


Abstract
Labeled annexin V is widely used to detect cell death in vitro and in vivo. Nearly all studies have been done with annexin V derivatized via amine-directed bifunctional agents; it was thought that these molecules retained full bioactivity compared with unmodified protein. We now show that this assumption is incorrect by measuring the affinity of annexin V for cells in vitro by quantitative calcium titration under conditions of low membrane occupancy. Methods: Annexin V was modified with 4 different amine-directed agents: the N-hydroxysuccinimide esters of hydrazinonicotinic acid, mercaptoacetyltriglycine, and biotin; and with fluorescein isothiocyanate. Results: In all cases, the membrane-binding affinity was decreased by derivatization, even at very low average stoichiometries. A statistical model based on the Poisson distribution accurately predicted the observed heterogeneity of derivatization as a function of average derivatization stoichiometry. This model also showed that multiply derivatized forms, which are the ones most likely to have compromised bioactivity, contributed disproportionately to the binding and imaging signals. The in vitro binding assay correctly predicted in vivo uptake in a mouse liver model of apoptosis for all proteins tested. The annexin V-128 protein, labeled at a single specific site at the N terminus, showed twice as much apoptosis-specific liver uptake as did all forms of annexin V derivatized randomly via amino groups. Conclusion: The membrane-binding activity of annexin V is much more sensitive to amine-directed chemical modification than previously realized. New annexin V molecules labeled by site-specific methods will greatly improve sensitivity for detecting cell death in vivo.
Since the initial report in 1998 (1), in vivo imaging of cell death with radiolabeled annexin V has become widely used (2). The method is based on the high-affinity binding of annexin V to membrane phosphatidylserine (PS) (3), which becomes exposed during apoptosis (4). The technique has been widely applied to the detection of apoptosis in animal models of cancer, myocardial infarction, stroke, transplant rejection, and other diseases (2). Imaging of cell death also has been demonstrated in several clinical studies (5–9), but improvements in the sensitivity of detection could greatly extend the clinical applicability of annexin V imaging.
Nearly all work in vivo has been done with annexin V modified with bifunctional agents attached to the protein through its amino groups. Researchers have used a variety of binding assays to verify the in vitro potency of chemically modified annexins and have generally concluded that annexin V molecules retain full membrane-binding activity up to an average derivatization stoichiometry of 2 mol/mol (1,10–17). However, it is challenging to accurately measure the biopotency of annexin V in vitro. As shown by Bazzi and Nelsestuen (18), the binding of annexins to membranes is negatively cooperative with respect to protein. This means that binding measurements made by the usual procedure of titrating cells with labeled protein until the membrane is saturated will overestimate the binding affinity of molecules bound at higher occupancies. Furthermore, in vitro binding measurements are usually made with calcium at 1.8 or 2.5 mmol/L rather than 1.25 mmol/L, the typical value for ionized calcium in vivo. Results obtained at higher calcium concentrations may not accurately predict in vivo binding because the affinity of annexin V binding to cells declines greatly over a calcium range of 2.5–1.25 mmol/L (19).
To overcome these problems, we recently developed a new method for measuring the membrane-binding affinity of annexin V (19). Because the binding reaction is calcium dependent, affinity can be determined by titrating calcium instead of protein. Provided that sensitive fluorescent or radioactive labels are used, this method allows measurements to be obtained under conditions of very low occupancy (<1% of membrane-binding sites occupied at saturation), thus avoiding the confounding effects seen as the membrane becomes more crowded. These conditions also are most likely to be relevant to the in vivo situation in imaging studies, in which only tracer doses of annexin V are used and target membranes are far from being saturated.
Recently, we developed a novel derivative, annexin V-128, which can be radiolabeled by direct chelation of Tc to a unique site at the amino terminus with the sequence Ala-Gly-Gly-Cys-Gly-His (11,20). In the course of comparative studies of the bioactivities of 99mTc-hydrazinonicotinamide (99mTc-HYNIC)-annexin V and 99mTc-mercaptoacetyltriglycine (99mTc-MAG3)-annexin V derivatives (21), we began to notice binding affinities seemingly lower than those obtained with 99mTc-annexin V-128. We therefore investigated whether the random derivatization of annexin V with common amine-directed agents for radioactive, fluorescent, and biotin derivatization would lower membrane-binding affinity more than would annexin V-128 labeled via a single specific site at the N terminus. We also investigated whether lower in vitro membrane-binding affinity would correlate with decreased uptake in apoptotic liver in vivo in a mouse model of cell death.
MATERIALS AND METHODS
Protein Production and Fluorescence Derivatization
Recombinant wild-type human annexin V was produced from plasmid pET12a-PAP1 in Escherichia coli strain BL21(DE3) as described previously (22) and derivatized with fluorescein isothiocyanate (FITC) as described previously (23). Recombinant annexin V-128 was produced from plasmid pJ128 in E. coli strain Tuner(DE3)pLacI as described previously (24) and derivatized with iodoacetamidofluorescein (IAF) as described previously (19). Annexin V-128 has an amino-terminal extension of Ala-Gly-Gly-Cys-Gly-His added to the wild-type sequence. The expression plasmids are available from the authors. FITC-annexin V and IAF-annexin V-128 were purified by anion-exchange chromatography at pH 8.0 with a MonoQ HR5/5 column (GE Healthcare) as described previously (23). The degree of derivatization in each fraction was determined from the ratio of absorbance measurements at 494 nm and protein measurement by the bicinchoninic acid assay (Pierce Chemical Co.).
