


Abstract
Transmembrane transfectors (carriers) are increasingly being viewed as helpful or even necessary to improve cellular delivery in connection with antisense tumor targeting and other applications requiring cell membrane transport of DNAs, RNAs, and other oligomers. We are investigating streptavidin as a convenient linker for biotinylated carriers and oligomers because it requires only simple mixing for preparation. The goal of this study was to evaluate antisense DNA–streptavidin–carrier nanoparticles for accumulation in cell culture and in xenograft-bearing mice. Methods: The 3 carriers were cholesterol, a 10-mer Tat peptide, and a 10-mer polyarginine peptide. A 20-mer DNA targeting the mdr1 messenger RNA coding for Pgp expression was used as the phosphodiester (PO) DNA as well as the phosphorothioate (PS) DNA. In all cases, the 99mTc radiolabel was on the DNA. The 8 nanoparticles were first tested in mdr1++ KB-G2 and TCO-1 cells and in mdr1+/− KB-31 cells in culture for evidence of improved accumulation and antisense targeting. Thereafter, the PS DNA–streptavidin–Tat, PO DNA–streptavidin–Tat, and PS DNA–streptavidin–cholesterol nanoparticles were administered intravenously to KB-G2 xenograft–bearing mice, and tissue distributions were measured. Results: In culture, the PO nanoparticles showed increased accumulation compared with the corresponding nanoparticles without the carrier in all 3 cell types; in contrast, with the PS nanoparticles, any similar carrier-mediated increase may have been obscured by the much higher protein-binding affinity of PS DNA. As evidence of antisense targeting, the Tat and cholesterol PS nanoparticles showed statistically significant accumulation at 23 h in cells in the descending order TCO-1, KB-G2, and KB-31, although there were no significant differences among the PO nanoparticles. In xenograft-bearing mice, the tissue accumulation of both forms of the PS nanoparticles greatly exceeded that of the PO nanoparticles and, including in the tumor, were similar to that obtained previously for naked PS DNA. Conclusion: The presence of the streptavidin linker had no obvious detrimental effect on the functions of the carriers and antisense DNAs. The higher protein-binding affinity of the PS nanoparticles than the PO nanoparticles was still apparent both in vitro and in vivo, the pharmacokinetics of the PS nanoparticles were similar to that of naked PS DNA, and the carriers improved cellular accumulation, at least for the PO nanoparticles. These observations, taken together with the higher accumulation of both forms of the antisense PS nanoparticles in mdr1++ KB-G2 and TCO-1 cells than in mdr1+/− KB-31 cells, suggest that further effort is justified to confirm that the antisense properties of the DNAs were not compromised by the presence of streptavidin.
The results of recent investigations suggest that radiolabeled DNAs and other oligomers for antisense targeting of tumor cells require transmembrane transfectors (carriers) for improved cell membrane transport. Fortunately, a large number of potential carriers have been identified, primarily for use in cell culture, but with increasing reports of successful in vivo use as well (1). However, at present, most of the potentially useful carriers can be attached to oligomers only by covalent conjugation, usually requiring involved organic synthesis, a process that can be technically challenging. Recent reports have described the use of streptavidin to alter the pharmacokinetics of the Tat peptide (2) and an antisense peptide nucleic acid oligomer (3). We considered that streptavidin could be used in this manner as a linker for radiolabeled antisense oligomer–carrier nanoparticles. In addition to a greatly simplified preparation stabilized by the high binding affinity of biotin for streptavidin, the advantages include the possibility of attaching up to 4 carriers or oligomers in any combination because streptavidin possesses 4 biotin-binding sites, because many potential carriers with biotin groups attached are already available commercially, and because other carriers with primary amines attached may be readily biotinylated. However, streptavidin may interfere with the transfection properties of the carrier, may unfavorably alter the pharmacokinetics of the oligomer within the nanoparticle, and may restrict the messenger RNA (mRNA)-binding properties of the antisense oligomer. The goal of this study was to evaluate DNA–streptavidin–carrier nanoparticles, first in culture, primarily to establish whether the carriers were still capable of increasing cellular accumulation, and then in xenograft-bearing mice, primarily to establish whether the pharmacokinetic properties of the DNA in the form of nanoparticles remained favorable for tumor targeting. As a secondary goal, the influence of streptavidin on the antisense properties of the DNA was also examined.
The 3 carriers used in this investigation have each been reported to increase cell membrane transport in culture. The Tat and polyarginine peptides are short cationic peptides with recognized effectiveness as carriers (4). The Tat peptide is believed to be capable of transfecting possibly all cell types by an as-yet-unknown mechanism (5) and has been used as a carrier for molecules such as proteins, peptides, and MRI contrast agents (6,7) as well as oligonucleotides (8). A polyarginine peptide with properties similar to those of Tat was recently reported to be an effective carrier (9–11). Finally, charge-neutral cholesterol was selected as a third carrier both because of recent reports of effectiveness (12) and to avoid anticipated difficulties (that were not realized) in the preparation of nanoparticles with anionic DNAs and cationic carriers.
