRI nude and BALB/c mice were supplied by Charles River. They were allowed free access to water and food.
Six-week-old mice were inoculated subcutaneously in the right shoulder with 2 × 106 PC-3 or MDA-MB-231 cells in 100 μL of Matrigel (BD Biosciences) or 5 × 106 NCI-H69 cells in 100 μL of phosphate-buffered saline with Ca2+ and Mg2+ (Invitrogen). At 3–5 wk after inoculation, when the tumor volumes reached 0.2–1.5 cm3, the mice were injected in a tail vein with 3.5–18 MBq of 18F-l-FEHTP or 18F-FDOPA. For experiments with AADC inhibition, mice were injected intraperitoneally with S-carbidopa at 25 mg/kg 60 min before tracer injection. For transport competition experiments, mice were injected intraperitoneally with l-tryptophan at 25 mg/kg 10 min before tracer injection. Anesthesia with 2%–3% isoflurane in oxygen–air was initiated 10 min before the PET scan, and animals were monitored as described previously (15).
PET scans were performed with a VISTA eXplore small-animal PET/CT camera (GE Healthcare) in list mode for dynamic analysis or static mode for whole-body scans (2 bed positions) (16). Static scans with 2 bed positions were started with the anterior body part containing the tumor 30 min after tracer injection. The complete 2 bed position scans lasted 30 min (15 min per bed position). Data were reconstructed with the 2-dimensional ordered-subsets expectation maximization protocol and analyzed with PMOD Version 3.2 software (PMOD Technologies Ltd.). A region of interest for the xenograft, a reference region of equal shape and volume on the contralateral side, and a region of interest for the brain were drawn in PMOD on the basis of the PET images. Tissue radioactivity was expressed as the standardized uptake value (SUV), that is, the decay-corrected radioactivity per cubic centimeter divided by the injected radioactivity dose per gram of body weight.
Ex Vivo Biodistribution, Metabolite Studies, and In Vitro Plasma Albumin Binding
After the PET scans, mice were sacrificed by decapitation under isoflurane anesthesia, and tissues were removed, weighed, and analyzed in the γ-counter. The accumulated radioactivity per gram of tissue was calculated as the decay-corrected radioactivity per gram of tissue divided by the injected radioactivity dose per gram of body weight.
For ex vivo metabolite studies, 14–32 MBq of 18F-l-FEHTP or 18F-FDOPA were injected via a lateral tail vein into xenograft-bearing 9- to 12-wk-old NMRI nude mice. Blood samples were withdrawn via the opposite tail vein, and animals were sacrificed by decapitation under isoflurane anesthesia at various time points. Xenografts were excised and homogenized in equal volumes of phosphate-buffered saline (Invitrogen) with a Polytron (Kinematica, Inc.). Proteins in the xenograft homogenates, plasma, and urine were precipitated with equal volumes of ice-cold acetonitrile and centrifugation. Supernatants of the xenograft homogenates were extracted again, and all supernatants were filtered and analyzed by reversed-phase thin-layer chromatography and ultra-performance liquid chromatography (Supplemental Material). Serum albumin binding was determined by equilibrium dialysis as described in the Supplemental Material.
RESULTS
Radiosynthesis of 18F-l-FEHTP and 18F-dl-FEHTP
Chiral HPLC analysis demonstrated that an l-enantiomeric pure product was obtained when an l-enantiomeric pure precursor was used for radiolabeling. In a typical experiment, a radiochemical yield of about 23% (decay corrected, corresponding to 15% not decay corrected) was achieved with a radiochemical purity of greater than 95%. The specific activities were in the range of 50–150 GBq/μmol at the end of synthesis. When 5-hydroxy-dl-tryptophan disodium salt was used as the precursor, similar radiochemical yield and radiochemical purity were obtained.
