


Abstract
Molecular imaging of gene expression is currently hindered by the lack of a generalizable platform for probe design. For any gene of interest, a probe that targets protein levels must often be generated empirically. Targeting gene expression at the level of mRNA, however, would allow probes to be built on the basis of sequence information alone. Presented here is a class of generalizable probes that can image pre-mRNA in a sequence-specific manner, using signal amplification and a facile method of delivery. Methods: Pre–trans-splicing molecules (PTMs) were engineered to capitalize on the phenomenon of spliceosome-mediated RNA trans-splicing. Using a modular binding domain that confers specificity by base-pair complementarity to the target pre-mRNA, PTMs were designed to target a chimeric target mini gene and trans-splice the Renilla luciferase gene onto the end of the target. PTMs and target genes were transfected in cell culture and assessed by luciferase assay, reverse-transcriptase polymerase chain reaction, Western blot, and rapid analysis of 5′ cDNA ends. PTMs and target genes were also assessed in vivo by hydrodynamic delivery in mice. Results: Efficiency and specificity of the trans-splicing reaction were found to vary depending on the binding domain length and structure. Specific trans-splicing was observed in living animals (P = 0.0862, Kruskal–Wallis test). Conclusion: Described here is a model system used to demonstrate the feasibility of spliceosome-mediated RNA trans-splicing for imaging gene expression at the level of pre-mRNA using optical imaging techniques in living animals. The experiments reported here show proof of principle for a generalizable imaging probe against RNA that can amplify signal on detection and be delivered using existing gene delivery methodology.
In recent years, a trend has emerged toward the production of highly specialized therapeutics that capitalize on genetic information (1). It is thought by some that this reflects the first steps toward personalized medicine. Given the advances in pharmacogenomics and the increasingly apparent role genes play in disease (2), this paradigm shift seems inescapable barring a regulatory reluctance to change. In concert with the advances in personalized treatment, improvements in individualized diagnostics must progress as well.
Among the genetic targets available for designing imaging probes, RNA is arguably the most attractive for this task. Unlike DNA, the copy number of RNA transcripts for any given gene reflects levels of expression, and unlike proteins, oligonucleotide probes can be designed on the basis of sequence knowledge alone. Two problems that have historically hindered RNA imaging in living subjects are sensitivity and delivery. Canonical mRNA imaging has relied on radiolabeled antisense oligonucleotides (RASONs) (3–9). Although a variety of different radionuclides and backbone chemistries have been used (e.g., morpholino, peptide nucleic acid), RASONs are fundamentally limited by their lack of signal amplification. Because of the potential for a low target number of any arbitrary mRNA species (50–1,000 copies per cell for mRNA and up to 106 copies per cell for pre-mRNA (10,11)), any mRNA imaging probe would benefit greatly from the incorporation of a mechanism to multiply the signal once recognition of the target has been achieved. In addition, some RASONs have been found to bind nonspecifically to serum proteins, others have difficulty crossing cellular membranes, and still others are susceptible to nucleases. All these factors make RASON delivery an arduous task.
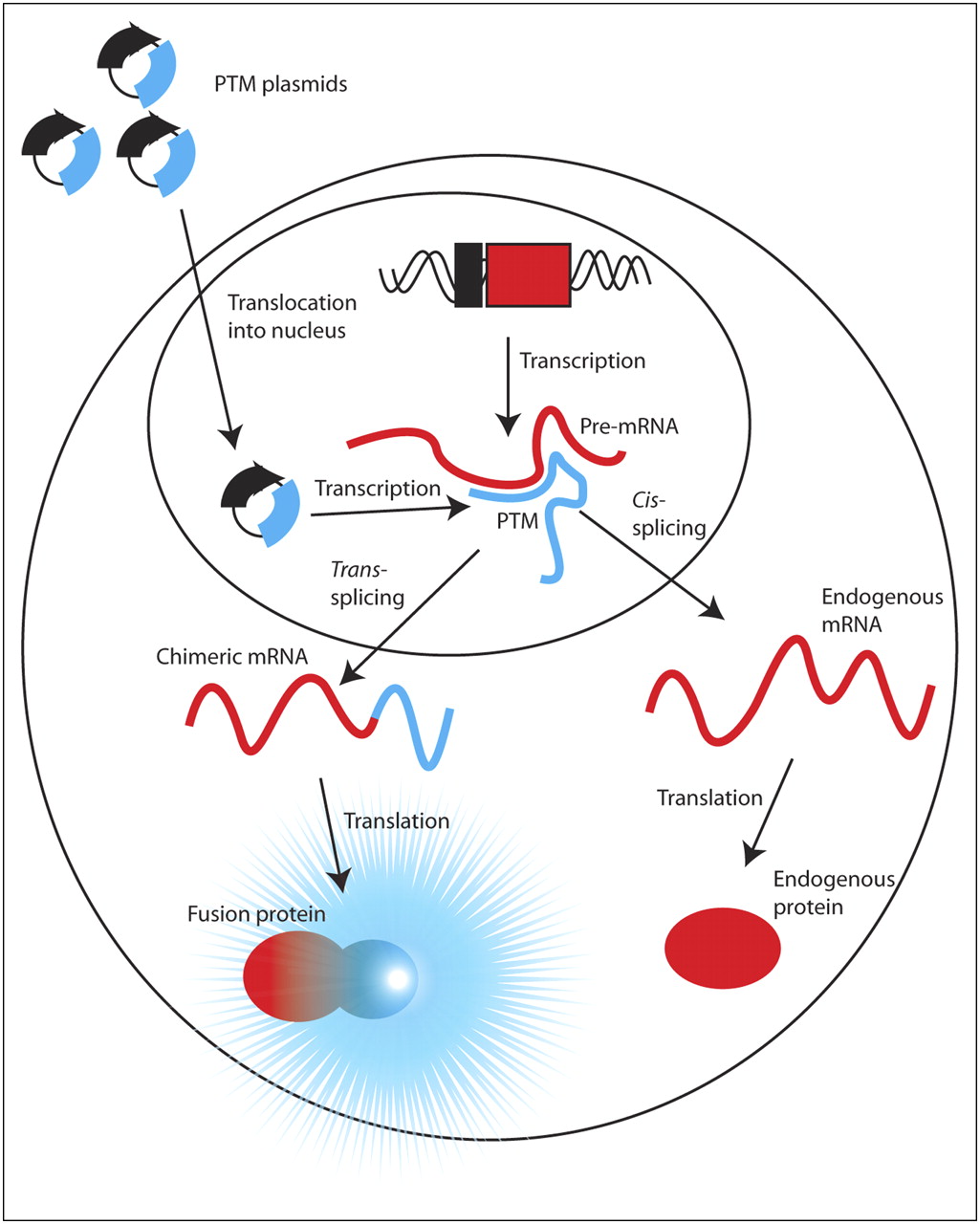
Spliceosome-mediated RNA trans-splicing (SMaRT) is a technique that attempts to circumvent some of these obstacles (12). SMaRT uses engineered pre–trans-splicing molecules (PTMs) that can introduce exogenous coding sequences into any pre-mRNA of interest. These PTMs contain sequence elements that can be recognized by mammalian splicing machinery and cause trans-splicing of the PTM coding domains into a subset of the target pre-mRNA transcripts. Thus, a fraction of the original transcripts is processed normally via cis-splicing to produce the endogenous protein, and the other portion that is trans-spliced is translated to produce a chimeric protein, containing one or more of the endogenous target exons appended to the genetic payload delivered by the PTM (Fig. 1). PTM specificity is conferred by a binding domain, which is complementary to intronic sequences in the gene of interest.