Cell-Binding Assay
The membrane-binding affinity of radioactive or fluorescent forms of annexin V was determined as described previously by the calcium titration assay with red blood cells (RBC) with exposed PS providing the membrane surface (19,20). A detailed protocol is available from the authors. Because the accuracy of the calculated affinity values depends on the concentrations of the calcium stock solutions, these values were verified by refractometry or by analysis in a clinical laboratory. There was excellent between-laboratory agreement in calculated affinity values for measurements obtained independently at University of Washington and University of Massachusetts for both 99mTc-HYNIC-annexin V and 99mTc-annexin V-128. RBC with exposed PS (4C Plus Cell Control, Normal, product 7547033) were obtained from Beckman Coulter.
Binding data were analyzed according to the following reaction scheme (19): where N is the number of calcium ions participating in complex formation, and K is the equilibrium constant governing the reaction. In principle, the equilibrium constant could be determined by titrating any of the 3 components (protein, membrane, or calcium). In practice, because of the negative cooperativity of binding with respect to protein, titrating calcium at very low ratios of protein to membrane provides the best means for accurately measuring the affinity of individual protein molecules for the membrane (19). Data from calcium titrations were fitted to the following equation by nonlinear least-squares analysis:
where N is the number of calcium ions participating in complex formation, and K is the equilibrium constant governing the reaction. In principle, the equilibrium constant could be determined by titrating any of the 3 components (protein, membrane, or calcium). In practice, because of the negative cooperativity of binding with respect to protein, titrating calcium at very low ratios of protein to membrane provides the best means for accurately measuring the affinity of individual protein molecules for the membrane (19). Data from calcium titrations were fitted to the following equation by nonlinear least-squares analysis: where B is the amount of protein bound at a given calcium concentration, Bmax is the amount bound at a saturating calcium concentration, N is the slope of the calcium titration curve, and EC50 is the midpoint of the calcium titration curve. The negative logarithm of the equilibrium constant of dissociation is calculated with the following formula (19):
where B is the amount of protein bound at a given calcium concentration, Bmax is the amount bound at a saturating calcium concentration, N is the slope of the calcium titration curve, and EC50 is the midpoint of the calcium titration curve. The negative logarithm of the equilibrium constant of dissociation is calculated with the following formula (19): where pK is the binding affinity value and [membrane] is the concentration of annexin V–binding sites, determined from the concentration of cells added to the assay times the number of binding sites per cell. For the assay conditions used here, log10[membrane] = −6.74 (19,20).
where pK is the binding affinity value and [membrane] is the concentration of annexin V–binding sites, determined from the concentration of cells added to the assay times the number of binding sites per cell. For the assay conditions used here, log10[membrane] = −6.74 (19,20).
Radiolabeling
Protocol for HYNIC-Annexin V.
HYNIC-annexin V was produced by conjugation with N-hydroxysuccinimide (NHS)-HYNIC as described previously (1) and stored frozen at −70°C in aliquots of 0.5 mL at 0.5 mg/mL in Tricine (Sigma) (114 mmol/L, pH 6.8). The preparation used in this study had a ratio of HYNIC to annexin V of 0.5 mol/mol, as determined by the method of King et al. (25). Lyophilized tin-Tricine reagent was reconstituted with 1 mL of degassed saline to give a final SnCl2·2H2O concentration of 128 μg/mL and a final Tricine concentration of 200 mmol/L (pH 7.1). Tricine buffer (100 μL, 114 mmol/L, pH 6.6) was added to 99mTc-pertechnetate solution (100 μL, about 740 MBq [20 mCi]). To this mixture was added HYNIC-annexin V solution (200 μL, 100 μg) and then 200 μL of tin-Tricine reagent. After 1 h of incubation at room temperature, the reaction mixture was purified by use of a PD-10 column (GE Healthcare) eluted with phosphate-buffered saline (PBS). The procedure yielded about 93% incorporation of Tc and a specific activity of about 7.4 MBq (200 μCi) per microgram of protein. About 3.7 MBq (100 μCi) of tracer (0.5 μg of protein) were injected per mouse.
Protocol for Biotin-HYNIC-Annexin V.
NHS-HYNIC was first reacted with annexin V at a 5:1 offering ratio in N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid) (HEPES) buffer (pH 7.6) for 60 min at room temperature. To the same reaction mixture, NHS-biotin was added, also at a 5:1 offering ratio, and the conjugation was extended for an additional 60 min. At the end of the 120-min conjugation period, an excess of glycine in HEPES buffer (pH 8.0) was added and reacted for 15 min. The reaction mixture was purified by use of a Sephadex G-25 column (GE Healthcare) equilibrated and eluted with Tricine (114 mmol/L, pH 6.8); the protein fraction was collected, divided into aliquots, and stored at −70°C until use. A vial of biotin-HYNIC-annexin V (50 μg of protein in 200 μL) was thawed. To this was added 25 μL of Tricine buffer (114 mmol/L, pH 6.6), 100 μL (88.8 MBq [2.4 mCi]) of diluted 99mTc-pertechnetate solution (10 μL plus 90 μL of Tricine buffer), and 8 μg of SnCl2·2H2O (4 μL of a 2 mg/mL solution in nitrogen-purged HCl at 0.05 mol/L). The reaction mixture was incubated for 1 h at room temperature in a nitrogen-purged tube and purified by use of a PD-10 column eluted with PBS. The procedure yielded greater than 99% incorporation of Tc and a specific activity of about 1.48 MBq (40 μCi) per microgram of protein. About 4.81 MBq (130 μCi) of tracer (3−4 μg of protein) were injected per mouse.
Protocol for MAG3-Annexin V.
