


Abstract
Developing a PET ligand for imaging of the peripheral benzodiazepine receptor (PBR; Translocator Protein [18 kDa] TSPO) is of great importance for studying its role in glial cells in the injured brain and in neurodegenerative disorders, such as Alzheimer's disease. The aim of this study was to synthesize and evaluate N-benzyl-N-ethyl-2-(7-11C-methyl-8-oxo-2-phenyl-7,8-dihydro-9H-purin-9-yl)acetamide (11C-AC-5216) as a PET ligand for imaging PBR in the primate brain. Methods: AC-5216 and its desmethyl precursor (compound 1) were synthesized starting from commercially available compounds. The radiosynthesis of 11C-AC-5216 was performed through the reaction of compound 1 with 11C-CH3I in the presence of NaH. The in vivo brain regional distribution was determined in mice (dissection) and a monkey (PET). Results: 11C-AC-5216 (800–1,230 MBq; n = 25) was obtained with a radiochemical purity of 98% and a specific activity of 85–130 GBq/μmol at the end of synthesis. After injection of 11C-AC-5216 into mice, a high accumulation of radioactivity was found in the lungs, heart, adrenal glands, and other PBR-rich organs. In the mouse brain, high radioactivity was observed in the olfactory bulb and cerebellum. Radioactivity in these regions was inhibited by nonradioactive AC-5216 or PK11195 but was not decreased by central benzodiazepine receptor–selective flumazenil and Ro15-4513. A PET study of the monkey brain determined that 11C-AC-5216 had a relatively high uptake in the occipital cortex, a rich PBR-dense area in the primate brain. Pretreatment with nonradioactive AC-5216 and PK11195 reduced the radioactivity of 11C-AC-5216 in the occipital cortex significantly, suggesting its high specific binding with PBR in the brain. Metabolite analysis demonstrated that 11C-AC-5216 was stable in vivo in the mouse brain, although it was metabolized in the plasma of mice and the monkey. Conclusion: 11C-AC-5216 is a promising PET ligand for imaging PBR in rodent and primate brains.
The peripheral benzodiazepine receptor (PBR; Translocator Protein [18 kDa] TSPO) (1) was identified as a peripheral binding site for diazepam and has been demonstrated to be functionally and structurally different from the classical central benzodiazepine receptor (CBR) (2–4). PBR, which was initially found in the peripheral organs, including the kidneys, nasal epithelium, lungs, and heart, and the endocrine organs, such as the adrenal glands, testes, and pituitary gland (3–5), was subsequently found in the central nervous system. PBR is mainly located in the glial cells of the brain, and PBR expression in vivo was found to be increased in microglia activated by brain injury (6,7). The increase in PBR density has thus been used as an indicator of neuronal damage and neurodenerative disorders, such as Alzheimer's disease (6–12). These findings have prompted the development of an imaging agent labeled with a radioisotope, which has made it possible to visualize the distribution of PBR in the animal brain, including the human brain (13–16).
The recent development of PET ligands specific for receptors has made it possible to quantitatively evaluate the density and pharmacologic action of receptors in addition to the receptor occupancy of therapeutic drugs, which provides information concerning the most effective doses of drug therapy (17). 11C-PK11195, a positron emitter–labeled ligand, was first developed to characterize PBR in the human brain (13,18). However, the relatively low uptake of 11C-PK11195 in the brain limited its wide application (11,19). To characterize PBR precisely using a PET ligand with advantages over 11C-PK11195, we developed 2 novel ligands, 11C-DAA1106 (20,21) and 18F-FEDAA1106 (22), for PBR imaging in the brain. These ligands exhibited more potent in vitro binding affinity for PBR than PK11195 and displayed remarkable selectivity against other receptors, including CBR (22–24). In vivo studies demonstrated that they had higher uptake and more specific binding in rodent and primate brains than 11C-PK11195 (20–22). Now, 11C-DAA1106 and 18F-FEDAA1106 are being used to investigate PBR in the human brain at our facility to elucidate the relationship between PBR and brain diseases (25,26). Subsequently, 11C-PBR28, an analog of 11C-DAA1106, was developed to localize and quantify upregulated PBR associated with cerebral ischemia in rats (27).
Despite the success of 11C-DAA1106 analogs, we are continuing to make efforts to develop new ligands for PBR imaging in the primate brain. In this study, we selected N-benzyl-N-ethyl-2-(7-methyl-8-oxo-2-phenyl-7,8-dihydro-9H-purin-9-yl)acetamide (AC-5216) (Fig. 1) (28) as a new candidate for use as a PET ligand for PBR. First, AC-5216 had a higher affinity for PBR prepared from rat brain (inhibition constant [Ki], 0.297 nM) than PK11195 (0.602 nM), a standard ligand for PBR (28). Moreover, AC-5216 exhibited negligible affinity for CBR and a large number of other receptors, monoamine transporters, or ion channels. Second, the binding site for AC-5216 in the PBR domain might be closer to that for PK11195 than to those for other PBR ligands, such as Ro5-4864 (28). In addition to Ro5-4864, the binding site for DAA1106 in the PBR domain might have an extra component that does not interact efficiently with PK11195 (23,24). Third, AC-5216 has moderate lipophilicity; a mean cLogP (P is the octanol/water partition coefficient) value of 3.5 was calculated with Pallas 3.4 software (Pallas GmbH), and a mean LogD (D is the octanol/water diffusion coefficient) value of 3.3 was measured in a phosphate buffer (pH 7.4):octanol system by the shaking flask method (n = 3; maximum range, ±5%). A moderate lipophilicity is a prerequisite for a favorable PET ligand, guaranteeing high uptake and low nonspecific binding in the brain. Compared with that of AC-5216, the lipophilicity of PK11195 is too high (cLogP: 5.1; LogD: 3.7); the cLogP and LogD values were determined and are presented as described for AC-5216. A high lipophilicity causes high nonspecific binding in the brain (19). Finally, this compound structurally has a methyl group on the amide moiety and is therefore suitable for labeling with 11C-CH3I without a change in its chemical structure or pharmacologic profile.
Synthesis scheme for AC-5216, desmethyl precursor (compound 1), and 11C-AC-5216. Reaction conditions included the following: a—BOP, DMF, room temperature, Et3N, 3 h; b—EtoH, reflux, 30 min; c—DMF, 100°C, 6 h; d—NaH, DMF, room temperature, 3 h; e—11C-CH3I, DMF, 30°C, 3 min.