Schematic of SMaRT imaging strategy. SMaRT PTMs encoding a reporter gene and cloned into plasmid DNA are delivered to nucleus of cell, where they are transcribed (blue lines). PTMs can intercept portion of pre-mRNA (red lines) of arbitrary gene via complementary nucleic acid binding and trans-splice coding domain into target. This hybrid mRNA is translated into chimeric protein composed of portion of target gene and reporter gene, which can elicit signal. Remaining pool of endogenous pre-mRNA can cis-splice and be translated into endogenous protein.
SMaRT has been used extensively for the therapeutic correction of mutated genes (13–17) and for the demonstration of imaging feasibility in vivo (18). In a previous study, a luciferase repair model was used in various applications to confirm the capacity of PTMs to be used for imaging gene expression (18). An artificial target gene was created containing the 5′ half of a luciferase reporter gene plus an intron from the human papillomavirus type 16 (HPV16) E6 oncogene. The PTM meanwhile contained a binding domain specific for the E6 intron, and its genetic payload encoded the remaining 3′ half of the luciferase gene. Thus, for any tissues in which both the artificial target and the PTM were expressed, trans-splicing occurred, producing a fully functional luciferase protein. Although this study was an important proof of principle to demonstrate that the trans-splicing reaction could be imaged, it was not a generalizable model, extendable to imaging any mRNA of interest in vivo. Because no other gene would contain the 5′ half of the luciferase gene, a different PTM had to be constructed that could remain inactive until trans-splicing occurred. The current work represents the next step along the pathway toward imaging endogenous mRNA levels. A PTM has been designed that encodes a full-length luciferase reporter gene, minus the ATG start codon. Thus, the reporter gene cannot be translated unless it trans-splices into a target that contains a start codon (Fig. 2B). The binding domain of the PTM is modular and confers specificity to the trans-splicing reaction, giving this technique the potential to be adapted to image expression of any arbitrary gene of interest at the level of pre-mRNA.

(A) Various target genes and PTMs used in study. Target gene consisted of HSV1-sr39tk coding sequence fused to intron from HPV16-E6 (TK). As a control, orientation of intron was reversed (TKas). PTMs were generated with binding domains of lengths 80 bp (H1), 160 bp (H2), and 240 bp (H3) complementary or homologous (H1s, H2s, H3s) to intron of target gene. These PTMs contained branch point (black circle), polypyrimidine tract (black rectangle), and dinucleotide 3′ splice site (AG), as well as Renilla luciferase gene without its start codon ((-AUG)luc). Two safety PTMs were constructed with complementary sequences to branch point–polypyrimidine tract–AG signals (white circle, white rectangle, UC) either upstream (S1) or downstream (S2) of 80-bp binding domain. (B) Target gene can undergo either cis-splicing, in which single-amino-acid codon plus stop signal are spliced onto HSV1-sr39tk gene, or trans-splicing, in which luciferase gene is spliced onto HSV1-sr39tk gene.
MATERIALS AND METHODS
Plasmid Construction
The backbone used for the construction of all expression vectors detailed here was modified from the pcDNA3.1 plasmid (Invitrogen). Specifically, a strong polyA signal and RNA polymerase II pause sequences were inserted upstream of the cytomegalovirus promoter to reduce cis-splicing from the vector backbone (19). The Renilla luciferase reporter gene used in all experiments was modified from the mammalian codon-optimized version marketed by Promega (Rluc). Two mutations were made to the protein primary sequence, M14I and M27I, to reduce the potential for cryptic translation initiation. The PTM binding domain was bracketed by NheI and SacII restriction enzyme sites, which were subsequently used to change the binding domain without affecting any other sequences. Several different PTMs were constructed with varying binding domain lengths. The 80-bp binding domain used against the HPV16-E6 intron was 5′CAGTTAATACACCTAATTAACAAATCACACAACGCTTTGTTGTATTGCTGTTCTAATGTTGTTCCATACACACTATAACA3′. The sequences of the 160-bp and 240-bp binding domains were 5′ CCACCGACCCCTTATATTATGGAATCTTTGCTTTTTGTCCAGATGTCTTTGCTTTTCTTCAGGACACAGTGGCTTTTGACAGTTAATACACCTAATTAACAAATCACACAACGCTTTGTTGTATTGCTGTTCTAATGTTGTTCCATACAAACTATAACAA3′ and 5′ CCACCGACCCCTTATATTATGGAATCTTTGCTTTTTGTCCAGATGTCTTTGCTTTTCTTCAGGACACAGTGGCTTTTGACAGTTAATACACCTAATTAACAAATCACACAACGCTTTGTTGTATTGCTGTTCTAATGTTGTTCCATACAAACTATAACAATAATGTCTATACTCACTAATTTTAGAATAAAACTTTAAACATTTATCACATACAGCATATGGATTCCCATCTCTATATAC3′, respectively. The original 80-bp binding domain was designed at Intronn, Inc., using a rational approach and further modified to eliminate cryptic cis-splicing. The longer binding domains were extensions of the original binding domain, modified at a single position of the new sequence to remove a potential 5′ splice site. The modification was made subsequent to the creation of the PTM using the Stratagene QuikChange II XL site-directed mutagenesis kit. Negative control PTMs were constructed for all binding domain lengths. This was accomplished by reversing the orientation of the binding domains such that they were homologous to portions of the HPV16-E6 intron, rather than complementary.
Several variants of the 80-bp binding domain PTM were also made containing a “safety” sequence either upstream or downstream of the 80-bp binding domain. This sequence was complementary to the branch point, polypyrimidine tract, and 3′ splice site of the PTM. The upstream safety (S1) was created by annealing complementary oligonucleotides and using SacI and NheI restriction sites. To construct the downstream safety (S2), the original PTM was first mutated to introduce a unique AgeI restriction site between the binding domain and the branch point. Then, complementary oligonucleotides were annealed and cloned into the plasmid using the SacII and AgeI sites.
The herpes simplex virus type 1 mutant thymidine kinase (HSV1-sr39tk) gene used in this study is based on a previously published sequence (20). The single mutation N376K was made for splicing-signal-sequence considerations (i.e., because U2-type GT-AG introns prefer an AG sequence upstream of the 5′ splice site, the 376 residue was altered to conform to this preference) (21). Before the stop codon of HSV1-sr39tk, nucleotides 227–525 from the HPV16 genome (K02718), corresponding to the E6*II intron (E6), were inserted, as well as a single coding glycine residue, also for splicing-signal-sequence considerations. All sequences were introduced by PCR using the PfuUltra DNA polymerase (Stratagene) and the manufacturer's protocol. Primers were generated by the Protein and Nucleic Acid Facility of Stanford School of Medicine. A negative control target gene was constructed by reversing the orientation of E6 (denoted as TKas).
Transfection of Plasmids in Cell Culture
All transfections were performed according the Superfect protocol (Qiagen), using the advised amount of DNA. In the cases where multiple plasmids were transfected, the total amount of DNA did not exceed the recommended limit.