MAG3-annexin V was produced by conjugation of annexin V with S-acetyl-NHS-MAG3 as described previously (21). A vial of frozen MAG3-annexin V (containing 20 μL of 20 μg of MAG3-annexin V) was thawed. To this was added 10 μL of a buffer (pH 9.2) containing sodium tartrate at 50 mg/mL, NaHCO3 at 0.5 mol/L, ammonium acetate at 0.25 mol/L, and ammonium hydroxide at 0.175 mol/L. To this was added 50 μL of 99mTc-pertechnetate in saline (92.5 MBq [2.5 mCi]) and 8 μg of SnCl2·2H2O (4 μL of a 2 mg/mL solution in nitrogen-purged HCl at 0.05 mol/L). The reaction mixture was mixed with a vortex mixer and incubated at room temperature for 1 h in a nitrogen-purged tube. The product was purified by use of a PD-10 column eluted with PBS. The incorporation of Tc was about 75%. About 2.96–3.33 MBq (80–90 μCi) of tracer (1.1–1.3 μg of protein, 2.59 MBq [70 μCi]/μg) were injected into each mouse via a tail vein.
In Vivo Studies
All mice used were adults (6–8 wk old) of the BALB/c strain and obtained from the breeding facility of the Department of Comparative Medicine, Stanford University. Experiments were usually done with groups of 5 mice per condition, but sometimes 4 or 6 mice per group were used because of shortages or excesses of mice available from the vivarium. Hepatic apoptosis was induced by intraperitoneal injection of cycloheximide (26) at a dose of 50 mg/kg and dissolved in 0.5 mL of PBS. All animals were anesthetized with a cocktail of ketamine (Fort Dodge Animal Health) (100 mg/kg intraperitoneally) and xylazine (Butler) (10 mg/kg intraperitoneally) before biodistribution studies. Animals were sacrificed 60 min after injection of the tracer, and a biodistribution assay was performed immediately after sacrifice. Tissue and organ samples were analyzed along with 4 samples of standard activity (1/100 the injected dose) by use of a scintillation well counter at an energy level of 140 keV and with an energy window of ±20 keV. Results are expressed as the average percentage injected dose (%ID).
RESULTS
Theoretic Analysis of Substituent and Radiolabel Distributions
We developed a theoretic model to understand how different degrees of derivatization would affect the distribution of derivatized species and the signal derived from the radiolabel. Recombinant human annexin V has a total of 23 amino groups potentially available for modification: a free amino terminus and 22 lysine residues (27) broadly distributed over the entire 3-dimensional surface of the molecule (28). At the low levels of derivatization typically used (i.e., ≤2 mol per mole of protein) (1,10–17), the chance that any one residue will be modified is low; therefore, the Poisson distribution should provide a suitable mathematic model: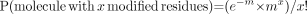 where P is probability and m is the mean number of modified residues in the protein preparation. Similar approaches based on the binomial distribution have long been used in the theoretic analysis of radioiodinated proteins (29).
where P is probability and m is the mean number of modified residues in the protein preparation. Similar approaches based on the binomial distribution have long been used in the theoretic analysis of radioiodinated proteins (29).
Figure 1 shows the results of this model for derivatization levels of up to 2 mol/mol. The lower curve shows the percentage of molecules with greater than 1 derivatization. The upper curve shows how in most cases the multiply derivatized species will contribute disproportionately to the radioactive signal. The upper curve will be applicable in situations in which the protein is modified directly with a carrier-free radiolabel (such as radioiodine or carrier-free 18F-fluorobenzoic acid [18F-FBA]-NHS ester)—molecules with 2 radioactive atoms will contribute twice the signal of molecules with 1 radioactive atom, and so on. It also will be applicable in most situations with HYNIC-derivatized protein, because molecules with 2 HYNIC groups have about twice the likelihood of chelating a Tc atom, those with 3 groups have about 3 times the likelihood, and so on. However, the percentage of radiation signal from each species will tend toward the lower curve when derivatization is performed with a radiolabeled precursor that has a high percentage of nonradioactive carrier. Overall, if multiply derivatized molecules are more likely to have decreased bioactivity, then the imaging signal could be noticeably compromised by even modest levels of derivatization.

Distribution of modified protein species as function of average degree of derivatization. Lower curve (△) was calculated on basis of Poisson distribution as described in text. Upper curve (○) was calculated from distribution of derivatized species multiplied by number of radioisotope atoms per species (see text).
We tested this model by comparing it with published experimental data. In a study by Grierson et al. (13), annexin V was reacted with the NHS ester of 18F-FBA at different offering ratios, and the distribution of modified protein species was analyzed by mass spectrometry. In Figure 2, the experimentally observed values are plotted along with the theoretic results calculated according to the Poisson distribution. There is a virtually perfect fit of the model to the data over the entire range of average derivatization stoichiometry, from 0.03 to 2.06 mol of 18F-FBA per mole of annexin V. Thus, the Poisson distribution can be used to predict the distribution of derivatized species for amine-modified annexin V up to at least 2 mol per mole of protein.

Theoretic versus observed derivatization distribution. Theoretic results for each degree of derivatization were calculated as described in text. Experimental data for annexin V derivatized with 18F-FBA for average stoichiometries of 2.06 (□), 0.89 (○), and 0.37 (△) were from Grierson et al. (13) (reprinted with permission from American Chemical Society). Theoretic data also closely fit experimental results for derivatization ratios of 0.21, 0.11, and 0.03 mol of 18F-FBA per mole of annexin V (not shown in graph).