The aim of this study was to synthesize and evaluate 11C-AC-5216 as a PET ligand for PBR in mouse and monkey brains. In this article, we report the chemical synthesis of AC-5216 and its desmethyl precursor (compound 1) starting from commercially available compounds; the labeling of 11C-AC-5216 through the N-11C-methylation of compound 1 with 11C-CH3I; the regional distribution in mice and the percentages of unmetabolized ligand in the mouse plasma and brain; and the uptake and specific binding to PBR in the monkey brain, as determined by PET. An evaluation of 11C-AC-5216 in tumor-bearing mice was published elsewhere (29).
MATERIALS AND METHODS
General
All chemicals and solvents were of analytic or high-performance liquid chromatography (HPLC) grade and were obtained from Aldrich and Wako Pure Industries. AC-5216 and its desmethyl precursor (compound 1) were synthesized in our laboratory. PK11195, flumazenil, and Ro15-4513 were purchased from Aldrich or ABX. These compounds (1 mg) were dissolved in distilled water (1.8 mL) and EtoH (0.2 mL) and used for in vivo studies. 11C was produced by 14N(p, α)11C nuclear reaction with a CYPRIS HM18 cyclotron (Sumitomo Heavy Industry). A dose calibrator (IGC-3R Curiemeter; Aloka) was used for all radioactivity measurements, unless otherwise stated. Reverse-phase HPLC was performed with a JASCO system (JASCO); effluent radioactivity was determined with an NaI(Tl) scintillation detector system. The animal experiments were performed according to the recommendations of the Committee for the Care and Use of Laboratory Animals, National Institute of Radiologic Sciences.
Chemical Synthesis
N-Benzyl-N-Ethyl-2-(8-Oxo-2-Phenyl-7,8-Dihydro-9H-Purine-9-yl)Acetamide (Compound 1).
The desmethyl precursor (compound 1) for radiosynthesis was prepared according to the procedures described in the patent (30), with minor modifications. The reagent 1H-benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate (BOP) (2.0 g, 4.4 mmol) was added to a mixture of 2-(5-ethoxycarbonyl)-2-phenylpyrimidin-4-ylamino)acetic acid (compound 2; 1.77 g, 4.5 mmol) and benzyl ethylamine (0.67 mL, 4.5 mmol) in N,N-dimethylformamide (DMF) (25 mL). The reaction mixture was stirred at room temperature for 3 h and then evaporated in vacuo to remove DMF. The residue was extracted with ethyl acetate (AcOEt) and water, and the organic layer was dried over Na2SO4 and evaporated. The crude product was purified by silica gel column chromatography (n-hexane:AcOEt, 2:1) to yield ethyl-4-2-(N-benzyl,N-ethylamino)-2-oxoethylamino-2-phenylpyrimidine-5-carboxylate (compound 3) (0.99 g, 53.9%). 1H nuclear magnetic resonance (NMR) (CDCl3, δ) (ppm) data were as follows: 9.05 (1H, br), 8.96 (1H, d, J = 11 Hz), 8.39 (1H, d, J = 6 Hz), 8.30 (1H, d, J = 6 Hz), 7.26–7.47 (8H, m), 4.65 (2H, d, J = 20 Hz), 4.51 (2H, dd, J = 4, 20 Hz), 4.39–4.45 (2H, m), 3.54 (1H, dd, J = 7 Hz), 3.39 (1H, dd, J = 7 Hz), 1.43 (3H, t, J = 7 Hz), and 1.22 (3H, dt, J = 7, 27 Hz). Mass spectrometry (MS) (Fast Atom Bombardment [FAB]) data were as follows: m/z 419 (M+H).
A mixture of compound 3 and 5 N NaOH (5 mL) in EtOH (50 mL) was heated for 30 min and then kept at room temperature for 12 h. After the EtOH was removed, 1 N HCl was added slowly to neutralize the reaction mixture. The obtained precipitate was collected and washed with water to yield 4-(2-N-benzy,N-ethylamino)-2-oxoethylamino)-2-phenylpyrimidine-5-carboxylic acid (compound 4). MS (FAB) data were as follows: m/z 391 (M+H).
Diphenyl phosphorylazide (0.22 mL, 1 mmol) was added to a solution of compound 4 (0.39 g, 1 mmol) and triethylamine (Et3N) (0.14 mL, 1 mmol) in DMF (5 mL). The reaction mixture was heated at 100°C for 6 h and then evaporated to remove DMF. The residue was extracted with AcOEt, and the organic layer was washed with brine and dried over Na2SO4. After the solvent was removed, the crude product was recrystallized from methanol to yield compound 1 as a white precipitate (26 mg, 32.5%). 1H NMR (CDCl3, δ) (ppm) data were as follows: 9.78 (1H, br), 8.30–8.34 (2H, m), 8.09 (1H, d, J = 4 Hz), 7.37–7.51 (5H, m), 7.26–7.30 (3H, m), 4.86 (2H, d, J = 19 Hz), 4.69 (2H, d, J = 4 Hz), 3.44–3.55 (2H, m), and 1.27 (3H, dt, J = 7, 58 Hz). High-resolution MS (FAB) data were as follows: calculated for C22H22N5O2, 388.1774; found: 388.1805.
N-Benzyl-N-Ethyl-2-(7-Methyl-8-Oxo-2-Phenyl-7,8-Dihydro-9H-Purin-9-yl)Acetamide (AC-5216).
A mixture of compound 1 (72 mg, 0.18 mmol), CH3I (13 μL, 0.21 mmol), and NaH (60% in mineral oil, 9 mg, 0.22 mmol) in DMF (1 mL) was stirred at room temperature for 3 h. After DMF was removed, the residue was quenched with AcOEt and washed with water and a saturated NaCl solution. After the organic layer was dried over Na2SO4, the solvent was removed to yield a crude product, which was recrystallized from methanol to yield AC-5216 as a white precipitate (59 mg, 82%). The melting point was 165.5°C–166.5°C (163°C–164°C (30)). 1H NMR (CDCl3, δ) (ppm) data were as follows: 8.35 (2H, d, J = 5 Hz), 8.24 (1H, d, J = 12 Hz), 7.37–7.46 (6H, m), 7.26 (2H, m), 4.87 (2H, d, J = 19 Hz), 4.65 (2H, d, J = 12 Hz), 3.43–3.51 (5H, m), and 1.24 (3H, dt, J = 7, 61 Hz). High-resolution MS (FAB) data were as follows: calculated for C23H24N5O2, 402.1930; found, 402.1933.
