


Abstract
We have previously shown that ex vivo counting of 131I-metaiodobenzylguanidine can identify regional reductions in sympathetic norepinephrine uptake in pigs with hibernating myocardium. However, nonneuronal uptake limited relative differences between regions and would preclude accurate assessment with conventional imaging. We therefore hypothesized that the superior specificity of the positron-emitting isotope 11C-hydroxyephedrine (HED) would facilitate the imaging of regional differences, and we designed this study to determine whether altered uptake of norepinephrine by sympathetic nerves in viable, dysfunctional myocardium can be imaged in vivo and to determine the temporal progression and stability of sympathetic dysinnervation in hibernating myocardium. Methods: Pigs (n = 15) were chronically instrumented with a 1.5-mm stenosis of the left anterior descending coronary artery, a procedure that we have previously shown to produce viable chronically dysfunctional myocardium with reduced resting flow, or hibernating myocardium, after 3 mo. Physiologic studies and HED PET were performed 1–5 mo later with the animals in the closed-chest sedated state. One animal with a myocardial infarct was analyzed separately. Results: After 3 mo, anterior hypokinesis developed (wall thickening, 32% ± 4% vs. 60% ± 4%, P < 0.001), with reductions in resting flow (subendocardial flow, 0.81 ± 0.11 vs. 1.20 ± 0.18 mL/min/g, P < 0.05) and a critical reduction in subendocardial flow reserve (subendocardial adenosine flow, 0.53 ± 0.20 vs. 3.96 ± 0.43 mL/min/g, P < 0.001). Extensive defects in HED uptake were found for hibernating myocardium, with regional retention ∼50% lower than that in normally perfused remote myocardium (0.035 ± 0.002 vs. 0.066 ± 0.002 min−1, P < 0.001). Relative HED uptake (left anterior descending coronary artery/remote) was lower in chronically instrumented animals than in control animals (n = 4, P < 0.001) and animals studied 1 mo after instrumentation (n = 2, P < 0.05). The regional reduction in sympathetic nerve function was persistent and unaltered for at least 2 mo after the development of hibernating myocardium. Conclusion: Hibernating myocardium is associated with persistent reductions in regional uptake of norepinephrine by sympathetic nerves. The inhomogeneity in sympathetic innervation in viable dysfunctional myocardium is similar to that occurring after myocardial infarction and may contribute to arrhythmic death in patients with ischemic cardiomyopathy.
Inhomogeneity in myocardial sympathetic innervation has been hypothesized to contribute to the development of sudden cardiac death after transmural myocardial infarction (1). Infarction results in denervation of the scar, and interruption of sympathetic nerves causes denervation in adjacent viable myocardium (2,3). Subsequent reinnervation occurs slowly and is accompanied by nerve sprouting (4), which may result in hyperinnervation and further exacerbate the heterogeneity in sympathetic function (5). The resultant increase in spatial dispersion of ventricular repolarization during sympathetic activation has been hypothesized to lead to lethal ventricular arrhythmias after myocardial infarction (6), and a similar mechanism may underlie the association of reduced norepinephrine-tracer uptake and cardiovascular mortality in patients with ischemic cardiomyopathy (7).
Sympathetic nerves are exquisitely sensitive to ischemia and may become dysfunctional after episodes of myocardial ischemia that do not result in irreversible myocyte injury (8,9). This fact is consistent with clinical studies showing that the area of denervation is larger than that of infarction and more closely correlates with the area at risk of ischemia (10). Furthermore, abnormal norepinephrine-tracer uptake has been described in patients without previous infarction (11). These data suggested to us that sympathetic nerve dysfunction may also contribute to the sudden death associated with hibernating myocardium in the absence of infarction (12). We initiated this line of investigation using the norepinephrine tracer 131I-metaiodobenzylguanidine (MIBG) in chronically instrumented pigs with hibernating myocardium (13), which have a high rate of spontaneous, arrhythmic sudden death (14). Although we found a significant regional reduction in MIBG uptake in hibernating myocardium using ex vivo tissue counting, regional differences in tracer uptake were limited by nonneuronal myocardial uptake and would preclude accurate imaging. Therefore, the present study was conducted with 11C-hydroxyephedrine (HED) and PET (11,15,16) to determine the feasibility of imaging the time course and stability of sympathetic dysinnervation in pigs with hibernating myocardium. Our results demonstrate profound reductions in myocardial HED retention in hibernating myocardium. These defects persisted for at least 2 mo after the development of hibernating myocardium and were much greater in magnitude than those demonstrated by ex vivo counting of MIBG (13).
MATERIALS AND METHODS
All experimental procedures and protocols conformed to institutional guidelines for the care and use of animals in research.
Initial Instrumentation
Studies were conducted on farm-bred pigs that were chronically instrumented to produce hibernating myocardium. The initial instrumentation and experimental protocol have been published in detail (17,18). Briefly, juvenile pigs (8.9 ± 0.5 kg, n = 15) were instrumented with a 1.5-mm Delrin (DuPont) stenosis on the proximal left anterior descending coronary artery (LAD). We have previously shown that after 3 mo, this instrumentation results in viable dysfunctional myocardium with reduced resting flow, consistent with hibernating myocardium (17,18).
Physiologic Study
Physiologic studies to document flow and function in hibernating myocardium were performed on animals that were in the closed-chest, propofol-sedated state (2–5 mg/kg/min intravenously) 118 ± 9 d after initial instrumentation (n = 10). A transducer-tipped catheter (Millar Instruments) was inserted into the left ventricular apex via the brachial artery using a sterile, percutaneous technique (14). Arterial pressure was monitored from the side port of the introducer. Selective coronary angiography was performed with hand injections of iodinated contrast material.