We have generally used the phosphorothioate (PS) form of DNA in antisense studies. Although this DNA has the advantage over the native phosphodiester (PO) form of increased stability against exo- and endonuclease degradation, PS DNA is also characterized by an affinity for serum and tissue proteins not shared by PO DNA (13). A high accumulation of PS DNA in both experimental and control cells has been reported (14); this accumulation may be attributable, at least in part, to the nonspecific protein-binding affinities of PS DNA. Although potentially an advantage in cell culture studies, this increased affinity may also be responsible for high background radioactivity levels of PS DNA used in vivo (1). Therefore, PO DNA blocked with biotin and streptavidin on the 3′ end and a mercaptoacetyltriglycine (MAG3) group on the 5′ end was included in this investigation as a potential alternative to PS DNA to reduce background radioactivity levels. In preparation for this investigation, an 18-mer PO DNA blocked with biotin and streptavidin on the 3′ end and a MAG3 group on the 5′ end was radiolabeled and incubated at a concentration of 20 nM in Dulbecco minimum essential medium containing 1% fetal bovine serum. Size-exclusion high-performance liquid chromatography (HPLC) analysis showed that radiolabeled low-molecular-weight degradation products were present after about 6 h of incubation; however, 87% of the DNA was still intact after 18 h of incubation.
The mdr1 mRNA was selected because of our continuing interest in this target (14–16) and because of the availability of 3 Pgp-expressing cell lines, KB-31, KB-G2, and TCO-1. KB-G2 was transfected from its parent, human oral epidermoid carcinoma cell line KB-31, and has been reported to overexpress the target mdr1 mRNA (17). TCO-1, an anaplastic thyroid carcinoma cell line established from chemotherapy-resistant cancer, was also shown by reverse transcription–polymerase chain reaction to express mdr1 mRNA (18). KB-31 was used as a limited expression control cell line (19) but with caution, because multidrug resistance may be easily and inadvertently acquired. Because its mdr1 expression may be equivocal, this cell line is described here as mdr1+/− to emphasize this fact. The base sequence of the antisense DNA was selected for this mRNA target and had been previously shown to provide inhibition of Pgp in MDM 3T3 cells when incubated at a 100 nM concentration (20). Indirect evidence (14,15) and direct evidence obtained by in situ transcription (21) of antisense targeting of this DNA to its mdr1 mRNA have been reported from these laboratories.
Because establishing the antisense function of the streptavidin nanoparticles was a secondary goal, no sense or scrambled control DNA was used in this investigation. However, accumulation specific to antisense targeting may be evident in the measurement of the accumulation of the anti-mdr1 antisense DNA in the KB-31 low-level-expression cell line compared with both the KB-G2 and the TCO-1 high-level-expression cell lines (14,15,18).
MATERIALS AND METHODS
All DNAs were obtained from Integrated DNA Technologies, Inc., with biotin on the 3′ end and a primary amine on the 5′ end attached via a 6-carbon alkyl linker and were HPLC purified. The base sequence was 5′-CCA-TCC-CGA-CCT-CGC-GCT-CC (15). S-Acetyl N-hydroxysuccinimide (NHS)–MAG3 was synthesized in-house (22), and its structure was confirmed by elemental analysis, proton nuclear magnetic resonance analysis, and mass spectroscopy. The Tat and polyarginine peptides were both 10-mer peptides purchased from 21st Century Biochemicals as the native l-isomer with biotin attached to the amine end also via a 6-carbon alkyl linker and were HPLC purified. The Tat amino acid sequence was biotin-glycine-arginine-lysine-lysine-arginine-arginine-glutamine-arginine-arginine-arginine. Cholesteryl choloroformate was obtained from Aldrich. Biotinyl-3,6-dioxaoctanediamine was purchased from Pierce. Biotinylated cholesterol was synthesized by reacting biotinyl-3,6-dioxaoctanediamine with cholesteryl choloroformate. The cell lines KB-31, KB-G2, and TCO-1 were gifts from Dr. Isamu Sugawara, Research Institute of Tuberculosis, Tokyo, Japan. Standard chemicals were obtained from various suppliers and used without purification. 99mTc-Pertechnetate was eluted from a 99Mo-99mTc generator (Daiichi Radioisotopes).