Radiochemistry
11C-CH3I for labeling was synthesized from cyclotron-produced 11C-CO2 as described previously (31). Briefly, 11C-CO2 was bubbled into 0.04 M LiAlH4 in anhydrous tetrahydrofuran (300 μL). After the evaporation of tetrahydrofuran, the remaining complex was treated with 57% hydroiodic acid (300 μL). 11C-CH3I was transferred under helium gas flow with heating into a reaction vessel containing compound 1 (0.6 mg), NaH (3–5 μL, 0.5 g/20 mL of DMF), and anhydrous DMF (300 μL) cooled to −15°C to −20°C. After the radioactivity reached a plateau, the reaction vessel was warmed to 30°C and maintained for 3 min. CH3CN:H2O (6:4, 500 μL) was added to the mixture to stop the reaction, and the radioactive mixture was applied to a semipreparative HPLC system. HPLC purification was completed by use of a Capcell Pack column (10 mm [inner diameter] × 250 mm; Shiseido) with a mobile phase of CH3CN:H2O (6:4) at a flow rate of 6.0 mL/min. The retention time (tR) for 11C-AC-5216 was 7.5 min, whereas that for unreacted compound 1 was 5.6 min. The radioactive fraction corresponding to the desired product was collected in a sterile flask, evaporated to dryness in vacuo, redissolved in 7 mL of sterile normal saline, and passed through a 0.22-μm filter (Millipore) for analysis and animal experiments.
Radiochemical Purity and Specific Activity Determinations
Radiochemical purity was assayed by analytic HPLC (Capcell Pack C18; 4.6 mm [inner diameter] × 250 mm; UV absorbance at 254 nm; CH3CN:H2O, 6:4). The tR for 11C-AC-5216 was 5.8 min at 2.0 mL/min. The identity of this product was confirmed by coinjection with authentic nonradioactive AC-5216. The specific activity of 11C-AC-5216 was calculated by comparison of the assayed radioactivity with the mass associated with the carrier UV peak at 254 nm.
Biodistribution Study of Mice
Whole Body.
A saline solution of 11C-AC-5216 (average of 8 MBq/200 μL; specific activity, 135 GBq/μmol) was injected into ddY mice (30–40 g, 9 wk, male) through the tail vein. Five mice were sacrificed at each time point (1, 5, 15, 30, and 60 min after injection) by cervical dislocation. The whole brain and liver, lung, heart, kidney, adrenal gland, intestine, spleen, testicle, and blood samples were quickly removed. The radioactivity present in these tissues was measured with an Autogamma scintillation counter (Packard) and expressed as the percentage injected dose per gram of wet tissue (%ID/g). All radioactivity measurements were corrected for decay.
Brain.
A saline solution of 11C-AC-5216 (8 MBq/200 μL; specific activity, 120 GBq/μmol) was injected into ddY mice through the tail vein. Five mice were sacrificed at each time point (1, 5, 15, 30, and 60 min after injection) by cervical dislocation. From the brain, the cerebellum, olfactory bulb, striatum, hippocampus, thalamus, hypothalamus, and cerebral cortex were dissected and weighed. The radioactivity present in these tissues was measured.
Blocking Study.
AC-5216, PK11195, flumazenil, or Ro15-4513 at a dose of 1 mg/kg each was mixed with 11C-AC-5216 (8 MBq/200 μL; specific activity, 110 GBq/μmol), and the mixture was injected into ddY mice (n = 5). At 15 min after injection, these mice were sacrificed, and the whole brains were removed quickly. Brain tissue samples (cerebellum, olfactory bulb, striatum, and cerebral cortex) were dissected and treated as described earlier.
PET Study of Monkey
A PET scan was performed by use of a high-resolution SHR-7700 PET camera (Hamamatsu Photonics) designed for laboratory animals; this camera provides 31 transaxial slices 3.6 mm (center-to-center) apart and has a 33.1-cm field of view. MRI of the brain was performed by use of a Gyroscan S15/ACS II scanner (1.5 T; Philips) with a 3-dimensional T1-weighted axial sequence. A male rhesus monkey (Macaca mulatta) weighing about 8 kg was repeatedly anesthetized with ketamine (Ketalar, Sankyo Co.; 10 mg/kg/h, intramuscularly) every hour throughout the session. Transmission scans for attenuation correction were subsequently obtained for 1 h with a 74-MBq 68Ge68Ga source. A dynamic emission scan in the 3-dimensional acquisition mode was obtained for 90 min (2 min × 5 scans, 4 min × 10 scans, and 10 min × 4 scans). All emission scan images were reconstructed with a Colsher filter of 4 mm, and circular regions of interest with a 5-mm diameter were placed over the occipital cortex by use of image analysis software (21). A solution of 11C-AC-5216 (90–120 MBq) was injected into the monkey, and sequential tomographic scanning was performed on a transverse section of the brain for 90 min. In pretreatment experiments, nonradioactive AC-5216 (0.1 or 1 mg/kg) or PK11195 (0.1, 1, or 5 mg/kg) was injected at 5 min before 11C-AC-5216 injection. Regions of interest were placed over the occipital cortex, frontal cortex, thalamus, striatum, and cerebellum by use of PMOD image analysis software (PMOD Technologies, Ltd.) with reference to the MR image of the monkey brain. Each PET image of 11C-AC-5216 was overlaid on the MR image of the monkey brain, and the time–activity curve for 11C-AC-5216 in each brain region was calculated.
Metabolite Assay
Mouse Plasma and Brain.
After intravenous injection of 11C-AC-5216 (50 MBq/200 μL) into ddY mice (n = 3), these mice were sacrificed by cervical dislocation at 1, 5, 15, 30, and 60 min. Blood samples (0.5–1.0 mL) and whole brains were removed quickly. The blood samples were centrifuged at 15,000 rpm for 1 min at 4°C to separate plasma (250 μL), which was collected in a test tube containing CH3CN (500 μL) and a solution of nonradioactive AC-5216 (1 mg/5.0 mL of CH3CN, 10 μL). After the tube was vortexed for 15 s and centrifuged at 15,000 rpm for 2 min for deproteinization, the supernatant was collected. The efficiency of extraction of radioactivity into the CH3CN supernatant ranged from 70% to 92% of the total radioactivity in the plasma. The cerebellum and forebrain, including the olfactory bulb, were dissected from the mouse brain and homogenized together in an ice-cooled CH3CN:H2O (1:1, 1.0 mL) solution. The homogenate was centrifuged at 15,000 rpm for 2 min at 4°C, and the supernatant was collected. The recovery of radioactivity in the supernatant was greater than 60% of the total radioactivity in the brain homogenate.
A portion of the supernatant (100–500 μL) prepared from the plasma or brain homogenate was injected into the analytic HPLC system and analyzed under the same conditions as those described earlier, except that the mobile phase was CH3CN:H2O (1:1). The ratio of 11C-AC-5216 (tR = 10.6 min) to total radioactivity (corrected for decay) on the HPLC chromatogram was calculated as (peak area for 11C-AC-5216/total peak area) × 100 and reported as a percentage.