Comparison of Binding Affinities of Different Tc-Labeled Forms of Annexin V
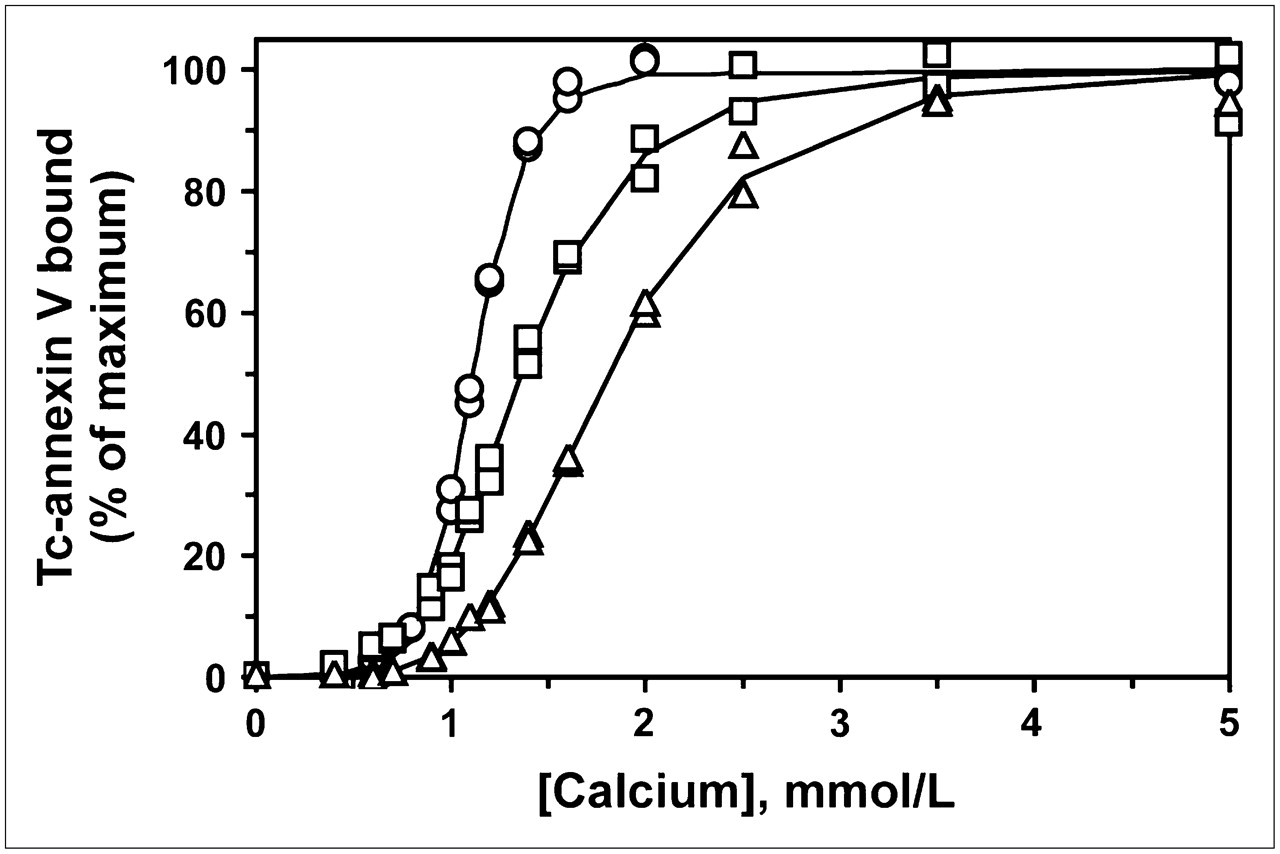
We analyzed the in vitro membrane-binding affinities of various Tc-labeled forms of annexin V. As shown in Figure 3, 99mTc-HYNIC-annexin V, prepared by amine-directed modification, had a lower binding affinity than did 99mTc-annexin V-128, which was labeled site specifically on the first few amino acid residues at the N terminus. Additional modification of 99mTc-HYNIC-annexin V with biotin further decreased its binding affinity (Fig. 3). The same pattern of decreased binding affinity was observed with 99mTc-MAG3-annexin V. Average pK values for all 4 proteins are given in Table 1. Thus, all proteins modified nonspecifically on amino groups through reaction with NHS esters over the pH range from 7.4 to 8.5 showed lower binding affinities than did annexin V-128, which was labeled site specifically at its N terminus.
RBC-calcium titration of 99mTc-annexin V-128 (○), 99mTc-HYNIC-annexin V (□), and biotin-99mTc-HYNIC-annexin V (△). Data for 99mTc-annexin V-128 were from Tait et al. (20), and those for 99mTc-HYNIC-annexin V were from Vanderheyden et al. (21) (reprinted with permission from Elsevier). pK values were calculated from slope (N) and midpoint of titration curve (EC50).
Uptake of 99mTc-Labeled Derivatives in Mouse Liver After Treatment with Cycloheximide*
The titration curves for 99mTc-HYNIC-annexin V and biotin-99mTc-HYNIC-annexin V represent composite values for the many different individual species present, which may have a wide range of affinities. Thus, the calculated pK value is an average value, weighted toward species with the highest probability of Tc labeling. It should be noted that the presence of some fraction of nonradioactive protein does not affect the results because under low-occupancy conditions, the pK value is essentially independent of the added protein concentration.