Biotinylated Cholesterol
Although both the Tat and the polyarginine peptides were biotinylated by the manufacturer, biotinylated cholesterol was prepared in-house for this investigation. In brief, 12 mg of cholesteryl choloroformate (26.7 μmol) in 60 μL of anhydrous chloroform was added to a 1.5 mL centrifuge tube containing 12 mg (32 μmol) of biotinyl-3,6-dioxaoctanediamine in 200 μL of anhydrous 1-methyl-2-pyrrolidinone (NMP). The mixture was vortexed to a clear solution. To the solution was added 4 μL of diisopropylethylamine, and the solution was agitated and placed at room temperature for 16 h. The raw product was precipitated as a white powder by the addition of 1 mL of ethyl ether. The white powder was dissolved in 200 μL of NMP and precipitated again by the addition of ethyl ether. The product was solubilized and precipitated 2 more times. After vacuum drying, the white powder was dissolved in 400 μL of chloroform and washed with 400 μL of 0.1 NH4OAc 4 times. The chloroform solution was collected, the solvent was removed by evaporation over nitrogen, and the product was lyophilized under vacuum to remove any remaining NH4OAc. The product was analyzed on a Thermo LCQ Classic quadrupole ion-trap mass spectrometer (Thermo Electron Corp.), with data being acquired in the positive-ion mode (Proteomics and Mass Spectrometry Facility, University of Massachusetts Medical School). The results compared favorably with the expected mass of 786 Da (787.1: MW+H+; 809.4: MW+Na+, electron ionization).
DNA Conjugation and Radiolabeling
All DNAs were conjugated with S-acetyl NHS–MAG3 via the 5′-derivatized amine as previously described (23). A solution of 1 mg of DNA in 200 μL of 0.2 M (pH 8.0) N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) buffer was added to a vial containing 1.0–1.5 mg of S-acetyl NHS–MAG3. The mixture was vortexed immediately and incubated for 1 h at room temperature. Thereafter, to the solution was added, in order, 50 μL of 2 M NH4OAc and 120 μL of freshly prepared SnCl2·2H2O:tartrate solution (20 mg/mL; disodium tartrate [100 mg/mL] in 0.5 M NH4HCO3, 0.25 M NH4OAc, and 0.18 M NH4OH; pH 9.2) with agitation. The mixture was heated at 100°C for 25 min, allowed to cool, and then purified on a 1- by 20-cm P4 column (BioRad) with 0.25 M NH4OAc as the eluant. The peak fractions were pooled, and the DNA concentration was quantitated by measuring the UV absorbance at 260 nm. The purified samples were stored at refrigerator temperatures. All solutions were passed through a 0.22 μm filter to ensure sterility.
MAG3-conjugated biotinylated PO DNA (molecular weight [MW] = 7,004) and PS DNA (MW = 6,873) were radiolabeled with 99mTc in the usual manner. Radiolabeling of each DNA was achieved by introducing about 18.5–37 MBq (20 μL) of 99mTc-pertechnetate generator eluant into a solution consisting of 15 μL (about 10 μg) of MAG3-conjugated oligomer in 0.25 M NH4OAc, 45 μL of 0.25 M NH4OAc, 15 μL of disodium tartrate·2H2O (50 μg/μl) in a pH 9.2 buffer, and 5 μL of fresh SnCl2·2H2O (4 μg/μl) in 10 mM HCl with ascorbate added to 1 mg/mL. The final pH was about 7.8. After the mixture was vortexed and then heated for 20 min in boiling water, labeling was confirmed by chromatography on a C18 SepPak column (Waters), in which the first elution with phosphate-buffered saline (PBS) removed radiolabeled pertechnetate and tartrate, the second elution with 40% acetonitrile removed radiolabeled DNA, and radiolabeled colloids were retained on the SepPak column. The radiochemical purity was consistently 90% or better.
Preparation of Nanoparticles
The streptavidin–Tat nanoparticles were prepared by dissolving streptavidin (MW = 60,000) and biotinylated Tat (MW = 1,736) in 0.5 M NaCl and 0.2 M NH4OAc at 10.0 μg/μl (streptavidin) and 0.5 μg/μl (Tat). To 20 μL of the streptavidin solution in a 0.5-mL nonstick centrifuge tube was added, very slowly, 11.6 μL of the Tat solution, with continuous vigorous agitation (molar ratio, 1:1). Using the Tat peptide radiolabeled with 99mTc via MAG3 and added at tracer levels during nanoparticle preparation, the streptavidin–Tat nanoparticles were analyzed by size-exclusion HPLC with an eluant of 20% acetonitrile in 1 M NaCl and 0.1 M NH4OAc at a flow rate of 0.6 mL/min. The appearance in the UV trace at 280 nm of a single peak corresponding to a single peak in the radioactivity trace was evidence of complete complex formation. Thereafter the biotinylated DNA was added to the streptavidin–Tat nanoparticles in the same fashion. Evaluation of the final product was accomplished by adding, at tracer levels, complementary DNA (cDNA) radiolabeled with 99mTc followed by size-exclusion HPLC analysis. The appearance in the UV trace at 280 nm of a single peak corresponding to a single peak in the radiochromatogram was evidence of successful nanoparticle preparation, since this assay is capable of resolving 2:1 and higher-order nanoparticles (Fig. 1). Radiolabeled polyarginine and cholesterol nanoparticles were both prepared and tested in the same fashion, with identical results. Radioactivity recoveries were measured in all radioactive HPLC analyses and were, in all cases, 90% or better. The specific radioactivity of each type of nanoparticles was between 29.6 and 37 MBq/mg.