Myocardial function was assessed with transthoracic echocardiography through a right parasternal window with the animals lying on their left side (19,14). From midventricular short-axis images, anatomic M-mode (System 5; GE Healthcare) was used to quantify wall thickness in the LAD-perfused anteroseptum and the normally perfused posterolateral walls. Regional function was assessed by calculation of regional percentage wall thickening (100 × [end-systolic wall thickness – end-diastolic wall thickness]/end-diastolic wall thickness). Fractional shortening (100 × [end-diastolic left ventricular diameter – end-systolic left ventricular diameter]/end-diastolic left ventricular diameter) was used to assess global function.
Resting myocardial perfusion was measured by injecting 2 × 106 fluorescent microspheres into the left ventricle while a reference withdrawal sample was taken from the arterial introducer (17,20). Subsequently, vasodilated flow was determined during adenosine infusion (0.9 mg/kg/min), with phenylephrine coinfused to maintain arterial pressure (17). The catheters were then removed, and the animals were allowed to recover before the PET studies.
PET Protocol
PET was performed on 12 pigs with hibernating myocardium (126 ± 9 d after initial instrumentation). One of these animals was studied serially at 2 and 3 mo after instrumentation. Additional studies were performed on 2 animals after 1 mo to assess the effects of instrumentation on uptake of norepinephrine by myocardial sympathetic nerves, on an animal with nontransmural infarction, and on 4 noninstrumented control animals. The pigs were sedated with a mixture of tiletamine hydrochloride (50 mg/mL) and zolazepam hydrochloride (50 mg/mL) (Telazol; Wyeth Holdings Corp.) and with xylazine (100 mg/mL) (0.022 mL/kg intramuscularly), with supplemental doses (0.011 mL/kg intramuscularly) as needed.
HED was synthesized by direct N-methylation of metaraminol free base with 11C-methyl iodide in dimethyl formamide. The mixture was heated at 100°C for 5 min with semipreparative reverse-phase high-performance liquid chromatography purification (15). Scanning was performed on an ECAT 951/31-R PET camera (Siemens/CTI) with a 10.8-cm axial field of view and a resolution of ∼5.9 mm3 in full width at half maximum. After a 5-min transmission scan with a retractable 68Ga/68Ge rod source had been obtained to confirm correct positioning, a 15-min transmission scan was obtained to correct for attenuation. HED (dose, 703 ± 2 MBq [19.0 ± 0.06 mCi]; nominal specific activity, 11.1 GBq/μmol [300 mCi/μmol]) was injected intravenously over 30 s while heart rate and blood pressure were monitored noninvasively. Imaging began at the time of tracer injection and continued for a total of 60 min, using the following image sequence: 6 × 30 s, 2 × 60 s, 2 × 150 s, 2 × 300 s, 2 × 600 s, and 1 × 1,200 s. All animals tolerated the imaging and recovered uneventfully.
Image Reconstruction
Emission data were corrected for attenuation and reconstructed using filtered backprojection and a Hann filter (cutoff frequency, 0.3 cycles per pixel). A summed HED image from 20 to 60 min after injection was used for generation of polar maps as follows. The images were automatically reoriented into short-axis sections (21). Maximum activity profiles were used to define the 3-dimensional left ventricular shape with combined cylindric and hemispheric (bottle-brush) sampling (22). The resulting set of 496 midmyocardial coordinates was used to generate polar maps, with each sector corresponding to midmyocardial voxels of ∼5 mm3. Regional tracer activity was assessed using a 17-segment model (23). The midanteroseptal, midanterior, apical anterior, and apicoseptal segments were assigned to the LAD perfusion territory, and the midinferior, midinferolateral, basal inferior, and basal inferolateral segments were assigned to the normally perfused remote myocardium. Regional HED uptake was expressed as a percentage of maximal regional uptake on a per-animal basis. An arterial blood time–activity curve was obtained from a user-defined region of interest (2,075 ± 312 voxels in the center of the left ventricular cavity). HED retention (min−1) was calculated by dividing the mean segment activity (kBq/mL) by the integrated arterial activity (kBq/mL/min) derived from the blood pool (24). Partial-volume recovery was assumed to be 100% in the myocardial segments.
Tissue Sampling
After imaging and physiologic studies had been completed, the animals were anesthetized with isoflurane (2%–4%, oxygen balance) and the heart arrested with potassium chloride. The left ventricle was cut into circumferential rings, with a midventricular ring divided into 12 wedges (17,20). Wedges were subdivided into 3 transmural layers (subendocardium, midmyocardium, and subepicardium). Samples were then digested and processed for microsphere flow analysis, as previously described (17,20,19). Adjacent rings were stained with triphenyltetrazolium chloride to exclude infarction (19).
Data Analysis
Data are presented as mean ± SEM, with P < 0.05 considered significant. Values in hibernating and normal remote regions were compared using paired t tests, and differences between animals with hibernating myocardium and noninstrumented controls were compared using unpaired t tests. Differences in segmental activity were compared using a 2-way ANOVA, with the Holm–Sidak test applied for post hoc comparisons.
RESULTS
By 3 mo after instrumentation, LAD occlusion or high-grade stenosis with regional myocardial dysfunction had developed in every animal. All animals were in good health at the time of study. Myocardial necrosis was absent, as determined by triphenyltetrazolium chloride staining, in all but one animal (necrosis involving 8% of the left ventricle), the results for which were excluded from the hibernating group and are presented separately.