In Vitro Cell Uptake and Efflux Studies
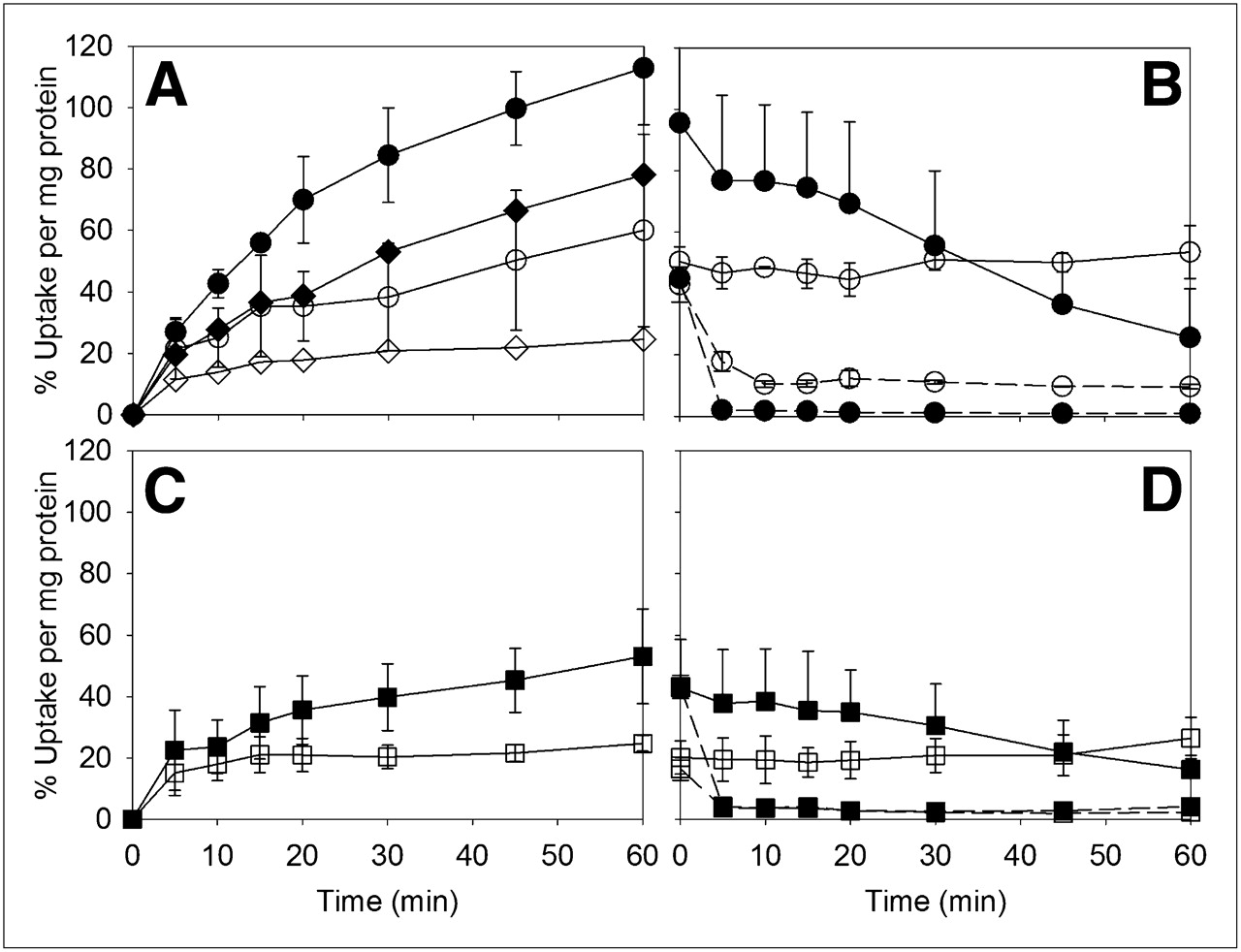
Figure 2 shows the rapid influx of 18F-l-FEHTP and 18F-FDOPA into NCI-H69 endocrine cells at 37°C and 4°C, reaching between 37% and 52% of the total added radioactivity per milligram of protein within 5 min. After the fast initial uptake, 18F-l-FEHTP radioactivity increased steadily, whereas 18F-FDOPA radioactivity decreased at 37°C but not at 4°C.