Comparison of Binding Affinities of Different Fluorescent Forms of Annexin V
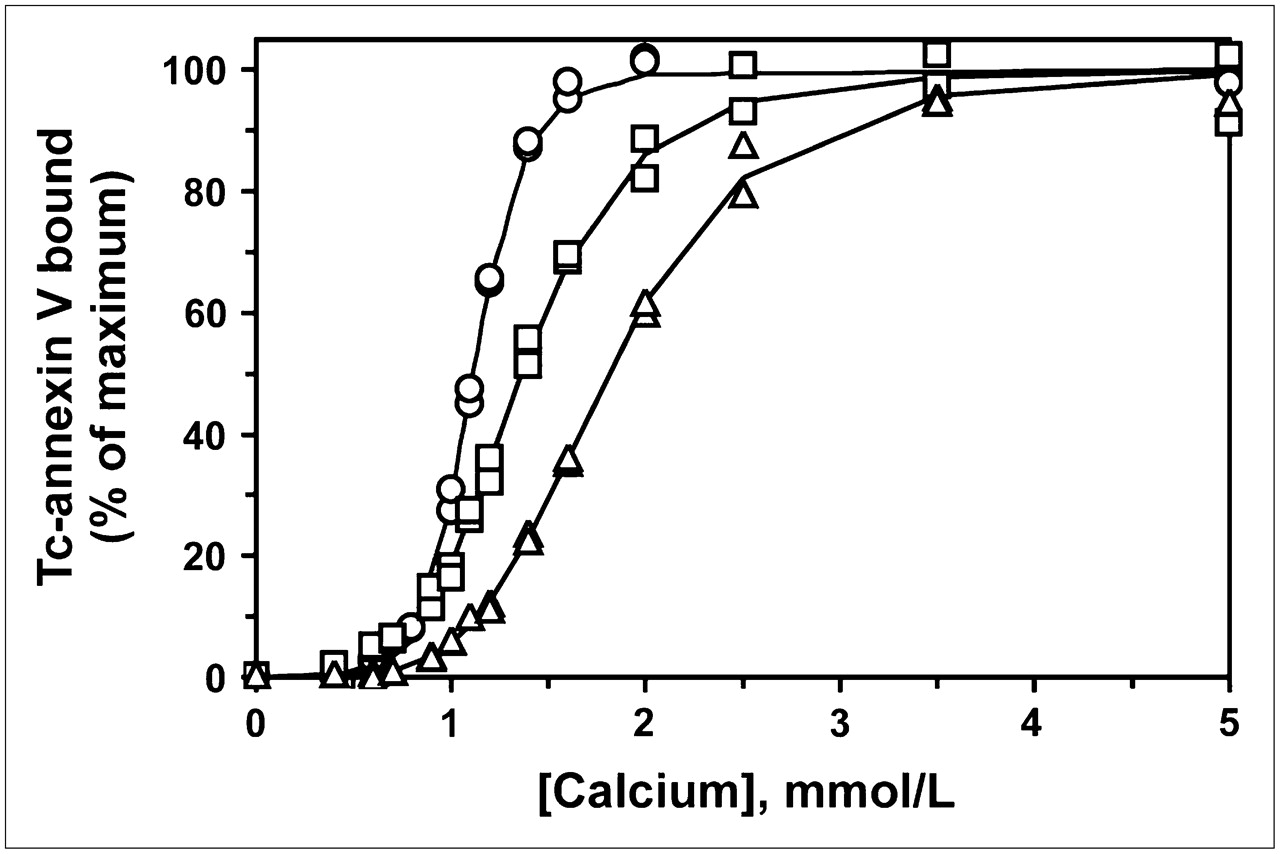
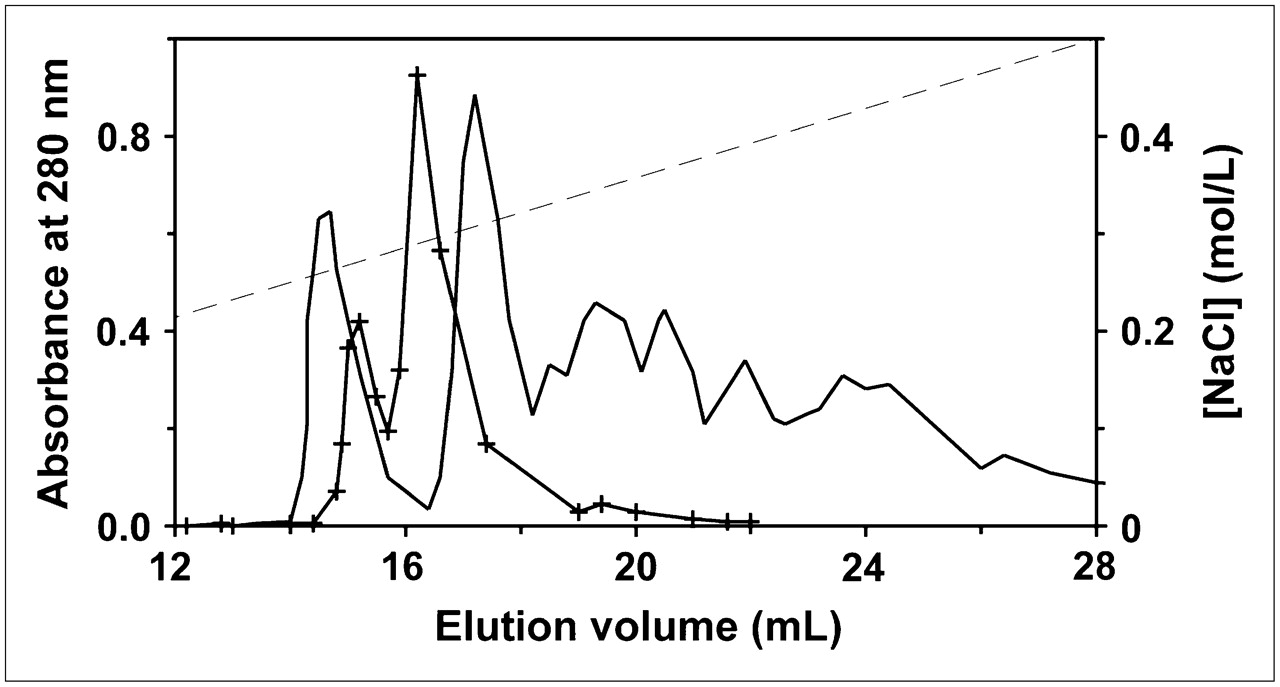
To determine whether these findings would extend to other reactive groups, we prepared annexin V derivatized with different amounts of fluorescein via reaction with FITC at pH 9.0. Because fluorescein modification introduces an additional negative charge near a neutral pH, it is possible to separate the different derivatized forms by anion-exchange chromatography (Fig. 4) (23). As predicted by the theoretic model, the reaction products were complex, consisting of multiple species with 0, 1, or more molecules of FITC per molecule of annexin V. In contrast, annexin V-128 modified with IAF at its single N-terminal cysteine yielded a single fluorescent product, as expected.