At the physiologic study, heart rate averaged 100 ± 7 bpm; systolic pressure, 133 ± 5 mm Hg; and left ventricular end-diastolic pressure, 25 ± 1 mm Hg. Echocardiography demonstrated a marked decrease in LAD-perfused anteroseptal wall thickening, as compared with the remote, normally perfused posterior wall (32% ± 4% vs. 60% ± 4%, P < 0.001), whereas global function was normal (fractional shortening, 27% ± 2%; normal value, >25%) (25). End-diastolic wall thickness was similar in the LAD and remote regions (11.7 ± 0.7 vs. 10.9 ± 0.06 mm, respectively, P = not statistically significant [NS]). Microsphere analysis demonstrated significant reductions in subendocardial (LAD, 0.81 ± 0.11, vs. remote, 1.20 ± 0.18 mL/min/g, P < 0.05) and full-thickness perfusion (LAD, 0.90 ± 0.12, vs. remote, 1.03 ± 0.15 mL/min/g, P < 0.05), consistent with hibernating myocardium. During vasodilation, subendocardial flow reserve was critically impaired, such that flow was unable to increase over resting values (adenosine flow: LAD, 0.53 ± 0.20, vs. remote, 3.96 ± 0.43 mL/min/g, P < 0.001). On a full-thickness basis, relative flow in the LAD region during adenosine vasodilation was only 27% of that in the normally perfused region (LAD, 1.26 ± 0.27, vs. remote, 4.59 ± 0.47 mL/min/g, P < 0.001).
11C-HED Uptake in Hibernating Myocardium
Hemodynamic parameters at the time of PET (heart rate, 84 ± 6 bpm; systolic pressure, 133 ± 4 mm Hg) were unchanged after HED administration (heart rate, 83 ± 6 bpm, P = NS; systolic pressure, 135 ± 5 mm Hg, P = NS). Reconstructed maps from a noninstrumented control animal, a representative animal with hibernating myocardium, and the animal with anterior infarction are shown in Figure 1. In contrast to the control animal, the instrumented animal showed an extensive area of reduced HED uptake in the anteroseptal, anterior, and apical regions consistent with sympathetic dysinnervation in the LAD distribution. An HED defect of similar size and severity was noted in the animal with infarcted myocardium.
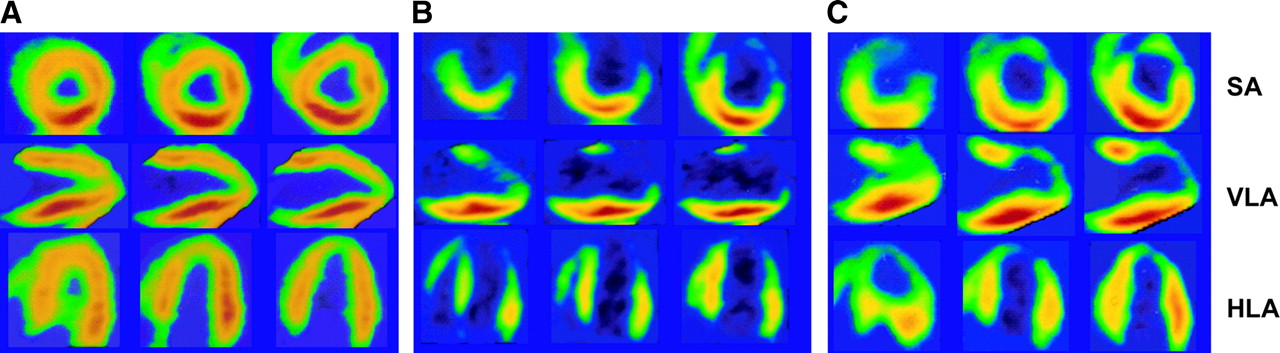
Reconstructed HED images of pigs with normal (A), hibernating (B), or infarcted (C) myocardium. Standard views demonstrate homogeneous HED uptake in a healthy control animal. In contrast, a severe and extensive HED defect involving LAD distribution is seen in the pig with hibernating myocardium, consistent with sympathetic dysinnervation. In the animal with subendocardial infarction in the LAD distribution, the defect in HED uptake is similar to that in hibernating myocardium. HED activity is color coded from red, with the greatest activity, to blue-black, with the least. HLA = horizontal long axis; SA = short axis; VLA = vertical long axis.
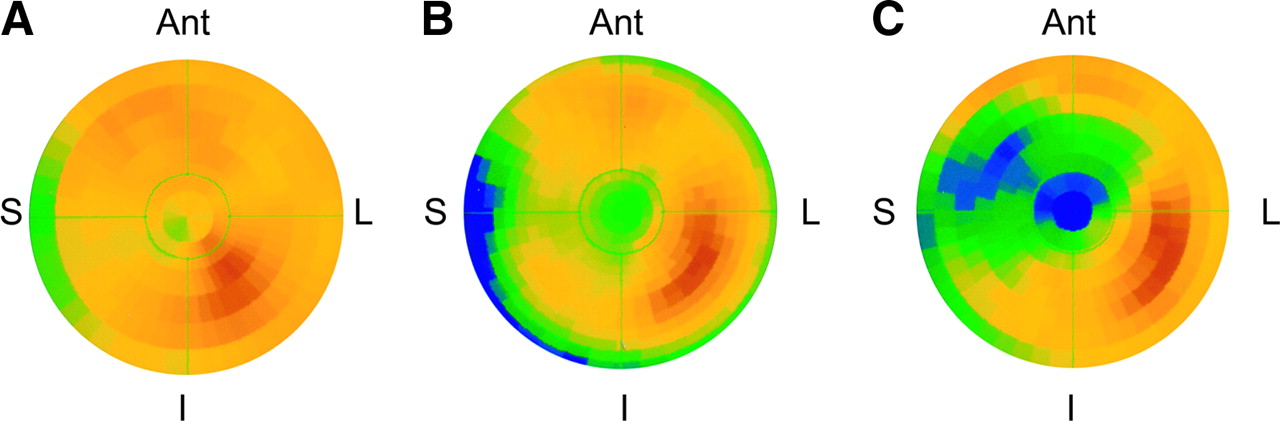
The polar map of the representative animal with hibernating myocardium in Figure 1 is shown in Figure 2, along with the averaged segmental data for all animals with hibernating myocardium. The averaged data show the percentage of maximal uptake in each of the 17 segments of the left ventricle (Fig. 2B). A 2-way ANOVA demonstrated significant differences in regional HED uptake between animals with hibernating myocardium and noninstrumented control animals (P < 0.001). The regions with significant reductions in HED uptake were consistent with the LAD perfusion territory and encompassed 8 of the 17 left ventricular segments (47%; shaded segments in Fig. 2B).