Rapid Analysis of 5′ cDNA Ends (RACE)
Total RNA was isolated using the RNeasy kit (Qiagen) from transiently cotransfected 293T cells. The RNA-ligase mediated RACE kit (Invitrogen) was used in conjunction with a gene-specific reverse primer, complementary to a portion of the Rluc gene. For each sample, 24 or 48 colonies were picked and sequenced. Human genome sequences were identified using the Basic Local Alignment Search Tool of the National Center for Biotechnology Information.
Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated from transiently transfected cells using the RNeasy kit. Primers were generated to bind to either side of a splice junction or exon–intron boundary for all pre-mRNA species (target, PTM, trans-spliced product). Real-time RT-PCR was performed using the Bio-Rad iScript 1-step RT-PCR kit with SYBR Green according to the manufacturer's protocol.
Western Blot Analysis
Protein samples were collected from transfected cells using passive lysis buffer (Promega) and run on NuPAGE 4–12% precast gradient gels (Invitrogen) using 2 μg per sample and 3-morpholino-propanesulfonic acid running buffer at 200 V for 50 min. Separated proteins were transferred onto polyvinylidene fluoride membranes using the XCell II blot module (Invitrogen) according to the manufacturer's protocol. The α-Rluc antibody (MAB4400; Chemicon) was diluted 1:1000 with phosphate-buffered saline tween-20 (PBST) and incubated with the membrane for 1 h at room temperature. Blots were washed with PBST and then incubated with goat antimouse antibody conjugated horseradish peroxidase (BioRad), diluted at 1:25,000 for 1 h. The membranes were washed again with PBST and then treated with ECL Plus (GE Healthcare) for 5 min before exposure to film. Blots were stripped with 100 mM β-mercaptoethanol, 2% sodium dodecylsulfate, and 62.5 mM Tris-HCl, pH 6.7, for 30 min at 50°C. The membranes were washed with PBST and then probed again with the α-HSV1-tk antibody (graciously provided by Margaret Black, Washington State University) at a 1:10,000 dilution and also α-tubulin at a 1:5,000 dilution as a loading control.
Luminometer, Protein Assays
Twenty-four hours after transfection, cells were aspirated, washed with phosphate-buffered saline, and lysed using Passive Lysis Buffer according to the manufacturer's protocol. Samples were assayed for luminescence by combining 20 μL of lysate with 100 μL of coelenterazine assay buffer (5 mM Tris, 0.5 mM ethylenediamine tetraacetic acid, 0.6 M NaCl, 50 mM KPO4, 0.1 g of bovine serum albumin per liter, 0.03 mM NaN3, pH 7.8) and then adding 1 μL of coelenterazine (0.5 μg/μL in 50:50 methanol:propylene glycol), stirring briefly in a vortex mixer, and collecting luminescence for 10 s in a 20/20n luminometer (Turner). Protein content was also measured to normalize light output for cell number and lysis efficiency using protein assay reagent (BioRad) according to the manufacturer's protocol.
Cell Culture
293T cells were cultured in Dulbecco's modified Eagle medium with high glucose, supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Imaging Index
An index was generated to compare various PTMs for imaging suitability. The data used contained measurements of absolute signal (AS) and signal to background (SB) from luciferase assay experiments. Also used were measures of PTM trans-splicing efficiency (EF) and specificity (SP), calculated from the RACE experiments. PTM trans-splicing efficiency and specificity were calculated as: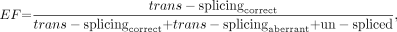
 Several alterations were made to the metrics before insertion in the index, including ranking, mathematic transformation, and normalization to the highest value for each metric independently. The index was then calculated by taking the product of a weighted average of EF and SB and a weighted average of SP and AS. The weights for EF and SP were the same (w1), as were the weights for AS and SB (1 − w1). Thus, the final index was calculated as:
Several alterations were made to the metrics before insertion in the index, including ranking, mathematic transformation, and normalization to the highest value for each metric independently. The index was then calculated by taking the product of a weighted average of EF and SB and a weighted average of SP and AS. The weights for EF and SP were the same (w1), as were the weights for AS and SB (1 − w1). Thus, the final index was calculated as: Plots of the index were generated for each alteration as a function of w1 using MATLAB, student edition, version 5.3 (The MathWorks, Inc.). Imaging index data are presented in Table 1.
Plots of the index were generated for each alteration as a function of w1 using MATLAB, student edition, version 5.3 (The MathWorks, Inc.). Imaging index data are presented in Table 1.
PTM Imaging Index Values for w1 = 0.25
Animal Experiments
Animal experiments were performed using nu/nu mice. Twenty micrograms of total plasmid DNA (10 μg of PTM plus 10 μg of target) were diluted in 1.8 mL of phosphate-buffered saline. The total volume was rapidly (5–7 s) injected into the lateral tail vein. Twenty-four hours later, the mice were imaged by injecting coelenterazine (2 μL of a 5 mg/mL stock dissolved in ethanol diluted in 98 μL of phosphate-buffered saline) into the lateral tail vein and then placing the animal supine in an IVIS-Spectrum or IVIS-100 (Caliper Life Sciences) and collecting photons for 1 min. All procedures were performed according to the guidelines set forth by the Stanford University Administration Panel on Laboratory Animal Care.
Statistics
PTMs were grouped with their appropriate negative controls for comparison, and statistical calculations were made. For example, the H1 PTM was compared against the H1s controls, whereas the H2 PTM was compared against the H2s samples. Various tests were used when comparing more than 2 groups, including ANOVA, Kruskal–Wallis, and the multiple-range test.
RESULTS
Selection of PTM for In Vivo Imaging Using Transient Cotransfection in Cell Culture
A model system was generated to demonstrate the ability of SMaRT to image gene expression at the pre-mRNA level. This consisted of an artificial chimeric target, which was composed of the HSV1-sr39tk gene fused to the HPV16-E6 intron (TK). The PTMs used to target this gene contained binding domains of various lengths (80, 160, and 240 bps), plus splicing signal sequences including a branch point, polypyrimidine tract, and acceptor AG dinucleotide. In addition, the PTM coded for the codon-optimized Renilla luciferase (Rluc) gene, minus the ATG start codon. This was to ensure that the reporter gene could be translated into an active enzyme if and only if it first trans-spliced into its target pre-mRNA, which would provide the translation initiation site. Also, PTMs with safety sequences (sequences that were complementary to the branch point–polypyrimidine tract–AG splicing signals) either upstream or downstream of the 80-bp binding domain were constructed (Fig. 2A).