(A–D) Uptake (A and C) and efflux after 60 min of uptake (B and D) of 18F-l-FEHTP (A and B) and 18F-FDOPA (C and D) in NCI-H69 cell cultures at 37°C (• and ▪) and 4°C (○ and □). (E) 18F-FDOPA uptake in presence of 80 μM S-carbidopa (▲ and ▵) and 0.3% DMSO as control (▪ and □) at 37°C (▲ and ▪) and 4°C (▵ and □). (F) 18F-l-FEHTP efflux in presence of 0.8 mM l-leucine in incubation buffer at 37°C (•) and 4°C (○). Symbols represent averages from 3 independent experiments each (except for 2 independent experiments with l-leucine). Error bars show SDs, except for experiments with l-leucine, for which error bars indicate values from 2 independent experiments.
When no amino acids were added to the incubation buffer, the efflux of 18F-l-FEHTP was slow compared with the uptake at 37°C and was negligible at 4°C (Fig. 2B). The release of 18F-FDOPA or its metabolites continued as observed during uptake at 37°C and was negligible at 4°C (Fig. 2D). The decrease in radioactivity after the initial fast uptake of 18F-FDOPA was almost abolished in the presence of 80 μM S-carbidopa (Fig. 2E). At this concentration, intracellular AADC is partially inhibited without a significant loss of viability of endocrine tumor cells (5,17,18). Figure 2F shows the rapid efflux of 18F-l-FEHTP at 37°C and 4°C after the addition of 0.8 mM l-leucine, a LAT1 substrate.
The accumulation (average ± SD) of 18F-l-FEHTP and 18F-FDOPA in PC-3 cells reached 113% ± 19%/mg and 53% ± 15%/mg, respectively, of added radioactivity at 37°C and 60 min (Fig. 3). Efflux was negligible at 4°C but was significant at 37°C. As observed for NCI-H69 cells, the addition of 0.8 mM l-leucine resulted in the rapid efflux of 18F-l-FEHTP and 18F-FDOPA within 5 min. The uptake of racemic 18F-dl-FEHTP was between 50% and 70% of that of 18F-l-FEHTP, indicating a lower level of uptake of the d-isomer than of the l-isomer (Fig. 3A).

Uptake (A and C) and efflux (B and D) in PC-3 cells of 18F-l-FEHTP (A and B, • at 37°C and ○ at 4°C connected with solid lines), 18F-dl-FEHTP (A, ◆ at 37°C and ♢ at 4°C), and 18F-FDOPA (C and D, ▪ at 37°C and • at 4°C connected with solid lines). Efflux of 18F-l-FEHTP (B) and 18F-FDOPA (D) was also studied with 0.8 mM l-leucine in incubation buffer (symbols connected with dashed lines). Symbols represent averages from 3 independent experiments each (except for 2 independent experiments with l-leucine and 1 experiment with 18F-dl-FEHTP). Error bars show SDs, except for experiments with l-leucine, for which error bars indicate values from 2 independent experiments.
Figure 4 shows the accumulation of 18F-labeled amino acids in MDA-MB-231 exocrine cells. 18F-l-FEHTP and 18F-FDOPA uptake reached 48% ± 3%/mg and 26% ± 4%/mg, respectively, of added radioactivity after 60 min of incubation at 37°C. Both amino acids also showed significant uptake at 4°C. When no amino acids were added to the incubation buffer, both amino acids showed moderate efflux at 37°C and no significant net efflux at 4°C.

Uptake (A and C) and efflux (B and D) of 18F-l-FEHTP (A and B) and 18F-FDOPA (C and D) in MDA-MB-231 cells at 37°C (• and ▪) and 4°C (○ and □). Symbols and error bars represent averages and SDs from 3 independent experiments each.
The LAT competitive inhibitor BCH blocked 18F-l-FEHTP uptake in all cell lines by greater than 95% at 10 mM. Blocking was less efficient for 18F-FDOPA, with 13% ± 6% and 10% ± 6% residual uptake at 37°C in NCI-H69 cells and PC-3 cells, respectively (Supplemental Material).
In Vivo Metabolism
18F-FDOPA and 5-hydroxy-l-tryptophan are decarboxylated in humans and laboratory animals (19). We found only radioactive metabolites and no 18F-FDOPA in the blood, brain, and tumor 60 min after 18F-FDOPA administration (data not shown). In contrast, mainly parent 18F-l-FEHTP was detected in the blood, brain, and xenografts 15 and 60 min after 18F-l-FEHTP administration. Some radioactive metabolites of unknown identity were detected in the urine, and traces were detected in the blood. The results of reversed-phase thin-layer chromatography of the tissue extracts are shown in Figure 5. The results were confirmed by ultra-performance liquid chromatography with 2 different mobile phases (data not shown).