Representative polar map and averaged segmental HED uptake in animals with hibernating myocardium. Polar map from an animal with hibernating myocardium (left) shows a large and severe defect in HED uptake in the anteroseptal, anterior, and apical regions. Schematic polar map (right) shows average HED uptake, as percentage of maximum uptake, in each of 17 segments in animals with hibernating myocardium. Shading indicates regions with significantly lower HED uptake in animals with hibernating myocardium than in noninstrumented control animals.
Figure 3 summarizes the HED retention and percentage of maximal uptake for the LAD and normally perfused remote regions of pigs with hibernating myocardium. HED retention in hibernating myocardium was 52% of values in the corresponding remote regions (0.035 ± 0.002 vs. 0.066 ± 0.002 min−1, P < 0.001, Fig. 3A) and averaged only 49% ± 3% of maximal activity in the heart (Fig. 3B). The relative reduction (LAD/normal) in animals with hybernating myocardium (0.52 ± 0.03) was significantly reduced as compared with control animals (0.79 ± 0.01, P < 0.001) and animals studied 1 mo after instrumentation (0.73 ± 0.10, P < 0.05). Hyperinnervation was not evident in the remote region of animals with hibernating myocardium, as regional retention was similar to that in noninstrumented control animals (0.066 ± 0.002 vs. 0.060 ± 0.008 min−1, P = NS).
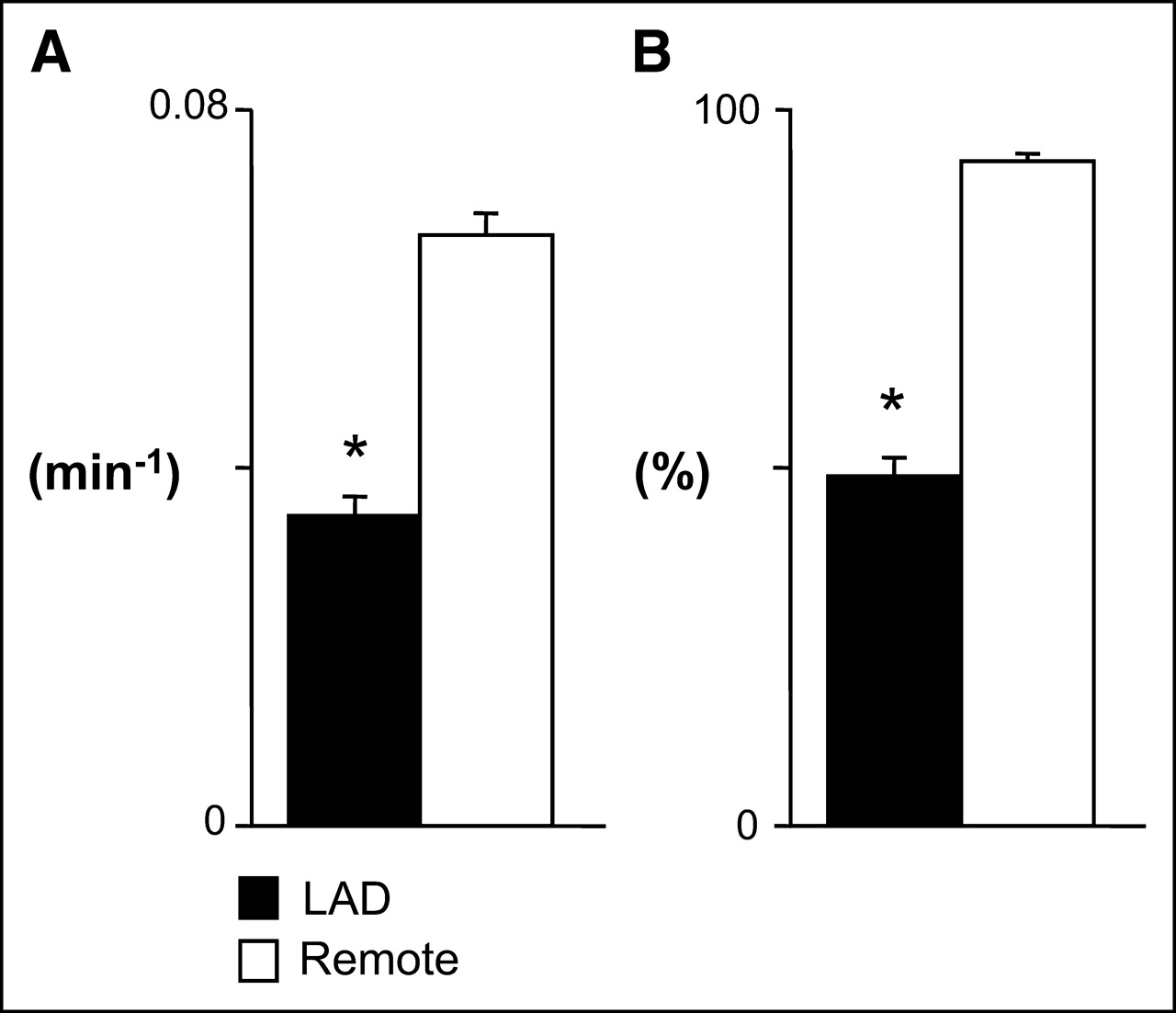
Average regional HED retention in pigs with hibernating myocardium. (A) In comparison with remote normally perfused myocardium, absolute HED retention was reduced by ∼50% in the hibernating LAD region (P < 0.001). (B) Similar relationship was evident when HED uptake was normalized to maximum segmental uptake in each animal. *P < 0.05 vs. remote.