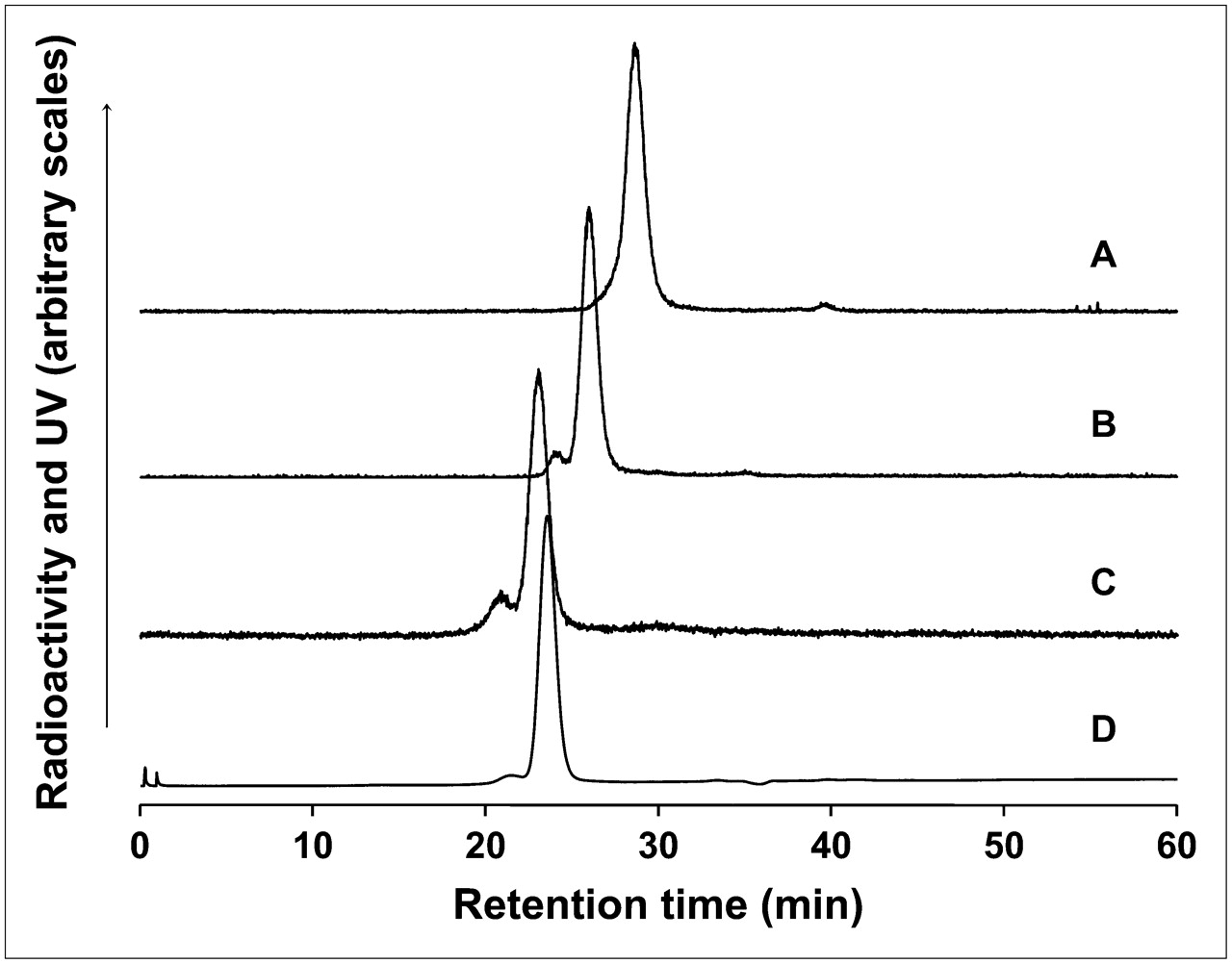
Size-exclusion HPLC radiochromatograms of free 99mTc-labeled PO-cDNA before (trace A) and after (trace B) addition to naked PS DNA (trace B) and after addition to PS DNA as the DNA–streptavidin–Tat nanoparticle (trace C). The retention time of the nanoparticle is confirmed by UV absorbance at 280 nm (trace D). The slight shoulder in the radioactivity profile of the DNA–streptavidin–Tat nanoparticle at higher MW is evidence that the contribution of higher-order products was no more than about 10%.