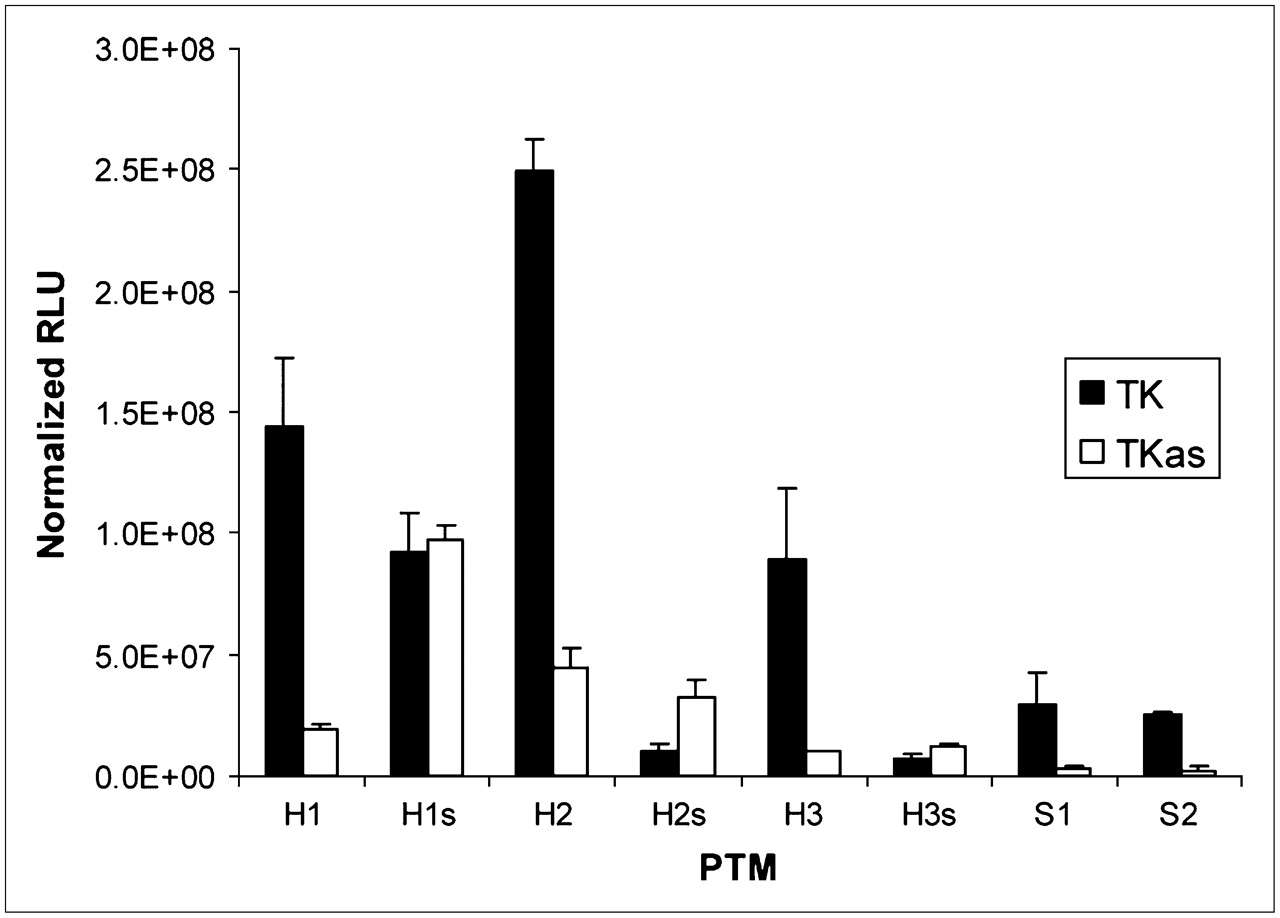
PTMs and target genes were cotransfected as separate plasmids into 293T cells and showed a range of signal-to-background ratios (i.e., the amount of light produced when the correct trans-splicing partners were cotransfected together, compared with the amount of light produced when the PTM binding domain and target were not complementary). As shown in Figure 3, the 80-bp binding domain PTM (H1) demonstrated a statistically significant 7.5-fold increase in signal when transfected with the correct target as opposed to the negative control target (P = 0.0249, Kruskal–Wallis, multiple-range test). However, the H1s PTM displayed a statistically indistinguishable signal when transfected with either target, compared with H1. The 160-bp binding domain PTM, H2, showed a greater absolute signal than the H1 PTM, but the signal-to-background ratio was only 5.6 (P = 0.0188). The 240-bp binding domain H3 PTM demonstrated a 9.1 signal-to-background ratio (P = 0.0156). The S1 and S2 safety PTMs demonstrated 9.3 and 9.8 signal-to-background ratios, respectively (P < 0.04, t test performed independently for each PTM); however, the absolute signal produced by the safeties was only about 20% of the signal produced by the original PTM.
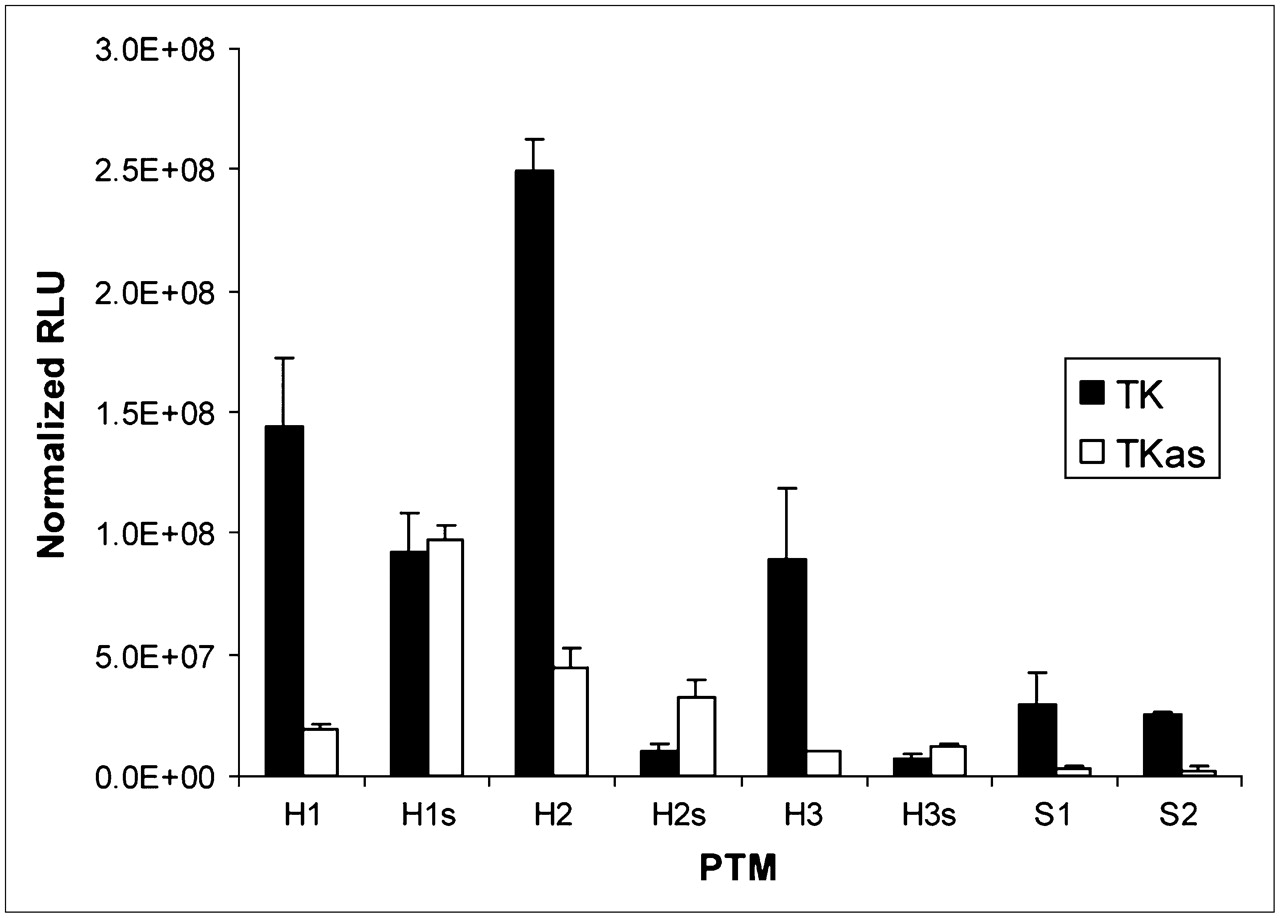
Cotransfection results. Units are reported in relative light units (RLUs) normalized for total protein content (mean ± SD). Each PTM was cotransfected with either target gene or control gene. Cells were lysed and assayed 24 h after transfection.