Monkey Plasma.
After intravenous injection of 11C-AC-5216 (250 MBq) into the monkey, arterial blood samples (1 mL) were collected at 2, 5, 15, 30, 60, and 90 min. All samples were centrifuged at 15,000 rpm for 1 min at 4°C to separate plasma (250 μL), which was collected in a test tube containing CH3CN (0.5 mL). The plasma was treated as described earlier.
RESULTS
Chemistry
AC-5216 and the desmethyl precursor (compound 1) for radiosynthesis were synthesized according to the reaction sequences shown in Figure 1. The key step in the synthetic process was the Curtius arrangement, which afforded compound 1 at a total yield of 18% starting from compound 2. The reaction of compound 1 with CH3I and with NaH as a base proceeded efficiently to produce AC-5216 as an authentic sample at a yield of 82%.
11C-AC-5216 was synthesized by N-11C-methylation of compound 1 with 11C-CH3I in the presence of NaH at 30°C for 3 min (Fig. 1). The radiochemical yield of 11C-AC-5216 was largely dependent on the amount of NaH used for this reaction. When more than 2 equivalents of NaH relative to compound 1 were used, a radioactive by-product peak (tR = 5.3 min) was observed in the HPLC purification chart in addition to the peak for the desired product 11C-AC-5216 (tR = 7.5 min). Under these conditions, 11C-AC-5216 was sometimes obtained only at a poor radiochemical yield (18–110 MBq starting from a total radioactivity of 2.7 GBq). Limiting the amount of NaH restrained the formation of the radioactive by-product. When less than 1 equivalent of NaH was used, 11C-AC-5216 was yielded as the main reaction product. Under the optimized reaction conditions, 11C-AC-5216 was successfully obtained with a radioactivity incorporation yield of 60% ± 26% (mean ± SD) (based on HPLC purification charts; n = 25). Radiosynthesis, semipreparative HPLC purification, and formulation were completed in an average synthesis time of 22 min (n = 25). At the end of synthesis, 11C-AC-5216 at 800–1,230 GBq was obtained as an injectable sterile normal saline solution after 15–20 min of proton bombardment (14.2 MeV on target) at a beam current of 15 μA. The final formulated solution was ≥98% radiochemically pure, as determined by analytic HPLC. No significant UV peak corresponding to compound 1 was observed in the final product solution. The specific activity of 11C-AC-5216 was 85–130 GBq/μmol, calculated at the end of synthesis. Moreover, the radiochemical purity of 11C-AC-5216 remained at greater than 95% after maintenance of this radioactive product at room temperature for 4 h, and it was stable for performing the animal evaluations.
Biodistribution Study of Mice