Cellular Accumulation Studies
All cells were growing in monolayers and were expected to be in the logarithmic phase at the time of study. Wells of 24-well plates were seeded with 0.3 × 105–0.4 × 105 cells per well. In triplicate, 0.5 mL of 99mTc-labeled oligomers, either as nanoparticles with a carrier or, as a control, nanoparticles without a carrier (i.e., DNA–streptavidin), was added to each well, and the cells were incubated with OptiMEM (serum-free) medium (Invitrogen Japan K.K. Tokyo) at 37°C in humidified 5% carbon dioxide. The DNA concentration was, at all times 50 nM and therefore was well below the levels of saturation of mdr1 mRNA in these cells (14). A 100 μL portion of each supernatant was removed from each well, and radioactivity was counted. The contents of each well were then aspirated, and the cells were rinsed twice with cold PBS. The cells were lysed, and aliquots of the cell lysate were collected. The cell-associated radioactivity in the lysis fraction was measured against a standard of the added radioactivity. After the radioactivity was counted, the protein concentration in the cell suspension was determined as described previously (14). No evidence of cellular toxicity by growth delay was observed in any cells studied in this investigation.
Animal Studies
All animal studies were performed with the approval of the Keio University School of Medicine Laboratory Animal Care and Use Committee. Nude mice (6 wk-old male BALB/c nu/nu; Sankyo Laboratory Inc.) were each injected subcutaneously in the left thigh with a 0.1 mL suspension containing 107 KB-G2 tumor cells. Animals were used 12 d later, when the tumors were about 1 cm in any dimension (weight, 0.8–1.2 g). In groups of 3, animals received, via a tail vein, 1 μg (3.0–3.7 MBq) of each type of DNA nanoparticle for each time point. Animals were sacrificed under halothane anesthesia, blood and other organs were removed and weighed, and radioactivity was counted in a NaI(Tl) well counter along with a standard of the injectate.
RESULTS
Preparation of Nanoparticles
Figure 1 shows size-exclusion HPLC evidence for the successful preparation of the PS DNA–streptavidin–Tat nanoparticle. The radiochromatogram of free 99mTc-labeled MAG3–PO cDNA showed only a single peak as evidence of successful radiolabeling (Fig. 1, trace A), and that peak shifted appropriately to a higher MW with the addition of the naked PS DNA (Fig. 1, trace B). A shift to an even higher MW occurred, as expected, when the radiolabeled cDNA was added to the PS DNA as the DNA–streptavidin–Tat nanoparticle (Fig. 1, trace C). That this peak may be attributed to the nanoparticle is evident in the UV trace at 280 nm (trace D) that shows that the retention time of streptavidin protein is approximately that of the radioactivity peak. Because only trace levels of radiolabeled cDNA was added in this analysis, most of the nanoparticle was unlabeled and would have a slightly lower MW than the labeled nanoparticle and therefore a slightly longer retention time. The slight shoulder in the radioactivity profile at a higher MW was at the expected retention time for 2:1 nanoparticles (i.e., DNA–streptavidin–2Tat or 2DNA–streptavidin–Tat), indicating that if higher-order nanoparticles were present, then they contributed no more than about 10% to the final product. The same results were obtained when the radiolabeled PO cDNA was used to evaluate the PO DNA–streptavidin–Tat nanoparticle.
Cellular Accumulation Studies
The histograms in Figure 2 (PO DNA) and Figure 3 (PS DNA) show the average percentage accumulation (n = 3) for each type of nanoparticle in each of the 3 cell types at 3, 7, and 23 h of incubation. For comparison, the figures also show the results for the corresponding PO or PS DNA carrier-free nanoparticles. As shown in Figure 2, at all 3 time points and for all 3 types of nanoparticles, there was an obvious increase in the accumulation of the PO DNA incubated as nanoparticles with a carrier as opposed to without a carrier. However, the results shown for the PS DNA in Figure 3 are very different. The accumulation of the PS DNA in all of the nanoparticle types was more than an order of magnitude higher than that of the PO DNA. An analysis of significance (Student t test, P < 0.05) identified no significant differences among the 3 cell types for the PO DNA nanoparticles at any time. However, at 23 h, the accumulation of the PS DNA incubated as nanoparticles with Tat and cholesterol or without a carrier was significantly different in cells in the descending order TCO-1, KB-G2, and KB-31. This analysis therefore provides evidence of cellular accumulation through an antisense mechanism for the PS DNA–streptavidin–Tat and PS DNA–streptavidin–cholesterol nanoparticles as well as the carrier-free nanoparticles.