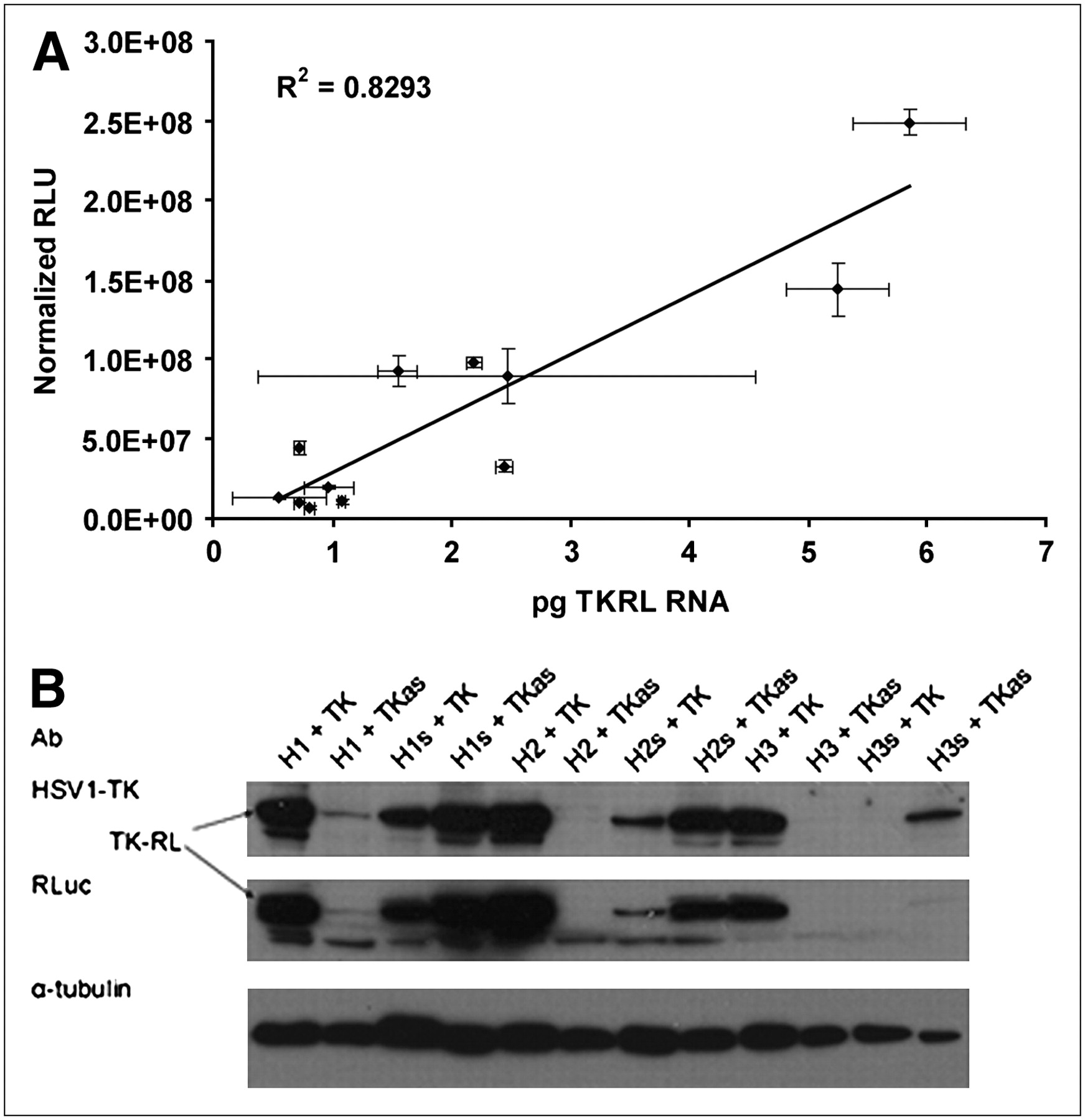
Real-time RT-PCR was performed on total RNA from cotransfected samples using primers on either side of the putative trans-spliced product. Product was quantified using in vitro transcribed trans-spliced product as a standard and then correlated to the absolute levels of signal shown in Figure 4A (R2 = 0.83). Western blot analysis, using both α–Renilla luciferase and α-HSV1-tk antibodies, showed bands for each cotransfection sample that corresponded to the 77-kDa weight of the chimeric TK–Renilla luciferase protein (Fig. 4B).