The initial in vivo evaluation of 11C-AC-5216 was performed in mice. Radioactivity was measured in 10 specific regions of the mice after injection of 11C-AC-5216. Table 1 shows the decay-corrected %ID/g values for all regions. An initial high concentration of radioactivity (>5 %ID/g) was found in the lungs, heart, kidneys, and adrenal glands. The highest concentration of 11C-AC-5216 radioactivity was observed in the lungs. On the other hand, a relatively high concentration of radioactivity was also found in the brain, the target tissue in this study.
Tissue Distribution in 5 Mice
The radioactivity distribution for 11C-AC-5216 in mouse brains is shown in Table 2. This ligand showed rapid penetration across the blood–brain barrier (BBB) into all brain regions at 1 min after injection. Uptake in the olfactory bulb and cerebellum was higher than 1.3 %ID/g at 5 min after injection. Radioactivity accumulated with time in these 2 regions, and the level peaked at 15 min and then declined until 60 min after injection. The highest radioactivity of 11C-AC-5216 (2.5 %ID/g at 15 min) was present in the olfactory bulb, and moderate radioactivity (1.5 %ID/g at 15 min) was also detected in the cerebellum; however, low uptake was found in other regions, such as the cerebral cortex and striatum.
Brain Regional Distribution in 5 Mice
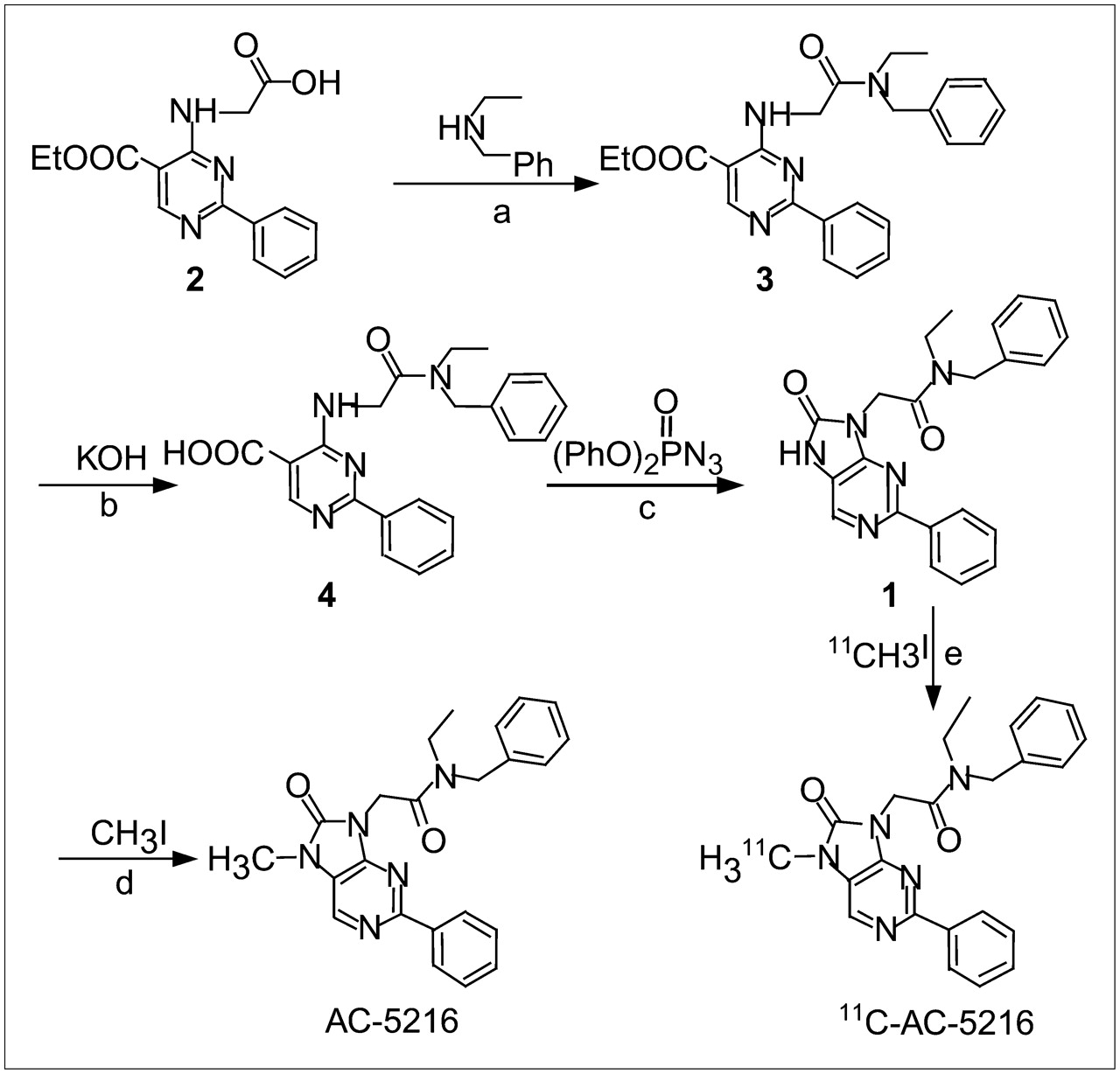
The in vivo selectivity and specificity of 11C-AC-5216 were tested by coinjecting nonradioactive AC-5216, PBR-selective PK11195, and CBR-selective flumazenil and Ro15-4513 at a dose of 1 mg/kg with 11C-AC-5216 (Fig. 2). AC-5216 produced a statistically significant reduction in radioactivity in the brain regions compared with the control group. The most significant reduced uptake was found in the olfactory bulb (18% of control) and in the cerebellum (27% of control). The striatum and cerebral cortex showed a moderate reduction (40%–50%) in the uptake of 11C-AC-5216. PBR-selective PK11195 (1 mg/kg) also produced a significant decrease in radioactivity in the olfactory bulb and cerebellum, to an extent similar to that obtained with the same amount of AC-5216. In contrast, CBR-selective flumazenil or Ro15-4513 coinjection did not have a clear inhibitory effect on the uptake of 11C-AC-5216 (Fig. 2). In some regions examined, flumazenil and Ro15-4513 produced an increase in total binding of less than 20%.
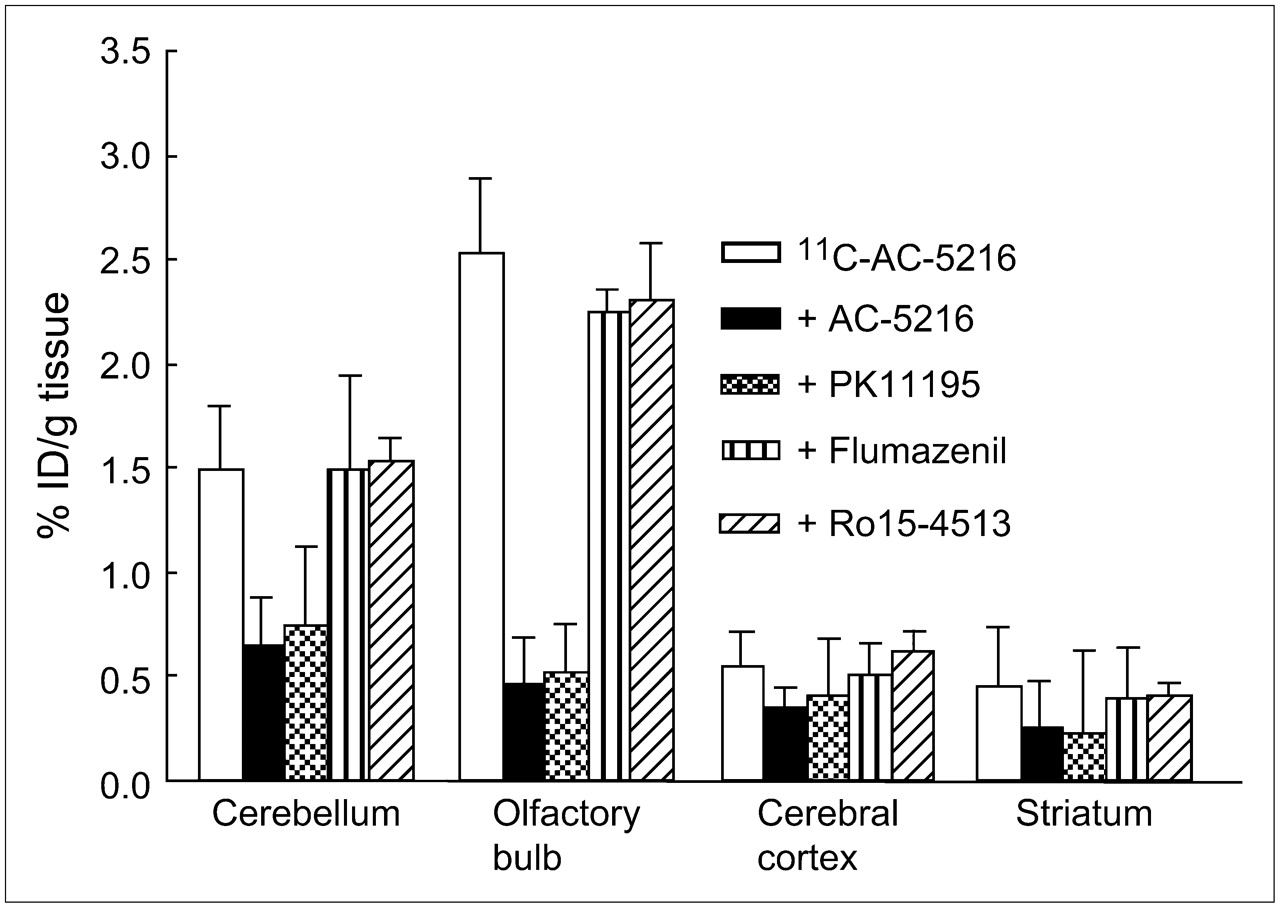
Effect of nonradioactive AC-5216 (1 mg/kg), PBR-selective PK11195 (1 mg/kg), and CBR-selective flumazenil (1 mg/kg) and Ro15-4513 (1 mg/kg) on 11C-AC-5216 radioactivity (mean ± SD, n = 5) in selected regions of mouse brains at 30 min after injection of 11C-AC-5216 (8 MBq).
PET Study of Monkey
The uptake of 11C-AC-5216 in the monkey brain was examined by PET. The time–activity curve and summation image of 11C-AC-5216 in the monkey brain are shown in Figures 3A and 3B, respectively. 11C-AC-5216 entered the brain and remained at almost the same level during the scan time of 90 min in all measured brain regions. Radioactivity in the occipital cortex was higher than that in other brain structures, such as the cerebellum, frontal cortex, striatum, and thalamus.