In vivo metabolism of 18F-l-FEHTP. 18F-l-FEHTP (14–32 MBq) was injected into xenograft-bearing NMRI nude mice. Tissues were extracted and analyzed by reversed-phase thin-layer chromatography. 18F-l-FEHTP reference sample had same retention time as main spot in all lanes. Minutes indicate time of sacrifice after 18F-l-FEHTP injection. No metabolites were detected in xenografts, and only traces of metabolites were detected in blood.
Small-Animal PET with Xenograft-Bearing Mice
On the basis of the favorable uptake kinetics of 18F-l-FEHTP in the cell experiments, we expected significant uptake into xenografts in vivo. Pilot dynamic PET scans with NMRI nude mice showed the accumulation of both 18F-l-FEHTP and 18F-FDOPA in PC-3 xenografts. The 18F-l-FEHTP SUV ratios in the xenografts and reference tissues decreased with time between 30 and 150 min after injection. The 18F-FDOPA SUV ratios did not decrease, in accordance with the APUD concept (Fig. 6). We chose the time window of 30–45 min after injection for static PET scans. No significant uptake of 18F-l-FEHTP into bone was observed, indicating that in vivo defluorination was negligible.

(A) Time–activity curves (SUVs) for 18F-l-FEHTP (circles) and 18F-FDOPA (squares) in PC-3 xenografts (black), brain (gray), and reference tissue (no fill). (B) Ratios of PC-3 xenograft activity to reference tissue activity (black) and brain activity to reference tissue activity (gray) for 18F-l-FEHTP (circles) and 18F-FDOPA (squares). Connected symbols represent time–activity curves from 1 mouse. 18F-FDOPA was used without S-carbidopa.
Figure 7A shows PET images (static) of NMRI nude mice bearing NCI-H69, PC-3, and MDA-MB-231 xenografts. 18F-FDOPA was administered after S-carbidopa preadministration. The levels of radioactivity accumulation in the xenografts and reference regions were similar for the 2 tracers. Figures 7B and 7C show the respective SUVs and SUV ratios. The MDA-MB-231 xenografts had a large gelatinous core (Supplemental Material), resulting in relatively low levels of average tumor uptake of 18F-l-FEHTP and 18F-FDOPA. However, PET sectional reconstruction (Fig. 7A) showed tracer accumulation in the periphery of the MDA-MB-231 xenografts.

PET analysis of xenograft-bearing mice with 18F-l-FEHTP and 18F-FDOPA. (A) Identical NCI-H69 (top row), PC-3 (middle row), and MDA-MB-231 (bottom row) xenograft–bearing NMRI nude mice were each scanned with 18F-l-FEHTP (left of scale bar) and 18F-FDOPA (right of scale bar). At 1 h before 18F-FDOPA scans, S-carbidopa (25 mg/kg) was injected intraperitoneally. Shown are transversal and coronal sections (SUV color scale) and maximum-intensity projections (MIP, gray scale). Tumors are indicated by arrows. (B) SUVs of xenografts and reference regions after 18F-l-FEHTP and 18F-FDOPA injections. PC-3, w/o carbid. = PC-3 xenograft–bearing BALB/c mice without S-carbidopa pretreatment (images not shown). (C) Ratios of xenograft SUVs to reference region SUVs for 18F-l-FEHTP and 18F-FDOPA. Data are averages and SDs from 3–7 animals each. There were no significant differences between 18F-l-FEHTP and 18F-FDOPA (P > 0.05; 18F-FDOPA after S-carbidopa pretreatment). There were significant differences between 18F-DOPA SUVs with and without S-carbidopa pretreatment (P < 0.01) but not between the xenograft-to-reference tissue SUV ratios with and without S-carbidopa pretreatment. Note that MDA-MB-231 tumors had gelatinous core.
18F-dl-FEHTP was tested in a PC-3 xenograft–bearing mouse. The xenograft and reference tissue SUVs were about half those of 18F-l-FEHTP, resulting in a similar xenograft-to-reference tissue SUV ratio as found with 18F-l-FEHTP (data not shown).