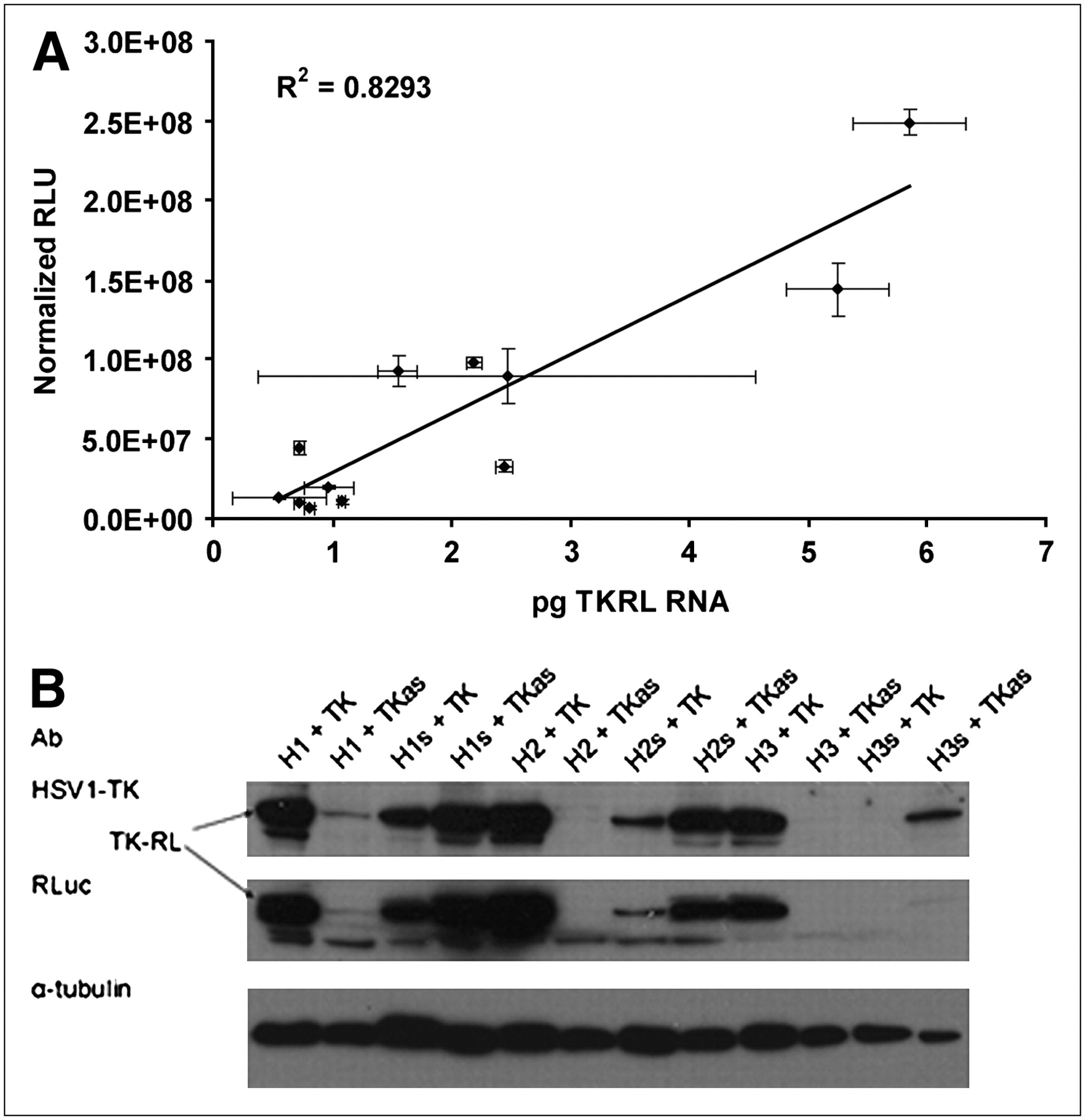
Cells were transfected with various combinations of PTM and target. Cells were lysed 24 h later, and both total RNA and total protein were collected for analysis. (A) Mass levels of trans-spliced mRNA quantified by real-time RT-PCR and correlated with absolute signals shown in Figure 3. (B) Western blot demonstrating qualitative agreement of trans-spliced protein levels with absolute signal and mRNA levels. RLU = relative light units.
Rapid analysis of 5′ cDNA ends (RACE) was also performed on cotransfected samples to more accurately quantify the percentage of correct trans-splicing events. Table 2 reports the RACE findings for each PTM. It was found that the longer binding domain PTMs (H2 and H3) were more prone to all types of splicing than was H1, showing increases in aberrant trans-splicing (except for H3), cis-splicing, and correct trans-splicing (with a subsequent drop in unspliced PTMs). The safety PTMs, on the other hand, showed different behavior, both compared with H1 and compared with one another. Both safety PTMs showed a decrease in aberrant trans-splicing, compared with H1; however, S1 demonstrated a concomitant decrease in correct trans-splicing and a subsequent increase in unspliced PTM, whereas S2 showed an increase in correct trans-splicing and a subsequent decrease in unspliced PTM.
Summary of RACE Data for All PTMs Tested
Animal Experiments
Before verifying that the trans-splicing reaction could occur in living subjects, an imaging index was created to assess the potential of each PTM and predict which one would be most useful for imaging purposes. To this end, a weighted average formula was created taking into account the absolute signal and signal-to-background ratios determined by the cotransfection luciferase assays, as well as the trans-splicing efficiency and specificity determined by RACE. By giving the RACE data one third of the weight of the luciferase assay data, it was found that the S2 PTM outperformed the other PTMs for each numeric metric (Table 1).
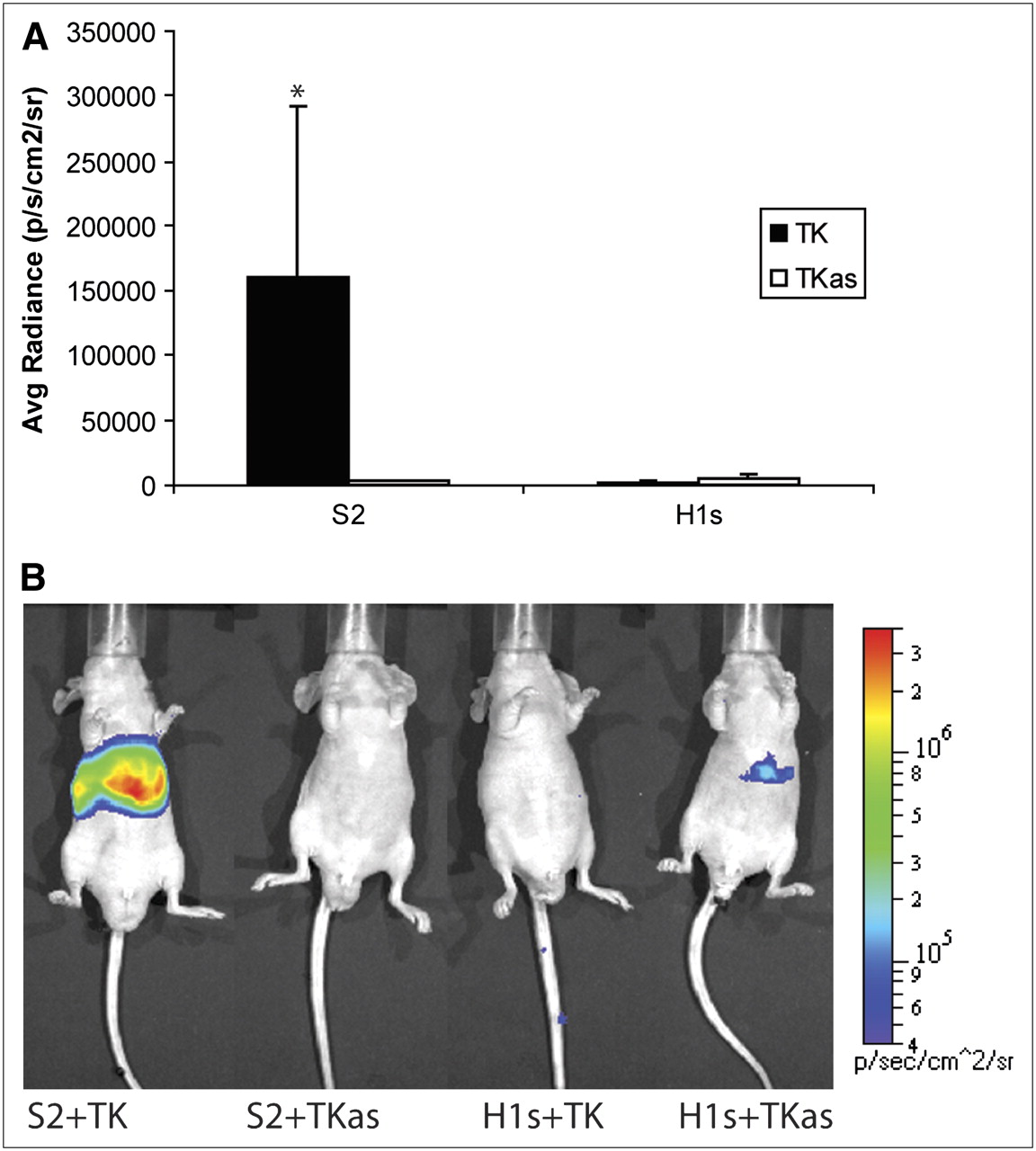
Both PTM and target were codelivered into nude mice via hydrodynamic tail vein injection. Twenty-four hours later, mice were injected with coelenterazine and imaged. Four different groups of mice were used (n = 3 each), comprised of 1 experimental group and 3 negative control groups as in the previous cell culture studies. Representative animal images are shown in Figure 5B. Significantly greater signal is seen from the livers of mice from the “S2 + TK” group than from the negative controls (P = 0.0862, Kruskal–Wallis test, Fig. 5A). Some signal was observed from the “H1s + TKas” group, although it was at least an order of magnitude lower than the “S2 + TK” group and not significantly above background levels.