Stability of HED Retention in Hibernating Myocardium
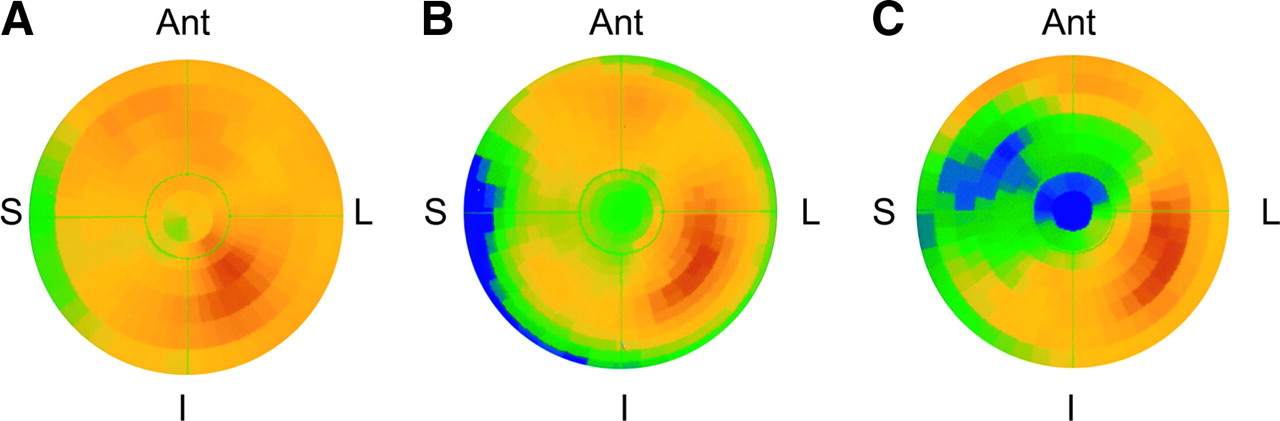
Figure 4 shows polar maps from PET studies performed 1, 2, and 3 mo after instrumentation, illustrating the progression of HED defects during the development of hibernating myocardium. The studies 1 mo after instrumentation were performed to confirm that instrumentation had no effect on uptake of norepinephrine by sympathetic nerves. Consistent with our previous findings with MIBG (13), HED uptake was preserved in the LAD distribution of these animals (Fig. 4). The images 2 and 3 mo after instrumentation were obtained serially in the same animal and show a clear progression in size and severity of the anteroapical HED defect (LAD vs. remote retention: 2 mo, 0.045 vs. 0.060 min−1; 3 mo, 0.030 vs. 0.060 min−1).
Polar maps showing the development of HED defects in hibernating myocardium. (A) Preserved HED uptake at 1 mo confirmed that LAD instrumentation did not affect regional sympathetic nerve function. (B and C) Polar maps serially obtained in the same animal at 2 mo (B) and 3 mo (C) after instrumentation show clear progression from a small, localized defect in apex at 2 mo (relative uptake [LAD/normal], 0.76) to a larger and more severe defect 3 mo after instrumentation (relative uptake, 0.50). Ant = anterior; I = inferior; L = lateral; S = septum.