Next, we investigated whether plasma l-tryptophan levels had an influence on 18F-l-FEHTP PET images. After the administration of tryptophan at 25 mg/kg, the xenograft SUVs and xenograft-to-reference tissue SUV ratios were lower by trend but without significance (P > 0.05). The SUV ratios for NCI-H69 xenografts were 1.71 ± 0.26 (with l-tryptophan preadministration, n = 4) versus 1.95 ± 0.30 (no l-tryptophan preadministration, n = 3, from Fig. 7C) and for PC-3 xenografts 1.57 ± 0.05 (with l-tryptophan preadministration, n = 4) versus 1.80 ± 0.39 (no l-tryptophan preadministration, n = 7, from Fig. 7C).
Influence of AADC Inhibition on 18F-FDOPA PET
The 18F-FDOPA PET scans shown in Figure 7A were performed after S-carbidopa pretreatment. Four PC-3 xenograft-bearing BALB/c mice were also scanned with 18F-FDOPA in the absence of S-carbidopa. The 18F-FDOPA SUVs in the xenografts and reference tissues were about 50% those after S-carbidopa pretreatment (Fig. 7B). However, the average xenograft-to-reference tissue SUV ratios in the 2 protocols were not significantly different (Fig. 7C). The xenograft and background 18F-l-FEHTP SUVs in the same mice were not significantly different from those in the NMRI nude mice (Fig. 7B). The 18F-l-FEHTP xenograft-to-reference tissue SUV ratio in the PC-3 xenograft–bearing BALB/c mice was 2.1 ± 0.3 (n = 4). S-carbidopa had no influence on 18F-l-FEHTP PET images (Supplemental Material).
Ex Vivo Biodistribution and In Vitro Serum Albumin Binding
Table 1 shows average uptake values (SUV) for 18F-l-FEHTP 70 min after injection. The levels of uptake (reported as percentage injected dose per gram of body weight [%ID/g]) were highest in the pancreas (29–55 %ID/g) and then the kidneys (6–18 %ID/g) and xenografts (5–9 %ID/g). The preadministration of tryptophan at 25 mg/kg had no significant influence on the biodistribution of 18F-l-FEHTP but resulted in a generally lower level of uptake in tissues with high LAT1 expression, that is, NCI-H69 xenografts, brain, and kidneys.
Ex Vivo Biodistribution* Data for 18F-l-FEHTP 70 Minutes After Injection
A high level of plasma protein binding could result in a high level of background radioactivity because of slow clearance from the blood. Plasma tryptophan is associated to 80%–90% with albumin (20). The bound fraction of 18F-l-FEHTP in 4% bovine serum albumin at 37°C was negligible, that is, 0.05 ± 0.01 (n = 4 dialysis cells). No binding was detected with human or rat plasma diluted 1/20 (data not shown).
DISCUSSION
18F-l-FEHTP synthesis was similar to the procedure recently described by Li et al. (7), with similar radiochemical yield and purity. The complete radiolabeling process was established in a fully automated module, resulting in reproducible quality of the product and reducing the radioactivity burden on the radiochemist. The crude product was purified by semipreparative HPLC with 35 mM acetate buffer containing 8% ethanol. The collected HPLC fraction was directly used for in vitro and in vivo experiments after neutralization with 8.4% sodium bicarbonate.
18F-l-FEHTP accumulated in cancer cells of endocrine, pseudoendocrine, and exocrine phenotypes in vitro and in vivo. Decarboxylation or another metabolic step was not involved in the accumulation, confirming that increased transport alone is sufficient for tumor imaging by PET with labeled large neutral amino acids (2).
Because 18F-l-FEHTP uptake into the cells was observed not only at 37°C but also at 4°C and because efflux was accelerated by the addition of l-leucine, we concluded that accumulation was mediated by the exchange of one or more amino acids rather than unilateral or concentrative transporters. The low temperature sensitivity of the cell uptake of 18F-l-FEHTP was in line with the results of an early study on l-tryptophan uptake into Ehrlich ascites tumor cells (21). Exchange transporters carrying neutral aromatic amino acids are SLC7A5 (LAT1), SLC7A6 (Y+LAT2), SLC7A7 (Y+LAT1), SLC7A8 (LAT2), SLC7A9 (B(0,+)AT), SLC43A1 (LAT3), and SLC43A2 (LAT4) (14,22). Na+-dependent symporters that may also be involved in the transport of l-tryptophan analogs are SLC1A5 (ATB0, ASCT2) and SLC6A14 (ATB0+) (14).