Anion-exchange chromatography of IAF-annexin V-128 (solid line with crosses) and FITC-annexin V (solid line) reaction mixtures. First peak in each chromatogram was attributable to underivatized protein; subsequent peaks were all fluorescence labeled. Broken line represents salt gradient.
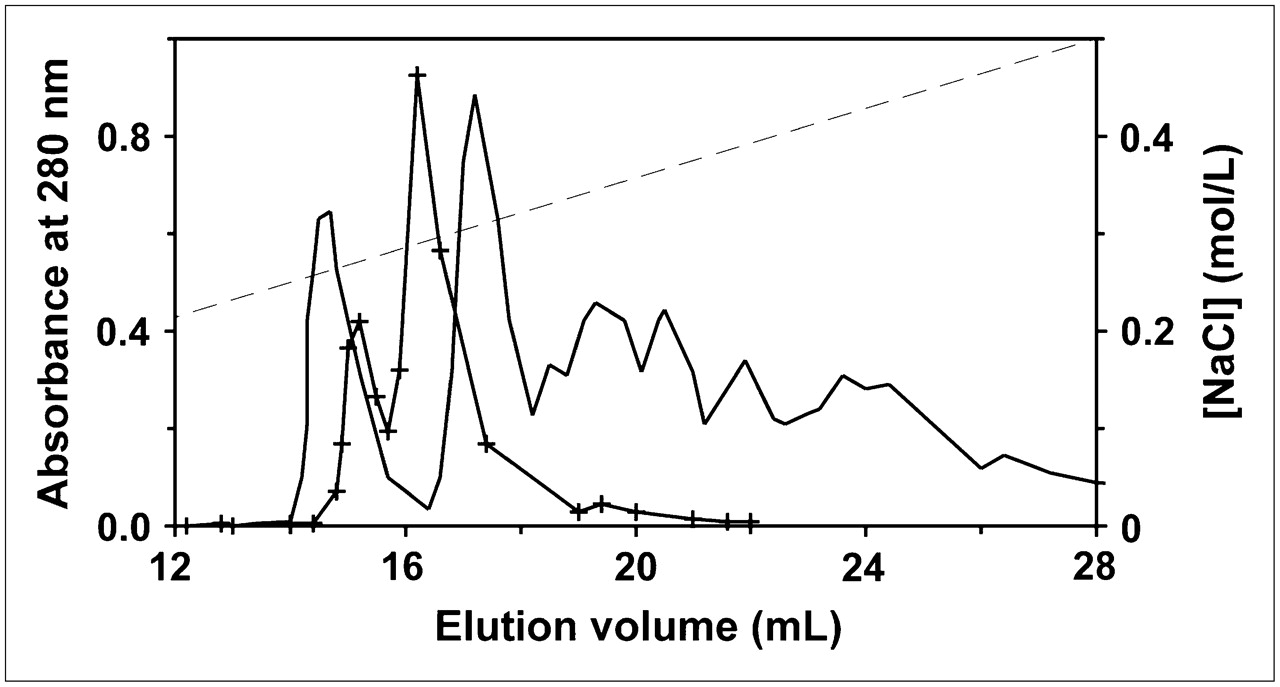
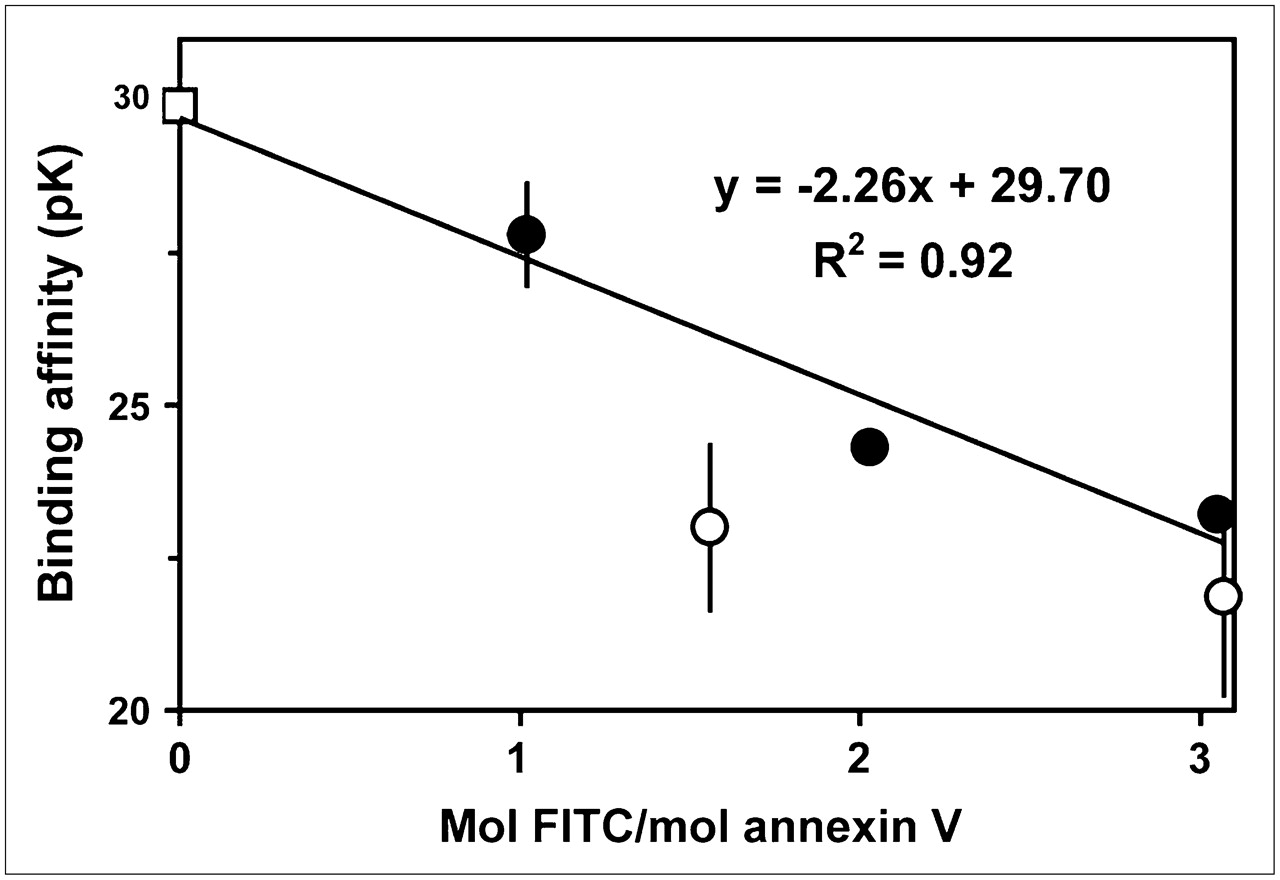
We measured the binding affinities of FITC-annexin V containing different amounts of fluorescein (Fig. 5). It is evident that affinity decreases progressively as more amino groups are modified. In addition, unpurified reaction mixtures, which contain the entire distribution of derivatized species, yield pK values below the curve obtained with purified materials with the same average derivatization stoichiometry. This result is consistent with the expectations of the model, in which multiply derivatized species with lower binding affinities contribute disproportionately to the fluorescence signal. It should be noted that this effect will be less pronounced for fluorescent labels than for radioactive labels, because the fluorescence signal often increases less than linearly with the degree of derivatization because of intramolecular self-quenching on protein molecules containing multiple fluorophores.
Effect of FITC derivatization on membrane-binding affinity of annexin V. After annexin V was reacted with FITC, fractions containing different levels of fluorescein were obtained by anion-exchange chromatography and assayed for activity by calcium titration (•). Unpurified reaction mixtures with indicated average degrees of derivatization also were assayed (○). For comparison, pK value for annexin V-128 derivatized with fluorescein via its N-terminal cysteine also is given (□). Results are means of 2−4 experiments; error bars indicate SDs (when they are larger than symbols).
In Vivo Uptake of Modified Proteins in Apoptotic Tissue
To determine whether amine-directed modification affected binding activity in vivo as well as in vitro, the biodistribution of the labeled proteins was determined by use of a mouse model of liver apoptosis. Proteins were labeled with Tc and then injected into control mice and mice treated with cycloheximide to induce apoptosis of the liver. Liver uptake was determined as the %ID (Table 