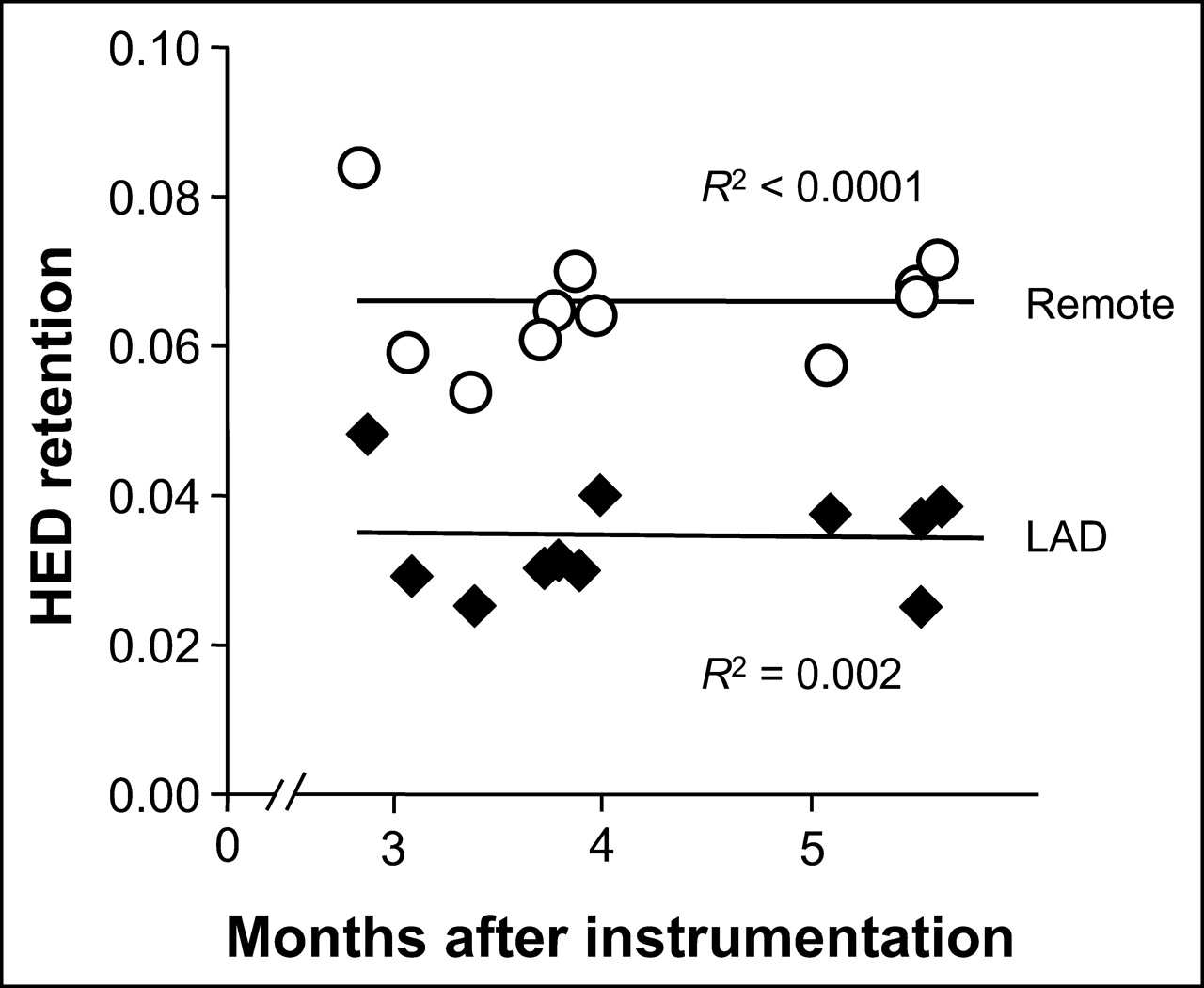
We have demonstrated previously that the physiologic features of hibernating myocardium (regionally reduced function, flow, and flow reserve) remain unchanged between 3 and 5 mo after initial instrumentation in this model (20). Accordingly, using regression analysis as a function of the time from initial instrumentation (range, 85–168 d), we evaluated whether any spontaneous improvement in regional HED retention was evident. As shown in Figure 5, neither the hibernating LAD nor the normally perfused remote region showed differences in absolute HED retention over time (R2 = 0.002 for LAD regions, P = NS; R2 < 0.0001 for remote regions, P = NS). Likewise, the relative retention of HED (LAD/normal) remained essentially constant at 0.52 ± 0.03 (R2 = 0.0001, P = NS). Therefore, these data suggest that in the absence of revascularization, spatial inhomogeneity in sympathetic dysinnervation persists in chronic hibernating myocardium.

Regression analysis of HED retention in animals with hibernating myocardium. Absolute regional HED retention was plotted against months after instrumentation for both hibernating LAD region (♦) and normally perfused remote myocardium (○). In both regions, retention remained essentially constant for at least 2 mo (P = NS for both regressions). Thus, after development of hibernating myocardium, HED retention does not improve or deteriorate over time.
DISCUSSION
This study provided several important new findings. First, hibernating myocardium is associated with profound regional reductions in norepinephrine reuptake as assessed noninvasively by PET with HED. The defects are large, are not the result of initial instrumentation, and approximate the area at risk of ischemia. Once developed, regional reductions in the retention of HED remain constant, with no spontaneous improvement or deterioration over time. Collectively, these findings indicate that repetitive episodes of ischemia leading to the development of hibernating myocardium result in progressive abnormalities in regional sympathetic norepinephrine uptake in the absence of histologic necrosis. The inhomogeneity in norepinephrine uptake may in part explain the high mortality and increased risk of sudden death in patients with hibernating myocardium (12,26,27).