Histogram showing average percent accumulation at 3 h (□), 7 h (▪), and 23 h ( ) of 99mTc-labeled PO DNA incubated as Tat, polyarginine (PolyA), and cholesterol (CHO) nanoparticles or carrier-free nanoparticle in KB-31, KB-G2, and TCO-1 cells (n = 3; error bars indicate 1 SD).
) of 99mTc-labeled PO DNA incubated as Tat, polyarginine (PolyA), and cholesterol (CHO) nanoparticles or carrier-free nanoparticle in KB-31, KB-G2, and TCO-1 cells (n = 3; error bars indicate 1 SD).

Histogram showing average percent accumulation at 3 h (□), 7 h (▪), and 23 h ( ) of 99mTc-labeled PS DNA incubated as Tat, polyarginine (PolyA), and cholesterol (CHO) nanoparticles or carrier-free nanoparticle in KB-31, KB-G2, and TCO-1 cells (n = 3; error bars indicate 1 SD). Note that scale differs from that in Figure 2.
) of 99mTc-labeled PS DNA incubated as Tat, polyarginine (PolyA), and cholesterol (CHO) nanoparticles or carrier-free nanoparticle in KB-31, KB-G2, and TCO-1 cells (n = 3; error bars indicate 1 SD). Note that scale differs from that in Figure 2.
Animal Studies
On the basis of the in vitro results shown in Figures 2 and 3, the 3 types of nanoparticles selected for study in animals were the PS DNA–streptavidin–cholesterol, PS DNA–streptavidin–Tat, and PO DNA–streptavidin–Tat nanoparticles. In groups of 3, nude mice bearing KB-G2 tumors growing subcutaneously in one thigh received one of the types of nanoparticles at 1 μg per animal via a tail vein and were sacrificed at 6 or 18 h. The results of these studies are shown in Table 1. As in the cell culture studies, the accumulation of the PS nanoparticles exceeded that of the PO nanoparticle because of the higher protein-binding affinity of the PS DNA (13). The presence of streptavidin did not interfere with this property.
Biodistribution in KB-G2 Xenograft–Bearing Mice of 99mTc-Labeled PS DNA–Streptavidin–Cholesterol, PS DNA–Streptavidin–Tat, and PO DNA–Streptavidin–Tat Nanoparticles with Sacrifice at 6 Hours and 18 Hours (n = 3)
DISCUSSION
In general, all in vivo applications of radiolabeled DNAs and their analogs (collectively, oligomers) are currently characterized to various degrees by the same limitations: poor pharmacokinetic delivery to the target after intravenous administration and, for applications with intracellular targets, poor transport through cell membranes. Attempts to increase delivery are now focusing on transmembrane transfectors (carriers), which are intended to improve cellular accumulation by chaperoning the oligomers through cellular membranes.
Fortunately, the number of candidate carriers available to transport oligomers into tumor cells is now exceedingly large. Among these, the most efficient are certainly the viral carriers (24). However, at present, no practical method for packaging within these vectors an antisense DNA radiolabeled with a relatively short-lived radionuclide, such as 99mTc, and in the concentrations required, has been reported. Among the nonviral carriers, cationic carriers such as cationic peptides (25) or cationic liposomes (26) have an advantage when used with anionic oligomers such as DNA because electrostatic attachment requires only simple mixing. However, when carriers are to be used with uncharged oligomers, such as peptide nucleic acids and phosphorodiamidate morpholino oligomers (MORFs), covalent conjugation is usually necessary for attachment, and the synthesis of these nanoparticles can prove difficult. Even the solid-phase synthesis of peptide nucleic acid–peptide carrier nanoparticle is not without its own difficulties and, in our experience has led to products of questionable quality.
Accordingly, in the present study we examined the use of streptavidin as a convenient linker for biotinylated carriers and radiolabeled, biotinylated oligomers as a means of avoiding difficult preparation. Although the use of streptavidin may limit repeat administrations because of immunogenicity, endogenous biotin is not expected to displace either biotinylated oligomers or biotinylated carriers. In preliminary studies, d-biotin at 56 mM was incubated with streptavidin–Tat for 1 h in 37°C PBS, with no evidence of displacement. Because the serum concentration of endogenous biotin in a mouse is about 10 nM (27), the displacement of Tat or other biotinylated agents from streptavidin by endogenous biotin should be negligible.
The use of streptavidin as a linker for carriers and oligomers functioned as anticipated in the preparation of the nanoparticles. As evidence of successful preparation, the addition of 99mTc-labeled cDNA at tracer levels to the unlabeled DNA–streptavidin–Tat nanoparticle resulted in an HPLC radioactivity profile demonstrating the absence of free DNA and therefore a successful preparation (Fig. 1).