(A) Time–activity curves for 11C-AC-5216 in occipital cortex, frontal cortex, cerebellum, striatum, and thalamus of monkey brain. Uptake was expressed as percentage injected dose per volume (%ID/mL). (B) Mean image of 11C-AC-5216 in brain of rhesus monkey. PET image was generated by averaging for whole scan and was overlaid on MR image.
Figure 4 shows the effect of pretreatment with AC-5216 (0.1 and 1 mg/kg) and PK11195 (0.1, 1, and 5 mg/kg) on radioactivity in the occipital cortex. Each compound was administered 5 min before the ligand injection. As shown in Figure 4A, uptake in the occipital cortex was dose dependently decreased by AC-5216. PK11195 at 1 and 5 mg/kg also inhibited the uptake of 11C-AC-5216 in a dose-dependent manner. Of the 3 PK11195 doses, 0.1 mg/kg did not have a significant inhibitory effect. There was a marked increase in initial uptake with both AC-5216 and PK11195 pretreatment. A similar inhibitory pattern was observed in the frontal cortex and cerebellum (data not shown). Because pretreatment with these radioactive ligands increased the initial uptake of 11C-AC-5216, time–activity curves were normalized to the maximum initial uptake, as shown in Figure 4B. The maximum uptake of 11C-AC-5216 was inhibited to 30%–40% of the control uptake by AC-5216 at 1 mg/kg and PK11195 at 5 mg/kg.

(A) Effects of compounds with affinity for PBR on 11C-AC-5216 binding in occipital cortex of monkey brain. (B) Radioactivity in occipital cortex was normalized to initial maximum uptake of ligand, which was designated as 100%.
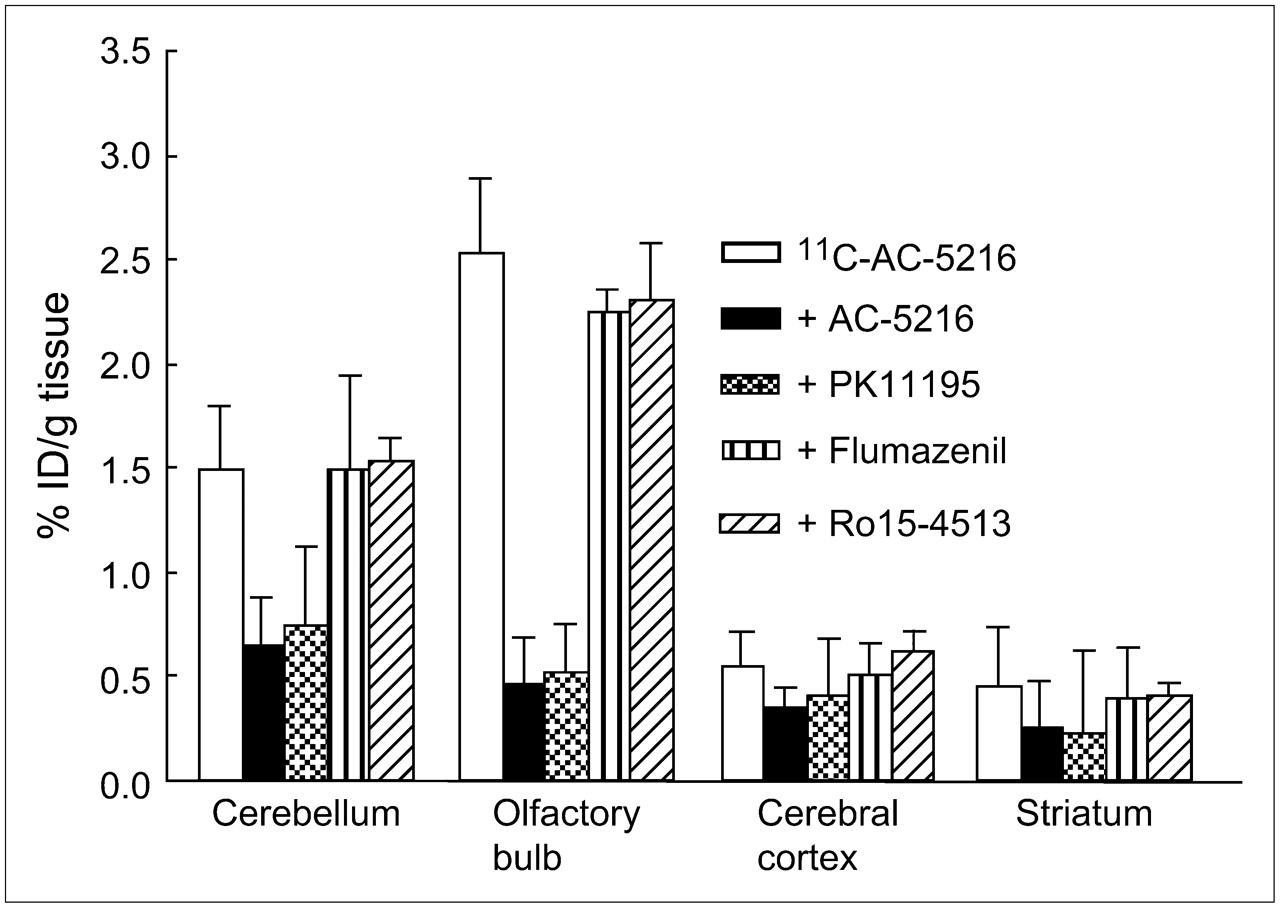
Figure 5 shows the percentages of unmetabolized 11C-AC-5216 in the plasma and brain homogenate of mice, as determined by HPLC. After injection into the mouse, the fraction corresponding to the unmetabolized ligand in the plasma decreased to 80% at 5 min and to 71% at 30 min. A major radioactive metabolite with a high polarity was observed on the HPLC chart. On the other hand, 11C-AC-5216 was detected in the brain homogenate as a minor (<10%) radioactive metabolite at 60 min after injection.

Percentages (mean ± SD, n = 3) of unchanged 11C-AC-5216 in mouse plasma and brain homogenate at several time points after injection of 11C-AC-5216 (37 MBq).