1). Compared with the 3 amine-modified proteins, 99mTc-annexin V-128 showed both the lowest uptake in control animals and the highest increase in uptake in cycloheximide-treated animals. Overall, 99mTc-annexin V-128 had the best performance, whether measured as the absolute increase in uptake or as the ratio of uptake in treated mice to uptake in control mice.
Correlation of the degree of apoptosis-specific uptake with the pK value showed that decreasing pK values correlated very well with decreasing in vivo uptake (r2 = 0.82) (Fig. 6). For comparison, earlier results (20) are included for 4 annexin V mutants that have a homogeneous reduction in binding affinity. The single-site mutants (annexin V-131 and annexin V-138) have less apoptosis-specific uptake than do the chemically modified annexins with pK values in a similar range. This difference can be explained by the homogeneity of the mutant annexin molecules, with a uniform reduction in binding affinity, whereas the chemically modified annexins are heterogeneous, with some fraction of the radiolabeled protein having an affinity only slightly lower than that of unmodified protein.

Correlation of binding affinity with in vivo uptake of 99mTc-labeled proteins in mouse liver after treatment with cycloheximide. Data are from Table 1. Error bars indicate SEMs.
DISCUSSION
We found that amine-directed chemical modification of annexin V substantially reduces its membrane-binding activity. This was true for modification reactions performed over a wide pH range (7.4–9.0) with 2 different reactive groups (isothiocyanate and NHS) that attached 4 different functional groups (HYNIC, MAG3, biotin, and fluorescein). Thus, the effect is general rather than reagent specific and therefore is likely to occur with most if not all amine-directed reagents that have been used to modify annexin V. Of course, this work does not invalidate earlier studies performed with amine-modified annexin V, which still demonstrated an affinity suitable for the detection of cell death in vivo. However, it may provide an explanation for findings that were seemingly negative—if the signal was insufficient because of either low absolute uptake or an insufficient target-to-background ratio, then relatively small effects may have been difficult to observe.