Alterations in Sympathetic Nerve Function in Hibernating Myocardium
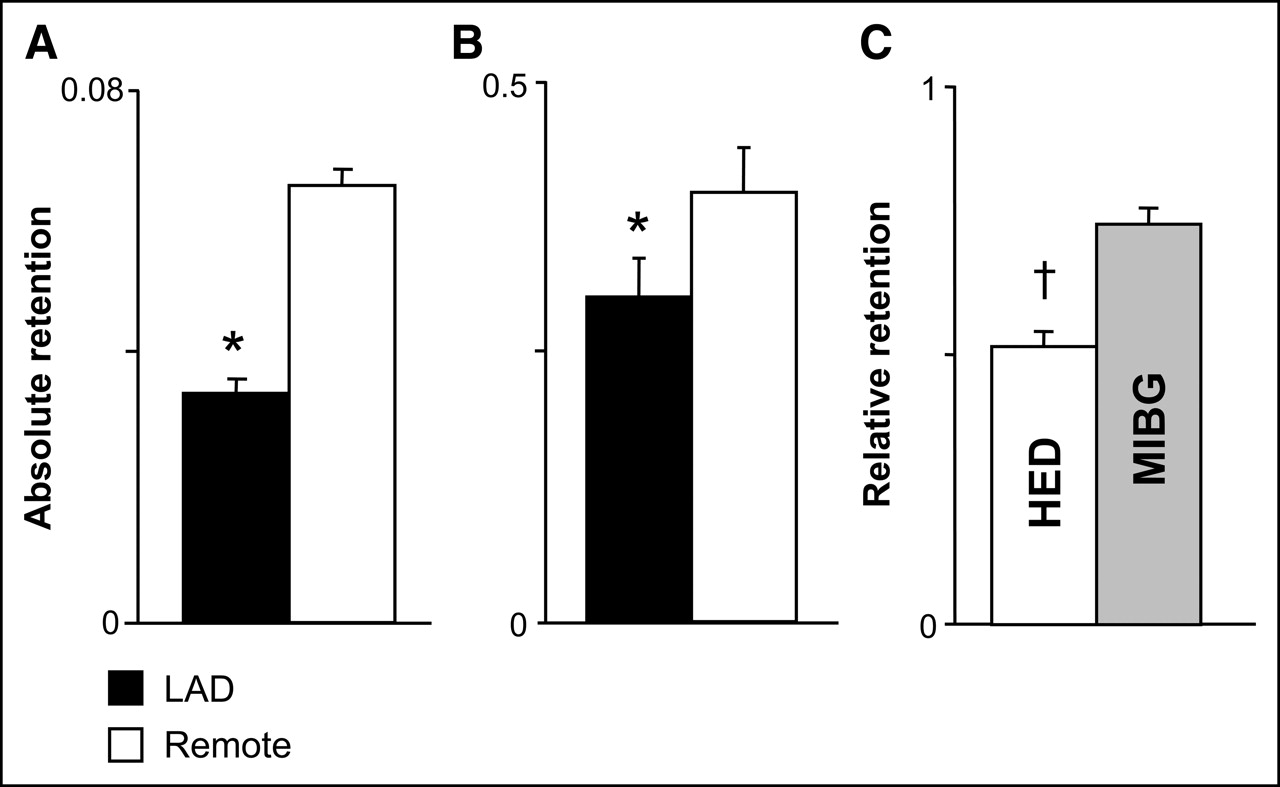
The reduced HED retention in hibernating myocardium confirms our previous findings using ex vivo tissue counting of the photon-emitting norepinephrine analog MIBG. We previously described a 27% reduction in MIBG uptake in hibernating myocardium—a reduction that was most pronounced in the subendocardium (13). Although the spatial resolution of PET is insufficient to assess transmural variations in presynaptic norepinephrine uptake, differences between innervated and dysinnervated myocardium are relatively greater using HED than using MIBG, because of differences in tracer kinetics or nonspecific binding. For example, after complete denervation using topical phenol, MIBG retention remained 46% of normal (28). In contrast, HED retention fell to 30% of normal in the surgically denervated transplanted heart (29). This greater regional difference is consistent with our findings of MIBG and HED retention in pigs with hibernating myocardium (summarized in Fig. 6). The relative retention (LAD/normal) of full-thickness MIBG (nominal specific activity, 30–63 MBq/μmol [800–1,700 μCi/μmol]; CIS-US, Inc.) in hibernating myocardium averaged 0.75 ± 0.03, compared with 0.52 ± 0.03 for HED (P < 0.001). Thus, the improved signal-to-noise ratio of HED imaged with PET suggests that this tracer is preferred for in vivo imaging of regional inhomogeneity in norepinephrine reuptake by myocardial sympathetic nerves.

Norepinephrine analog retention in hibernating myocardium. (A and B) Absolute HED retention (A) in hibernating LAD and remote myocardium from the present study is compared with full-thickness MIBG retention (B) from our previous study (13). (C) Difference in relative retention (LAD/normal) was significantly greater, with HED reflecting improved specificity as a result of less nonspecific uptake of this tracer than of MIBG (gray bar). *P < 0.05 vs. remote. †P < 0.05 vs. MIBG.
Although studies on dogs have demonstrated that coronary artery dissection can be associated with regional sympathetic denervation, LAD instrumentation is not responsible for the HED defects in our study, as supported by 3 separate lines of evidence. First, LAD dissection has no effect on regional tissue catecholamine levels in pigs (30), supporting the notion of no significant interruption of myocardial sympathetic nerves in this species. Second, we have previously shown normal norepinephrine uptake using MIBG in pigs that underwent identical dissection of the proximal LAD (13). Finally, regional HED uptake in pigs studied 1 mo after placement of an LAD stenosis was identical to that in noninstrumented control animals. Thus, defects in HED retention in hibernating myocardium develop as the physiologic significance of the stenosis progresses over time.
Ischemia-Mediated Sympathetic Nerve Dysfunction
Transmural myocardial infarction (28,31) and cardiac transplantation (29) result in myocardial sympathetic denervation. Serial studies using HED have suggested that nerve regeneration requires months or years (29), and in the case of the transplanted heart, reinnervation is inhomogeneous and primarily restricted to the anterior wall of the left ventricle (32). In contrast to denervation, reversible dysfunction or neural stunning can occur after brief, less severe episodes of myocardial ischemia (33). For example, Gutterman et al. (8) and Pettersen et al. (9) demonstrated impaired sympathetic vasoconstriction after 15 min but not 7 min of ischemia in dogs. Not surprisingly, the area of dysinnervation corresponded closely to the area at risk of ischemia (16,33) and was consistent with reports of dysinnervated myocardium in viable tissue surrounding areas of infarction (10). In further support of this observation, Bülow et al. have recently reported, in patients with multivessel coronary disease, regional HED defects that closely correlated to areas with inducible ischemia (11).