This investigation examined both PO and PS DNAs and cholesterol, Tat, and polyarginine carriers. Both in cell culture and in animals, higher radioactivity accumulation was observed for the PS nanoparticles than for the PO nanoparticles. Although the lower radioactivity levels for the PO DNA may have been due to nuclease degradation of the PO DNA, recent results show that PO DNAs protected at both ends, like the PO DNA used in the present study, are stable against degradation over prolonged periods (Yi Wang, unpublished observations, 2006). In our view, a more likely explanation for the higher radioactivity levels for the PS DNA is the protein-binding affinity of this chemical form of DNA (13), resulting in high levels in cell culture (Fig. 3) and high levels in blood in vivo (Table 1), exactly as was observed earlier for naked PS DNA (1,14). As shown here, substituting the PO DNA for the PS DNA in each nanoparticle resulted in decreased accumulation both in culture and in vivo.
Although the addition of each carrier to the PO DNA–streptavidin nanoparticle improved cellular accumulation at all 3 time points (Fig. 2), no similar improvement was observed for the PS DNA nanoparticles (Fig. 3). This apparent absence of carrier-mediated increased cellular accumulation may have been related to the 10-fold-higher accumulation of the PS DNA nanoparticles compared to the PO DNA nanoparticles. If the carrier-mediated accumulation of the PS DNA nanoparticles is similar to that of the PO DNA nanoparticles, as is reasonable, then this accumulation represents only a small and probably unobservable fraction of the total PS nanoparticle accumulation attributable primarily to nonspecific protein binding. These data confirm that cholesterol, Tat, and polyarginine functioned as carriers within the nanoparticles for the PO DNA and possibly the PS DNA as well.
In addition to confirming that carrier function was preserved in the nanoparticles, the results of the cell studies may provide evidence as to whether the DNAs accumulated through an antisense mechanism. In this regard, the accumulation of the anti-mdr1 DNA nanoparticles should increase in cells in the descending order TCO-1, KB-G2, and KB-31 in approximate proportion to the mdr1 expression level. Naked PS DNA accumulated in the same order after 20 h (14) through an antisense mechanism, as demonstrated better by indirect evidence (14,15) and by direct evidence from in situ transcription (21). When incubated for 23 h as both Tat and cholesterol nanoparticles as well as carrier-free nanoparticles, the PS DNA showed the expected statistically significant differences in accumulation in TCO-1, KB-G2, and KB-31 cells (Fig. 3). That evidence of antisense accumulation was not apparent at either 3 or 7 h of incubation may indicate that the clearance of excess radiolabeled DNA over 10–24 h is required. It is also possible that the protein-binding affinity of the PS DNA encouraged nonspecific protein-mediated, carrier-independent accumulation at levels that obscured carrier-mediated accumulation at early times, before specific binding to the target mRNA became evident. The reason for the anomalous results obtained with the polyarginine nanoparticles was not pursued. Recent studies with rhodamine 123 accumulation (a substrate of Pgp) measured by flow cytometry provided definitive evidence that the incubation of KB-G2 cells with antisense nanoparticles significantly decreases Pgp expression, almost certainly through an antisense mechanism (data not presented).
In contrast to the observations for the PS DNA nanoparticles, no obvious pattern among cell types was observed for the PO DNA nanoparticles at any time, suggesting that the accumulation was mediated by mechanisms other than an antisense mechanism. Ongoing fluorescence microscopy studies with KB-G2 cells of fluorescence-labeled PS and PO DNAs incubated naked and as Tat nanoparticles have shown migration to the nucleus in all cells incubated with PS DNA and a uniform absence of nuclear migration in identical studies with PO DNA (Xinrong Liu, unpublished observations, 2007). Because nuclear migration is increasingly being viewed as an indication of antisense targeting (21), these results, taken together, may indicate that the PO DNA under the conditions of these cell studies has limited access to the mdr1 mRNA target.
The xenograft-bearing animal studies were performed with the 3 types of nanoparticles judged from the cell studies to be the most instructive. Thus, the PS DNA–streptavidin–Tat, PS DNA–streptavidin–cholesterol, and PO DNA–streptavidin–Tat nanoparticles were administered at 1 μg, and the animals were sacrificed at either 6 or 18 h. A statistical comparison of the results in Table 1 for the PS DNA–streptavidin–Tat and PS DNA–streptavidin–cholesterol nanoparticles showed no significant differences (Student t test, P < 0.05) at 18 h but significantly higher PS DNA–streptavidin–Tat nanoparticle accumulation in all tissues at 6 h. At both time points, the exceptions were the stomach and large intestine. Because results for stomach and intestine samples often vary significantly with subtle changes in preparation quality, accumulation in these organs may be difficult to interpret. An examination of the tissue-to-blood ratios showed that the significant differences at 6 h might be explained by the higher level of the Tat nanoparticles in blood at this time leading to increases in the levels in organs simply because of the blood therein. Similarly, the absence of significant differences at 18 h might be explained by identical levels in the blood at this time. Only the liver-to-blood and spleen-to-blood ratios were anomalous and only at 18 h, suggesting that at this time, there was higher accumulation of the Tat nanoparticles than the cholesterol nanoparticles in these organs. These results suggest that some differences in pharmacokinetics between the 2 carriers were manifested despite the presence of streptavidin in the PS DNA nanoparticles.