The new annexin V-128 derivative shows a striking 2-fold improvement in the detection of cell death in vivo over that obtained with standard amine-derivatized forms of annexin V (Table 1). When combined with a lower body background because of the faster clearance of nonspecifically bound tracer (20), this property is likely to result in dramatic improvements in the ability to detect apoptosis in disease states. These results also have important implications for the further development of annexin V derivatives for the detection and targeting of exposed PS in vivo. This is particularly true for clinical studies, in which sensitivity and the target-to-background ratio are at a premium. As a corollary, homogeneous preparations with maximum affinity may yield lower radiation doses than heterogeneous preparations containing a fraction with a lower binding affinity because a lower total dose will be needed to yield the same absolute amount of radioactivity on the target. We recommend that the future development of annexin V–based imaging agents be based on site-specific modifications on regions and residues that have been verified to be uninvolved in regulating binding activity. Several such approaches already have been demonstrated—for example, the addition to the N terminus of annexin V of an oligopeptide sequence that can be used for the direct chelation of Tc as well as the chemical attachment of bifunctional groups (11). Another approach is to attach a polyhistidine sequence to the N terminus for radiolabeling via a 99mTc-tricarbonyl complex (30). Direct iodination of tyrosine residues also has been reported (17,31,32). Other N-terminal epitopes, such as the N-terminal peptide derived from human ribonuclease I, also can be used (33). It is also likely that cysteine residues can be introduced at selected internal sites within the protein sequence, as has been done extensively with annexin B-12 (34).
Alternatively, perhaps chemically conjugated protein still can be used, provided that the 1:1 stoichiometric complex is purified and shown to retain full binding activity. It is highly desirable to remove multiply derivatized molecules, because under most circumstances, they contribute disproportionately to the imaging signal (Fig. 1) and are the molecules most likely to have diminished binding activity. Provided that the purification procedure does not affect the chemistry of the chelation group, this approach would be feasible for the attachment of groups, such as 99mTc-HYNIC or 99mTc-MAG3, for which the nonradioactive precursor can be prepared in large quantities without time constraints. However, it is probably impractical for the attachment of substituents labeled with a positron emitter (such as 18F-FBA) because of their short half-life. It would be especially important to verify the bioactivity of doubly modified proteins, because of the additional load of the second modifying group. The theoretic model shown in Figure 1 provides some guidance in the choice of optimal conditions to maximize the production of singly modified proteins, provided that the assumptions of the model are met (i.e., a large number of potential derivatization sites, similar reactivities of most sites, and low average levels of derivatization).
An important outcome of our recent work, including this study, is a new in vitro bioactivity assay that is able to predict the in vivo behavior of annexin V derivatives (19,20). As far as we know, this is the only in vitro binding assay that has been validated as a predictor of the in vivo uptake of annexin V. We recommend the use of the RBC-calcium titration assay for future assessment of the bioactivity of annexin V derivatives. It is relatively simple to perform, does not require highly specialized equipment, and can be used on any derivative with a detectable label (such as radioactive or fluorescent). When the calculated affinity value falls below about 27, in vivo uptake probably will be compromised. Although the assay does not resolve the individual affinities of different species in a complex mixture, the overall average pK value is still quite predictive of in vivo behavior (Fig. 6; Table 1).
Why was the effect of chemical modification not detected in previous studies? Some previous assays relied on titrating cells to saturation with increasing amounts of labeled protein at a fixed calcium concentration (3,11,14). There are 2 reasons why such assays are likely to be less valid predictors of in vivo behavior. First, the apparent affinity of annexin V for a membrane is strongly influenced by the calcium concentration used in the assay (19). Because the concentration of in vivo ionized calcium is only 1.25 mmol/L, target uptake may be much more sensitive to chemical modifications than it appears to be when in vitro binding assays are performed at higher calcium concentrations. Second, as noted previously, the binding of annexins is negatively cooperative (18), which means that an average affinity constant obtained by titration to saturation with labeled protein will overestimate the binding affinity of molecules binding later. These factors probably explain why 99mTc-HYNIC-annexin V had the same apparent dissociation constant as 99mTc-annexin V-117 (equivalent to annexin V-128) in our earlier study, which relied on protein titration to membrane saturation at a calcium concentration of 2.5 mmol/L (11). Some other binding assays have used phospholipid-coated surfaces (16,17) containing 20% or more PS; in these systems, the affinity of annexin V is so high (23,35,36) that it cannot be accurately measured by titration of protein or phospholipid, making it impossible to detect moderate reductions in binding affinity. Finally, competitive binding assays will underestimate the effect of chemical modification because the underivatized fraction will contribute to the measurement; the results also are not weighted according to the degree of derivatization. All of these factors will make it more difficult to detect mild to moderate reductions in binding affinity or the presence of a heterogeneous population of annexin molecules with a range of binding affinities.
Acknowledgments
This work was supported by National Institutes of Health grants CA-102348, CA-042045, and EB-000898.
Footnotes
-
COPYRIGHT © 2006 by the Society of Nuclear Medicine, Inc.
References
- Received for publication December 14, 2005.
- Accepted for publication March 22, 2006.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Isostere 18F-protein post-translational editing enables dynamic tracking of neurodegeneration biomarkers
- PNAS Plus: From the Cover: Differential fates of biomolecules delivered to target cells via extracellular vesicles
- Time Course of Paclitaxel-Induced Apoptosis in an Experimental Model of Virus-Induced Breast Cancer
- Imaging of Apoptosis
- In Vivo Detection of Apoptosis
- Entropic and Enthalpic Contributions to Annexin V-Membrane Binding: A COMPREHENSIVE QUANTITATIVE MODEL
- Molecular Position of Radiolabels and Its Impact on Functional Integrity of Proteins