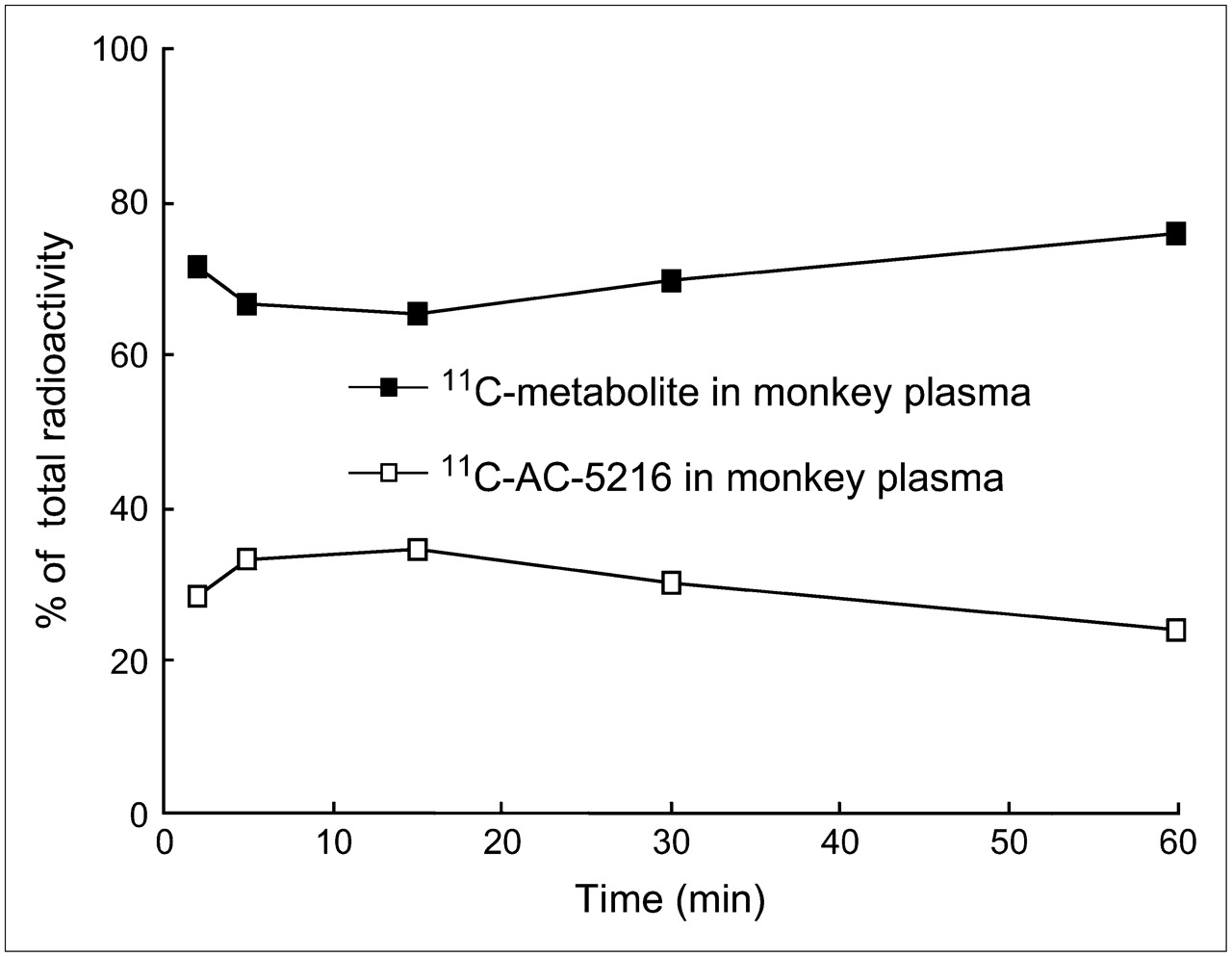
Metabolite analysis was also performed for monkey plasma (Fig. 6). After injection into the monkey, 11C-AC-5216 in the plasma rapidly decreased to 38% at 5 min and remained at a similar level until the end of this PET scan. The radioactive metabolite found in monkey plasma was the same component as that found in mouse plasma.

Percentage of unchanged 11C-AC-5216 in monkey plasma at several time points after injection of 11C-AC-5216 (250 MBq).
DISCUSSION
As a candidate for use as a PET ligand for PBR in the primate brain, AC-5216 may have several advantages over other ligands. A previous in vitro study demonstrated that AC-5216 had an excellent in vitro pharmacologic profile (28). It had a remarkable affinity for PBR, with significant selectivity for PBR over CBR and other receptors, monoamine transporters, or ion channels. PBR is known to be a heterotrimeric complex of 3 different subunits, and some PBR ligands have exhibited species differences in their binding affinities. For example, Ro5-4864 exhibited a much lower affinity for human PBR than for rodent PBR (3). In contrast to Ro5-4864, AC-5216 bound to rat glioma cells (IC50, 3.04 nM) and human glioma cells (2.73 nM) with similar affinities, and PK11195 also had a similar affinity for PBR from a variety of species (28). Therefore, the binding site for AC-5216 in the PBR domain might be closer to that of PK11195 than to those of other PBR ligands, such as Ro5-4864 (28). Further, compared with PK11195, AC-5216 has more suitable lipophilicity, which is a prerequisite for a favorable PET ligand, guaranteeing high uptake and low nonspecific binding in the brain. Although 11C-DAA1106 (21), 18F-FEDAA1106 (22), and 11C-PBR28 (27) have been successfully used for PBR imaging in the brain, the binding site for DAA1106 might contain an extra component that does not interact efficiently with PK11195 (23,24). Therefore, AC-5216 might be closer to PK11195 as a pure PBR ligand than DAA1106 and its analogs. More recently, 11C-DPA-27, with a structure different from that of 11C-DAA1106 analogs, was developed to visualize PBR in a rat model of neuroinflammation (16). However, to our knowledge, no data about its distribution in the primate brain have been published to date.
As expected, the in vivo distribution pattern for 11C-AC-5216 was in agreement with previous findings on PBR distribution in the peripheral systems of rodents (3,5,20). The highest radioactivity of 11C-AC-5216 was found in the lungs (Table 1), and this radioactivity was higher than that of 11C-PK11195 (32). Uptake in the lungs and heart may be related to mitochondrial contents containing PBR. High radioactivity of 11C-AC-5216 was also found in the brain, the target tissue in this study. The radioactivity was about 1.5- to 2-fold higher than that of 11C-PK11195 in the mouse brain at the corresponding time points (32). These results revealed that 11C-AC-5216 could pass through the BBB and enter the brain. 11C-AC-5216 displayed relatively high radioactivity in the olfactory bulb and cerebellum of the mouse brain (Table 2); these 2 areas of the rodent brain are PBR rich (3–5). The radioactivity distribution pattern was consistent with the binding sites for 3H- or 11C-DAA1106 (20,23) and 3H- or 11C-PK11195 (32) in the rodent brain. These results indicated that 11C-AC-5216 bound in vivo to PBR in the mouse brain.