Our findings of regionally impaired norepinephrine reuptake in pigs with hibernating myocardium are consistent with these reports and extend them in several important aspects. First, dysinnervation in hibernating myocardium unequivocally occurred in the absence of infarction, as necrosis was excluded by vital staining in every animal. Nevertheless, the extent and severity of the HED defect in the animal with infarction was indistinguishable from the defects in hibernating myocardium (Fig. 1). Second, our data demonstrate that, once developed, the reductions in norepinephrine reuptake in hibernating myocardium remain unchanged for at least 2 mo, with no evidence of improvement or deterioration. This finding is consistent with the stability of the other physiologic features of hibernating myocardium in this model (20).
Although the time course over which sympathetic nerve function normalizes after reversible ischemia is unknown, Dae et al. noted normal MIBG imaging results in 6 of 11 dogs studied 11 ± 3 d after a 2-h LAD occlusion (34). Thus, the alterations in regional MIBG uptake in their model were likely functional rather than due to sympathetic denervation, and a similar mechanism is likely responsible for our findings. However, the correlation of reductions in HED uptake with sympathetic nerve density, nerve sprouting, and regional sympathetic responses to afferent and efferent neural stimulation will require further studies.
Clinical Implications of Sympathetic Innervation and Sudden Death in Chronic Ischemic Heart Disease
Inhomogeneity in sympathetic innervation due to irreversible myocardial injury has been hypothesized to be a substrate responsible for the increased risk of sudden death after myocardial infarction (1). Our results extend the scope of this potential risk factor to viable myocardium by demonstrating abnormal sympathetic innervation in the absence of infarction. The mortality from arrhythmic sudden death in this model of hibernating myocardium with preserved left ventricular function (14) is comparable to the increased mortality associated with medically managed patients with viable dysfunctional myocardium and relatively preserved left ventricular function (27). Some speculate that myocardial sympathetic remodeling from chronic or repetitive ischemia may be a major substrate for arrhythmogenesis. This possibility may be particularly significant in the subset of patients who present with sudden death as their first and only manifestation of coronary artery disease. The implications of inhomogeneous sympathetic innervation may be even more profound in patients with ischemic cardiomyopathy than in pigs, as dysinnervation can occur in both viable and nonviable myocardium (35). As a result, the amount of dysinnervated myocardium will exceed the average 20% of the left ventricle identified as infarcted using 18F-FDG PET, gadolinium MRI (36), or postmortem pathologic examination (37). Thus, viable dysinnervated myocardium may be a more important substrate for sudden death than is scarred and infarcted myocardium and may account for the increased mortality associated with viability in patients with ischemic cardiomyopathy (12). Prospective studies to test this hypothesis by evaluating myocardial viability and sympathetic innervation using HED are currently under way.
CONCLUSION
Hibernating myocardium is associated with persistent reductions in regional uptake of norepinephrine by sympathetic nerves. The inhomogeneity in sympathetic innervation in viable dysfunctional myocardium is similar to that occurring after myocardial infarction and may contribute to arrhythmic death in patients with ischemic cardiomyopathy.
Acknowledgments
The technical assistance of Rebecca Benz, Anne Coe, Paul Galantowicz, Elaine Granica, Deana Gretka, and Amy Johnson was greatly appreciated. This work was supported by grants from the Department of Veterans Affairs; the American Heart Association; the National Heart, Lung and Blood Institute (HL-55324, HL-61610, and HL-76252); the Albert and Elizabeth Rekate Fund; the Mae Stone Goode Trust; the Thomas J. Frawley Research Fellowship; and the John R. Oishei Foundation.
Footnotes
Received Jan. 26, 2005; revision accepted Mar. 29, 2005.
For correspondence or reprints contact: James A. Fallavollita, MD, Biomedical Research Building, Room 361, Department of Medicine/Cardiology, SUNY at Buffalo, 3435 Main St., Buffalo, NY 14214.
E-mail: jaf7{at}buffalo.edu
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Nuclear Imaging of the Cardiac Sympathetic Nervous System: A Disease-Specific Interpretation in Heart Failure
- Structural and Physiological Imaging to Predict the Risk of Lethal Ventricular Arrhythmias and Sudden Death
- 123I-MIBG SPECT for Evaluation of Patients with Heart Failure
- Risk Stratification for Sudden Cardiac Death: A Plan for the Future
- Altered Cardiac Innervation Predisposes to Ventricular Arrhythmia: Targeted Positron Emission Tomography Identifies Risk in Ischemic Cardiomyopathy
- Regional Myocardial Sympathetic Denervation Predicts the Risk of Sudden Cardiac Arrest in Ischemic Cardiomyopathy
- Recent Insights Into the Role of the Autonomic Nervous System in the Creation of Substrate for Atrial Fibrillation: Implications for Therapies Targeting the Atrial Autonomic Nervous System
- Tracing Cardiac Metabolism In Vivo: One Substrate at a Time
- 123I-mIBG Scintigraphy to Predict Inducibility of Ventricular Arrhythmias on Cardiac Electrophysiology Testing: A Prospective Multicenter Pilot Study
- Myocardial Sympathetic Innervation in Patients with Symptomatic Coronary Artery Disease: Follow-up After 1 Year with Neurostimulation
- Abnormal Sympathetic Innervation of Viable Myocardium and the Substrate of Ventricular Tachycardia After Myocardial Infarction
- Regional Desensitization of {beta}-Adrenergic Receptor Signaling in Swine With Chronic Hibernating Myocardium
- When the Heart Loses Its Nerves...and How Nuclear Imaging Helps to Understand