A statistical comparison of the results shown in Table 1 for the PS DNA–streptavidin–Tat and the PO DNA–streptavidin–Tat nanoparticles showed that a significantly higher accumulation of both types of the PS nanoparticles than the PO nanoparticles occurred in all tissues other than the stomach and large intestine at both time points. An examination of the tissue-to-blood ratios indicate that the higher accumulation of the PS nanoparticles might have been attributable to higher levels in the blood, probably because of the higher protein-binding affinity of the PS DNA for serum proteins. That the tissue-to-blood ratios for the liver, spleen, and bone were anomalously high further suggests that the increased accumulation in these organs was attributable to the higher protein-binding affinity of the PS nanoparticles for these organs. These results suggested that the high protein-binding affinity of the PS DNA was manifested despite the presence of streptavidin in the nanoparticles.
Although naked PS DNA was not included in this investigation, the results of a recent similar xenograft-bearing animal study may be used to compare the PS DNA–streptavidin nanoparticles with naked PS DNA (16). At both time points, when stomach and large intestines are omitted, the values for the PS DNA–streptavidin–Tat and PS DNA–streptavidin–cholesterol nanoparticles are never more than about a factor of 4 of that for the naked PS DNA. Therefore, streptavidin and the carriers had a fairly modest influence on the pharmacokinetics of PS DNA. Furthermore, the levels in tumors in animals receiving the PS DNA–streptavidin–Tat nanoparticle and those receiving the PS DNA–streptavidin–cholesterol nanoparticle were similar at both times. These results show that, in addition to apparently preserving the properties of the PS DNA and the carriers, the presence of streptavidin or the carriers within the nanoparticles had only a modest influence, at best, on the pharmacokinetics of PS DNA. In particular, the presence of streptavidin did not encourage rapid accumulation in normal tissues, such as the liver.
Recently, an investigation of the influence of streptavidin on the properties of the same 3 carriers in MCF-7 cells in culture but constructed with a 25-mer antisense MORF against the survivin mRNA was conducted (28). A statistically significant increase in the accumulation of each type of nanoparticle compared with that of naked MORF was observed, with increases of about 9-fold for Tat and 11-fold for polyarginine after 12 h of incubation. The increase was lowest but still significant for cholesterol. In addition, the same relative accumulation was observed for the carrier-free MORF–streptavidin nanoparticle control versus the naked MORF control, providing evidence that streptavidin was not responsible for the increased accumulation. Finally, the higher cellular accumulation over time of the antisense MORF–streptavidin–Tat nanoparticle control relative to that of the sense MORF–streptavidin–Tat nanoparticle control was statistically significant at all times other than 3 h, providing evidence for antisense binding of the nanoparticles and therefore escape from entrapment.
CONCLUSION
An advantage of the use of streptavidin as a linker is the simplicity of preparation of oligomer–carrier nanoparticles in contrast to covalent conjugation and therefore the ease with which different nanoparticles may be made and tested. Obviously, the same approach may be used to link tumor-targeting agents, such as antitumor antibodies and antitumor peptides, to antisense oligomers with or without carriers. Evidence was obtained that the function of the carriers was at least partially preserved when they were bound to streptavidin in the nanoparticles and that the pharmacokinetic properties of the antisense DNA were not markedly changed. Although indications from this investigation suggest that adding radiolabeled antisense PS DNA to cells as streptavidin–carrier nanoparticles does not restrict the targeting of mRNA, further investigations are needed to firmly establish this point.
Acknowledgments
The authors thank Dr. Isamu Sugawara (Research Institute of Tuberculosis, Tokyo, Japan) for providing the Pgp-expressing cell lines for this investigation. This research was supported in part by the Office of Science (BER); the U.S. Department of Energy (grant DE-FG02-03ER63602); the National Institutes of Health (grant SR21CA100092); and Grants-in-Aid for Scientific Research (16591225 and 17659374) and the 21st Century Center of Excellence (COE) Program entitled “Establishment of Individualized Cancer Therapy Based on Comprehensive Development of Minimally Invasive and Innovative Therapeutic Methods (Keio University)” from the Japanese Ministry of Education, Science, Sports and Culture.
Footnotes
-
COPYRIGHT © 2007 by the Society of Nuclear Medicine, Inc.
References
- Received for publication December 26, 2006.
- Accepted for publication August 23, 2007.





{kind=link}
{kind=link}
{kind=link}