The distribution pattern for 11C-AC-5216 in the monkey brain (Fig. 3) was heterogeneous, and higher radioactivity was found in the occipital cortex, a finding that matched the reported distribution of PBR in a previous in vitro study of the postmortem human brain (12). The radioactivity of this ligand in the occipital cortex was 3–4 times higher than that of 11C-PK11195, which was measured in the same monkey under the same experimental conditions by our group (21). This distribution pattern was also observed in PET studies of the monkey brain with 11C-DAA1106 (21) and 18F-FEDAA1106 (22). Radioactivity peaked at about 5 min after injection, and the binding seemed to be irreversible because of slow washout from the brain.
Successful blocking of the accumulation of 11C-AC-5216 radioactivity in mice (Fig. 2) and a monkey (Fig. 4) with PBR-selective PK11195 demonstrated that the uptake in receptor-rich regions was PBR mediated. In addition, the obvious blockade of ligand uptake by AC-5216 suggested that the binding of 11C-AC-5216 was highly in vivo specific in mouse and monkey brains.
The in vivo PBR selectivity of 11C-AC-5216 was investigated with CBR-selective flumazenil and Ro15-4513 (Fig. 2). In mouse brains, coinjection of these compounds did not significantly reduce the accumulation of radioactivity compared with that seen in the control experiment. The reason for the increased uptake of 11C-AC-5216 in several brain regions in the presence of these compounds is unclear. However, such effects of central nervous system drugs are not uncommon and have been attributed to additional ligand availability from the periphery or blood flow changes.
A radioactive metabolite of a radioligand in plasma that enters the brain can confound PET studies of neuroreceptors, whether or not the metabolite binds to the target receptor. Metabolite analysis of 11C-AC-5216 was thus performed for plasma and brain samples by analytic HPLC. HPLC analyses of mouse (Fig. 5) and monkey (Fig. 6) plasma demonstrated that 11C-AC-5216 was metabolized to form the same radioactive metabolite in both species. This metabolite was much more polar than 11C-AC-5216, as estimated from the retention order on a reverse-phase HPLC column. In contrast to the findings in mouse plasma, only 11C-AC-5216 was detected as the main radioactive component in the mouse brain homogenate. Thus, although 11C-AC-5216 was extensively metabolized in plasma, its radiolabeled metabolite appeared not to cross the BBB to enter the brain. This result demonstrated that most specific binding to PBR in the mouse and monkey brains was attributable to 11C-AC-5216 itself and was not influenced by its radiolabeled metabolite.
In the PET study of a monkey, the uptake of 11C-AC-5216 was markedly decreased by pretreatment with AC-5216 and PK11195, despite the initial increases in uptake (Fig. 4). These initial increases in uptake obtained with AC-5216 and PK11195 could be explained by the preclusion of 11C-AC-5216 from peripheral organs, such as the lungs, because the most significant accumulation of radioactivity occurred in the mouse lungs. The uptake in the occipital cortex was reduced by AC-5216 at 1.0 mg/kg to about 30% of the control uptake, suggesting specific binding in vivo in the monkey brain. As shown in Figure 4, the percentage of reduction in 11C-AC-5216 uptake obtained with AC-5216 at 1.0 mg/kg was similar to that obtained with PK11195 at 5.0 mg/kg. This result confirmed that the binding site for AC-5216 in the PBR domain might be close to that for PK11195, consistent with the in vitro pharmacologic result (28). On the other hand, although the uptake of 11C-DAA1106 and 18F-FEDAA1106 in the monkey brain could also be inhibited by PK11195, the percentage of reduction obtained with PK11195 was lower than that obtained with DAA1106, suggesting that the binding site for DAA1106 in the PBR domain was not perfectly consistent with that for PK11195. Therefore, 11C-AC-5216 may be more favorable than 11C-DAA1106 for elucidating PBR in the brain precisely.
In the same PET study of a monkey, the time–activity curve for 11C-AC-5216 was similar to that for 11C-DAA1106 (21), and both ligands showed little dissociation over 90 min. This property is a significant disadvantage for a useful PET ligand. Moreover, 11C-AC-5216 showed strong blood flow dependence in a rodent tumor model (29), and blood flow might have a negative effect on the estimation of PBR in the brain. However, because 11C-DAA1106 showed much more dissociation in the human brain (25) than in the monkey brain, we expected that 11C-AC-5216 would also show much more dissociation in the human brain. At our facility, a method of quantitative analysis for estimating the binding potential of PBR in the human brain with 11C-DAA1106 (25) and 18F-FEDAA1106 (26) has been devised. With this analytic method, the binding potential of PBR in the human brain could be elucidated with 11C-AC-5216.
CONCLUSION
11C-AC-5216, a potent and selective PET ligand for PBR in the brain, was successfully labeled with 11C by alkylation of the corresponding desmethyl precursor (compound 1) with 11C-CH3I, with a high yield of incorporation of radioactivity. The in vivo distribution demonstrated that 11C-AC-5216 bound to PBR in the brain with high specificity and selectivity. The specific binding of 11C-AC-5216 in vivo could be saturated by pretreatment with nonradioactive AC-5216. PK11195 effectively blocked the specific binding of 11C-AC-5216 in the brain. 11C-AC-5216 underwent metabolism in mouse and monkey plasma to yield a hydrophilic metabolite, but no radioactive metabolite was found in the mouse brain. Therefore, 11C-AC-5216 is a promising PET ligand with good imaging properties for the quantification of PBR in the human brain.
Acknowledgments
The authors are grateful to Makoto Takei (Tokyo Nuclear Service Co., Ltd.) for radiochemistry assistance. We also thank the staff of the National Institute of Radiological Sciences for support in the cyclotron operation, radioisotope production, and animal experiments.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication May 9, 2007.
- Accepted for publication July 31, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Translocator Protein Ligand Protects against Neurodegeneration in the MPTP Mouse Model of Parkinsonism
- Current status and future perspectives: TSPO in steroid neuroendocrinology
- Detection of Microglial Activation in an Acute Model of Neuroinflammation Using PET and Radiotracers 11C-(R)-PK11195 and 18F-GE-180
- In Vivo Positron Emission Tomographic Imaging of Glial Responses to Amyloid-{beta} and Tau Pathologies in Mouse Models of Alzheimer's Disease and Related Disorders
- 18F-FEAC and 18F-FEDAC: PET of the Monkey Brain and Imaging of Translocator Protein (18 kDa) in the Infarcted Rat Brain
- Quantification of Translocator Protein (18 kDa) in the Human Brain with PET and a Novel Radioligand, 18F-PBR06
- Quantitative Analysis of Peripheral Benzodiazepine Receptor in the Human Brain Using PET with 11C-AC-5216