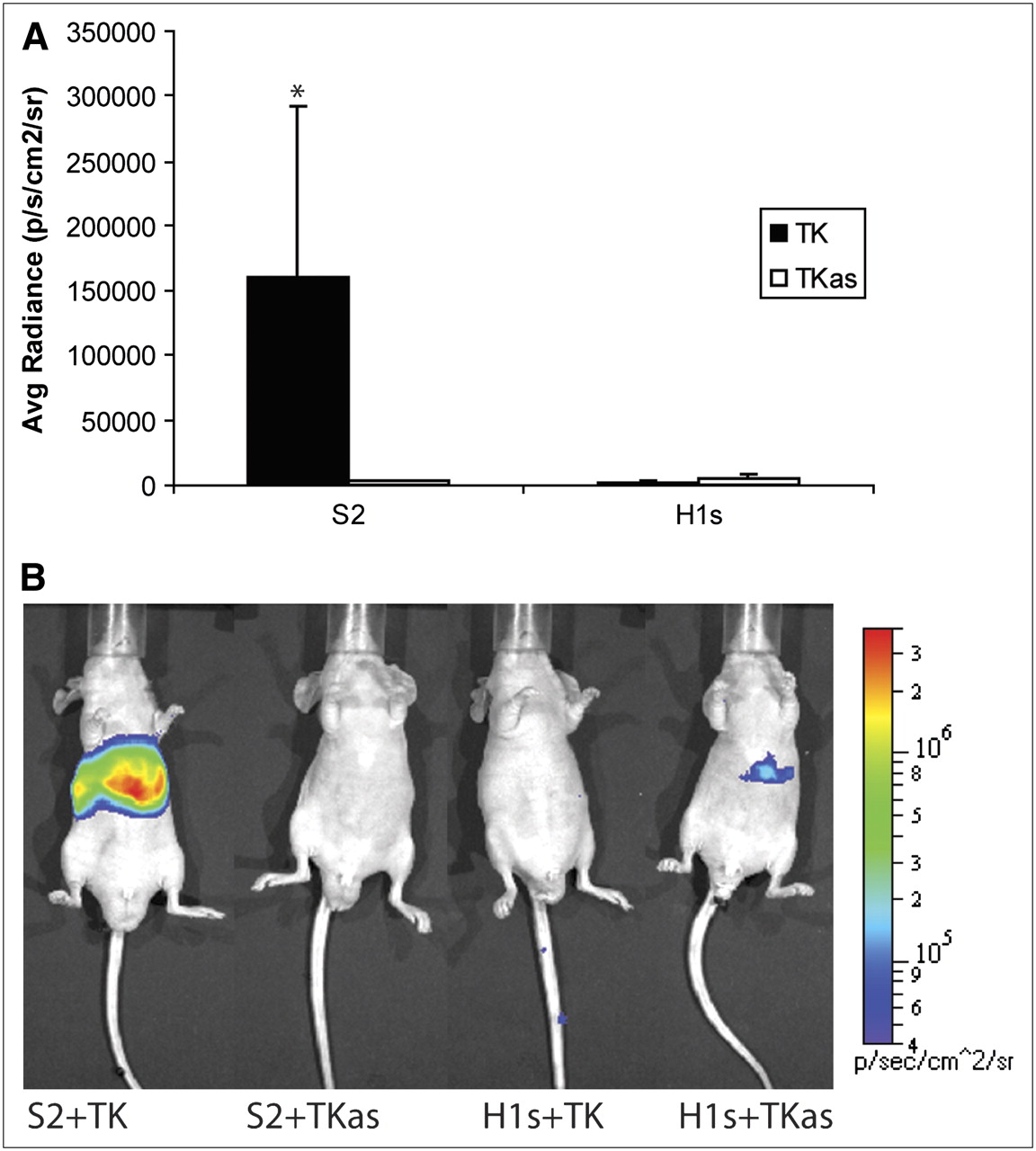
Summary of in vivo experiments. Nude mice (n = 12) were injected hydrodynamically with combination of PTM and target plasmids. Labels on abscissa refer to constructs shown in Figure 2. Twenty-four hours later, they were injected with coelenterazine and imaged for 1 min. ROIs of equal size for each mouse were drawn over entire liver, and average radiance was calculated. (A) Average radiance for each mouse (mean ± SEM). Values are reported in photons/s/cm2/steradian. *P = 0.0862, Kruskal–Wallis test. (B) Representative images of mice injected with PTM and target gene plus negative controls. Scale covers 2 orders of magnitude; units are reported in photons/cm2/s/steradian.
DISCUSSION
To demonstrate the imaging possibilities of a generalizable SMaRT probe, a model system was created composed of an artificial target gene and a PTM with a modular binding domain. Several different factors were investigated to determine their impact on trans-splicing efficiency and specificity. Because the binding domain of the PTM was thought to drive the specificity of the reaction, its length and secondary structure were varied. Although the cotransfection experiments demonstrated relatively good signal-to-background ratios when compared against cotransfection with a control gene for the 80-bp binding domain PTM, swapping the orientation of the binding domain produced similar results regardless of which target was cotransfected, suggesting that this binding domain was either too short or too promiscuous to confer an adequate specific signal. Consistent with earlier predictions (22), doubling the binding domain length increased the efficiency of the trans-splicing reaction, most likely because of the increased availability of complementary sequences to the target. Because RNA molecules generally possess some secondary structure, increasing the length of the binding domain increases the chances that at least some portion of the target intron will be available for binding (23). Conversely, this also means that there exist a greater number of targets with some level of complementarity to a portion of the binding domain. Thus, the 160-bp binding domain PTM showed an increase in absolute signal but a decrease in signal-to-background ratio, compared with the original PTM. The 240-bp binding domain PTM, however, showed a similar absolute signal, compared with H1, but a better signal-to-background ratio. This is most likely because the longer binding domain itself has some secondary structure, limiting the number of nucleotides available for binding to the target but also limiting its complementarity to aberrant targets. These results suggest that there exists some optimal length for the binding domain that balances efficiency and specificity, pointing to the probability that longer binding domains are better for imaging purposes.
The safety PTMs represented an attempt at rationally designing some secondary structure into the binding domains. Unlike the longer binding domains, whose secondary structure could not be definitively predicted, we could assume with reasonable certainty that by inserting an exactly complementary sequence to the splicing signals, PTM would take on a particular conformation (24). The results from the cotransfection experiments demonstrated that the safety PTMs increased the signal-to-background ratio; however, they had the unintended consequence of compromising the efficiency of the trans-splicing reaction. It is thus likely that the allosteric interaction of the safety PTMs and target pre-mRNA was insufficient to cause a substantial conformational change due to energetics.