The cell uptake of 18F-l-FEHTP was inhibited to greater than 95% by 10 mM BCH in all tested cell lines. BCH is a substrate and competitive inhibitor of Na+-independent transport of large neutral amino acids, with typical transporter-inhibitor constants Ki values of less than 1 mM for SLC7A5 (LAT1) and SLC7A8 (LAT2) (23,24). SLC7A6, SLC7A7, SLC1A5, and SLC6A14 were not directly inhibited by BCH, and no amino acid efflux was observed for SLC7A7 after the addition of l-leucine (25–29). SLC7A9, SLC43A1, and SLC43A2 were only moderately inhibited by 10 mM BCH (30–32).
Considering the highly efficient transport inhibition by 10 mM BCH in our experiments with 3 tumor cell lines of different phenotypes, we concluded that 18F-l-FEHTP was almost exclusively taken up by LAT1 or LAT2. The latter is, however, not typically overexpressed in cancer cells (33). 18F-FDOPA influx into MDA-MB-231 nonendocrine cells was almost completely blocked by BCH, also suggesting LAT1 or LAT2 as the major uptake mechanism. These results were in agreement with the findings of Neels et al. (5).
18F-FDOPA radioactivity was released from NCI-H69 endocrine cells during uptake experiments at 37°C but not at 4°C, suggesting an energy-driven mechanism. According to the APUD concept, a higher level of accumulation of 18F-FDOPA radioactivity would be expected at 37°C than at 4°C because of accumulation via enzymatic decarboxylation. The unexpected energy-dependent efflux of radioactivity was inhibited by the AADC inhibitor S-carbidopa, indicating that it was related to 18F-FDOPA decarboxylation. The observed AADC-dependent efflux may have resulted from the release of the decarboxylation product 6-18F-fluoro-l-dopamine. In contrast to the 18F-FDOPA results, the kinetics and extent of 18F-l-FEHTP uptake were temperature independent, excluding the involvement of any energy-dependent mechanism, such as AADC.
18F-l-FEHTP and 18F-FDOPA accumulated significantly in NCI-H69 and PC-3 xenografts, with no significant difference between the tracers or the tumor models at scan times between 30 and 45 min after injection. MDA-MB-231 xenografts were not ideal for quantitative comparisons because of their large gelatinous core. However, both tracers accumulated in the intact periphery of the xenografts.
Our data indicated that 18F-l-FEHTP was not decarboxylated in vivo. In contrast to the findings obtained with 18F-FDOPA, the AADC inhibitor S-carbidopa had no influence on 18F-l-FEHTP PET images. Furthermore, no metabolites of l-FEHTP were detected in tumor homogenates, and only traces of metabolites were found in blood. Although the 18F-FDOPA SUV ratios for PC-3 xenografts and reference tissues did not decrease between 30 and 150 min, in line with the APUD concept, the respective SUV ratios of 18F-l-FEHTP decreased with time. The observed metabolic stability of 18F-l-FEHTP was in contrast to the findings obtained with 11C-5HTP (5,19). The latter was almost quantitatively decarboxylated within minutes in human carcinoid (endocrine) liver tumors and metastases (19).
The methylation of l-3,4-dihydroxyphenylalanine at catechol 3-OH resulted in the loss of its properties as a substrate or inhibitor of AADC, suggesting that binding to the active site was abolished (34). On the basis of these findings and our observations with 18F-l-FEHTP, it may be speculated that O-alkylation in the aromatic system of AADC substrates changes their electronic or steric properties in a way that disfavors binding to the active site. Structure–activity relationship, site-directed mutagenesis, or molecular modeling studies are required to elaborate the structural properties of AADC substrates.
Our findings indicated that the mechanism of 18F-l-FEHTP accumulation was more similar to that of 18F-FET than to that of 11C-5HTP. 18F-FET is taken up by transport systems but is not metabolized in tumor cells (2). In mice with peripheral xenografts, 18F-l-FEHTP and 18F-FET showed similar distribution and kinetic behaviors, including brain uptake, with higher relative kidney radioactivity of 18F-l-FEHTP than of 18F-FET (35). In vitro 18F-FET uptake was less efficiently inhibited by BCH than we observed for 18F-l-FEHTP (2), indicating that the latter may be more selective for LAT1 than 18F-FET. However, only a direct comparison will provide a definitive conclusion.
l-tryptophan is a substrate of indoleamine 2,3-dioxygenase (IDO). Intracellular IDO activity may result in the trapping of polar 18F-l-FEHTP metabolites or in indirect 18F-l-FEHTP accumulation via intracellular l-tryptophan depletion. Significant IDO activity was shown for MDA-MB-231 cells after interferon-γ stimulation (12). We did not detect any radiometabolites in MDA-MB-231 xenografts 15 min after injection and therefore did not further investigate this potential route of 18F-l-FEHTP radioactivity accumulation.
In view of the evaluation of LAT1 substrates and inhibitors as potential anticancer drugs (36–38), 18F-l-FEHTP is a candidate PET tracer for staging tumors according to their LAT1 expression and activity and therefore their susceptibility to LAT1-targeted drugs. In addition, the in vivo efficiency of LAT1 inhibitors may be monitored directly by PET. Further studies are required to show the correlation between tumor LAT1 activity and 18F-l-FEHTP uptake kinetics in vitro and in vivo and to evaluate the direct and indirect effects of other amino acid transporters and IDO on this correlation.
We found relatively high levels of 18F-l-FEHTP and 18F-FDOPA radioactivity in all tissues, with notably high activity in the pancreas. Serum albumin binding was excluded as a cause of high levels of 18F-l-FEHTP background activity. The latter may be assigned to the relatively high LAT1 expression in healthy rodent tissue. Mice have relatively high LAT1 expression levels in most tissues, with particularly high levels in the pancreas. Humans have high LAT1 expression levels in protective endothelial and epithelial barriers and in proliferating cells but negligible expression in other tissues (22,25,39,40). Taking these species differences into account, we expect lower levels of background radioactivity in humans and therefore higher SUV ratios between tumors with increased LAT1 activity and healthy tissues.
CONCLUSION
We have characterized the biochemical and pharmacologic properties of 18F-l-FEHTP, an 18F-labeled aromatic l-amino acid analog with promise for tumor imaging. We identified 18F-l-FEHTP as a substrate for LAT1/2 transport but not for decarboxylation by AADC. Therefore,18F-l-FEHTP is a potent PET probe for the LAT1 activity of malignant lesions, independent of the AADC activity of tumors and healthy tissues. It may be used in the future to stage tumors for susceptibility to LAT1-targeted drugs.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Petra Wirth for assistance with animal experiments and Bruno Mancosu, Judith Frässdorf, Anass Johayem, and Zoran Vujicic for assistance with tracer development and for tracer production. We thank Olga S. Fedorova and Holger Siebeneicher for their contributions to tracer development. We thank Sandra Borkowski, Sabine Zitzmann-Kolbe, Keith Graham, and Ludger Dinkelborg from Bayer Schering Pharma for fruitful discussions. No potential conflict of interest relevant to this article was reported.
Footnotes
Published online Feb. 13, 2012.
- © 2012 by the Society of Nuclear Medicine, Inc.
REFERENCES
- Received for publication August 8, 2011.
- Accepted for publication October 25, 2011.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Assessment of Tryptophan Uptake and Kinetics Using 1-(2-18F-Fluoroethyl)-L-Tryptophan and {alpha}-11C-Methyl-L-Tryptophan PET Imaging in Mice Implanted with Patient-Derived Brain Tumor Xenografts
- Epileptic Activity Increases Cerebral Amino Acid Transport Assessed by 18F-Fluoroethyl-L-Tyrosine Amino Acid PET: A Potential Brain Tumor Mimic
- 18F-Fluorodihydroxyphenylalanine PET/CT in Patients with Neuroendocrine Tumors of Unknown Origin: Relation to Tumor Origin and Differentiation