The RACE analysis provided interesting clues as to the nature of the various PTMs and their safety-modified counterparts. The RACE data showed that by lengthening the binding domains, all types of splicing (i.e., cis-splicing and specific and nonspecific trans-splicing) were increased. One of the unintended consequences of lengthening the binding domains was that cryptic 5′ splice sites were introduced upstream of the branch point. This had the effect of allowing cis-splicing to occur within the PTM itself. In addition, the RACE data suggested that the safety PTMs increase signal-to-background ratios by different mechanisms. Because S1 had approximately the same percentage of correctly trans-spliced products but far fewer aberrant trans-spliced products, the signal-to-background ratio was most likely increased by limiting partial complementarity to other targets. On the other hand, S2 greatly increased the number of correctly trans-spliced products whereas the number of aberrant trans-spliced products remained relatively constant. Because the absolute signal for S2 was less than that for H1, it is unlikely that the safety makes the trans-splicing more efficient; rather, the safety probably confers greater specificity by limiting the pool of aberrant trans-splicing targets to those species that cannot produce a functional fusion protein. Thus, even though the percentage of aberrant trans-splicing is unchanged, the type of target is altered. All the RACE results reflected percentages of different types of splicing products but were not normalized for overall splicing efficiency. Thus, the differences in percentages per splicing category between PTMs do not necessarily reflect similar changes in absolute numbers of molecules. For example, although a greater percentage of H3 splicing products was correctly trans-spliced, compared with H2 and H1, the absolute number of correctly trans-spliced products was lower because of the decreased efficiency of H3 splicing.
The animal experiments validated that the observations of trans-splicing in cell culture could be readily translated to an in vivo setting. Surprisingly, the signal-to-noise ratio was better in vivo than in cell culture. This could be due, in part, to the possibility that the background levels of trans-splicing are below the threshold of detection using the current small-animal imaging instrumentation. Also, some of the nonspecificity issues pertaining to the shorter (80-bp) binding domain were not seen in living mice. This can potentially be explained by the fact that the cell culture experiments were performed using cancer cells, grown in an artificial environment. It is reasonable to conjecture that these cells have intrinsically abnormal splicing patterns and that this led to the nonspecificity observed by some of the PTMs tested. The murine hepatocytes, however, require a more regulated splicing system and most likely do not harbor cancerous mutations that could lead to a disruption in splicing. The other surprising finding from the animal experiments was the signal observed in the “H1s + TKas” group. In this group, the PTM binding domain and target intron are complementary; however, the splicing signal sequences have been abolished from the target gene. This implies that only one of the pre-mRNA species needs to be recognized by the spliceosome in order for trans-splicing to occur, albeit at a much lower efficiency.
The model system used in this study relied on a chimeric target consisting of the HSV1-sr39tk gene fused to the HPV16-E6 intron. The original target gene used, however, was a portion of the HPV16 genome containing the oncoproteins E6 and E7. Because the HPV16 E6 gene is known to contain intronic sequences that are associated with oncogenicity, it was believed that this would be an appropriately relevant target (25). However, once an expression vector was constructed coding for the fusion of the 48 residues of E6* to the N terminus of Renilla luciferase (the predicted trans-splicing product), it was found that this fusion severely diminished the enzymatic activity of the luciferase (data not shown). Because other studies had dealt extensively with creating multimodality reporter genes, it was known that HSV1-sr39tk could be fused to the N terminus of Renilla luciferase without a complete abolition of activity (26). It is unknown exactly why the E6-Rluc fusion caused a reduction in activity, although similar results have been observed with other N-terminal Renilla fusions and thus may be a function of the luciferase enzyme. Alternatively, because the HPV16 E6 protein is known to associate with membranes, it is also possible that the fusion protein was posttranslationally modified such that it was unable to interact with its substrate.
SMaRT imaging has several advantages over more traditional RASON-based imaging. Because the PTMs used in SMaRT can be encoded by plasmid DNA and expressed using the transcriptional machinery of the cell, they can be delivered using any available methods for gene delivery. Recent advances in viral vectors such as gutless adenoviruses and site-specific integrating adeno-associated viruses, as well as nonviral vectors in combination with endosomolytic agents, have made gene delivery more clinically realistic (27–29). Although these gene delivery mechanisms may be suboptimal for particular applications, they present more tractable problems than do the hurdles of nonspecific binding and membrane translocation associated with the delivery of RASONs. The inherent signal amplification of SMaRT also offers advantages over RASONs. After the PTM selectively splices into its target, the newly formed hybrid RNA molecule can serve as a template for multiple translation initiation events, thus producing more than one signal-generating moiety (luciferase enzyme) per recognition event. For RASONs, the number of signal-generating moieties (i.e., radioisotopes) cannot exceed the number of target molecules. Additionally, SMaRT probes can be activated, in that signal is generated only after correctly splicing into the target gene. This is an advantage over RASONs that are always “on” and emit a signal regardless of whether they bind to the target gene. One disadvantage of SMaRT imaging is that it is unable to differentiate between differentially spliced isoforms of the same gene. RASONs, on the other hand, can be designed to target any exon–intron or exon–exon junction.
This study provides clues and important direction toward the applicability of SMaRT for pre-mRNA imaging. By engineering PTMs that incorporate secondary structure and optimizing the amount of PTM relative to target, it seems feasible that these probes can move beyond the current model system and image true endogenous targets. Because endogenous targets are typically expressed at much lower levels than are transiently transfected genes, it might be possible to further reduce the amount of imaging probe introduced into the system by making changes to the reporter protein. Recently published reports detail mutated forms of Renilla luciferase that are brighter and more stable (30). Applying some of these mutations to the PTMs might enable lower amounts of PTM to be delivered while retaining the same level of absolute signal. Conversely, the signal-to-noise ratio could also be increased by selecting highly expressed targets such as albumin, casein, myosin, etc. This, however, is applicable only to a handful of genes and not generalizable to any given gene of interest.
Moreover, the Renilla luciferase gene could be exchanged for another, more robust and clinically relevant, reporter gene. Indeed, one of the strengths of the SMaRT PTM as an imaging probe is the modularity of the reporter protein. Although Renilla luciferase was used exclusively in this study, this choice was made mainly out of convenience. Other reporter genes such as HSV1-sr39tk or transferrin receptor could be used just as easily by substituting the cDNA for these genes in place of luciferase. This generalizability of the SMaRT imaging platform allows it to be adapted for other modalities such as PET or MRI and thus could potentially be scaled for clinical use. Although this reporter gene/reporter probe paradigm is not as desirable for clinical applications as are directly radiolabeled oligonucleotides in terms of simplicity, it still provides a mechanism for the translation of SMaRT imaging into the clinical arena.
CONCLUSION
The ability to image gene expression at the level of pre-mRNA has been demonstrated for a model system. The evidence presented here argues strongly for the possibility of creating a generalizable platform for molecular imaging probes targeting a pre-mRNA of choice. Such molecules would be invaluable tools for researchers and clinicians alike.
Footnotes
-
COPYRIGHT © 2008 by the Society of Nuclear Medicine, Inc.
References
- Received for publication September 26, 2007.
- Accepted for publication December 19, 2007.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}