


Abstract
Bombesin (BN), a 14-amino-acid peptide, shows high affinity for the human gastrin-releasing peptide receptor (GRP-r), which is overexpressed on several types of cancer, including prostate, breast, gastrointestinal, and small cell lung cancer. Thus, radiolabeled BN or BN analogs may prove to be specific tracers for diagnostic and therapeutic targeting of GRP-r–positive tumors in nuclear medicine. This study evaluated a novel BN analog labeled with the positron emitter 68Ga for receptor imaging with PET. Methods: DOTA-PEG2-[d-Tyr6,β-Ala11,Thi13,Nle14] BN(6–14) amide (BZH3) (DOTA is 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid; PEG is ethyleneglycol (2-aminoethyl)carboxymethyl ether) was synthetized using the Fmoc strategy and radiolabeled with either 67Ga or 177Lu for in vitro and biodistribution experiments. 68Ga for PET was obtained from a 68Ge/68Ga generator. In vitro binding, internalization, and efflux were determined using the pancreatic tumor cell line AR42J. Biodistribution of the peptide as a function of time and dose was studied in AR42J tumor-bearing mice. Results: In vitro assays demonstrated a high affinity of 67Ga-BZH3 (dissociation constant = 0.46 nmol/L), a rapid internalization (70% of total cell-associated activity was endocytosed after a 15-min incubation), and an intracellular retention half-life (t1/2) of the 67Ga activity of 16.5 ± 2.4 h. Biodistribution indicated a dose-dependent uptake in the tumor and a prolonged tumor residence time (t1/2 ∼ 16 h). Clearance from GRP-r–negative tissues was fast, resulting in high tumor-to-tissue ratios as early as 1 h after injection. Replacing 67Ga by 177Lu, a therapeutic radionuclide, for peptide labeling resulted in a slightly reduced (∼20%) tumor uptake and tumor residence time of 177Lu-BZH3. In contrast, 177Lu decline in the pancreas was significantly accelerated by a factor of ∼3 compared with that of 67Ga. PET of mice with 68Ga-BZH3 clearly delineated tumors in the mediastinal area. Conclusion: The promising in vivo data of 68Ga-BZH3 indicate its potential for an improved localization of GRP-r–positive tumors and also suggest its application in patients. PET may also be favorably used for GRP-r density determination, a prerequisite for therapeutic applications.
Bombesin (BN) is an amphibian neuropeptide of 14 amino acids that shows—just as its mammalian homolog gastrin-releasing peptide (GRP)—high affinity for the human GRP receptor (GRP-r). Aside from its physiologic actions on the central nervous system (1) and the release of gastrointestinal hormones (2), BN acts as an autocrine growth stimulator in a variety of human neoplasms (3–5). Autoradiographic examination of malignant tissues using radioiodine-labeled BN or BN analogs demonstrated a high GRP-r expression in prostate, gastrointestinal, breast, and small cell lung cancer specimens compared with the surrounding normal tissues (6–12), suggesting the potential of GRP-r for a specific tumor targeting. Consequently, various radiolabeled BN analogs have been investigated and proposed for use in nuclear medicine (13–20). In particular, 90Y, 188Re, and 177Lu have been used to radiolabel BN analogs for potential radiotherapy applications, whereas 99mTc and 111In labeling has been used for γ-camera imaging of GRP-r–positive tumors.
PET is the most efficient imaging method in nuclear medicine because of its option of an absolute activity determination, its better contrast resolution, and its higher detection efficiency compared with conventional γ-cameras. Thus, we have focused on the labeling of BN analogs with the short-lived positron emitter 68Ga (half-life [t1/2] = 68 min; β+, 88%), which is obtained from a 68Ge/68Ga radionuclide generator. In the present study, we evaluated a novel BN analog (BZH3) derivatized with the macrocyclic chelator 1,4,7,10-tetraazacyclododecane-N,N′,N″,N‴-tetraacetic acid (DOTA). This ligand forms M3+ metal chelates of high in vivo stability and can be labeled with 68/67Ga, 111In, 90Y, and 177Lu and, thus, is suitable for diagnostic and therapeutic applications as well.
MATERIALS AND METHODS
Synthesis and Radiolabeling of BZH3
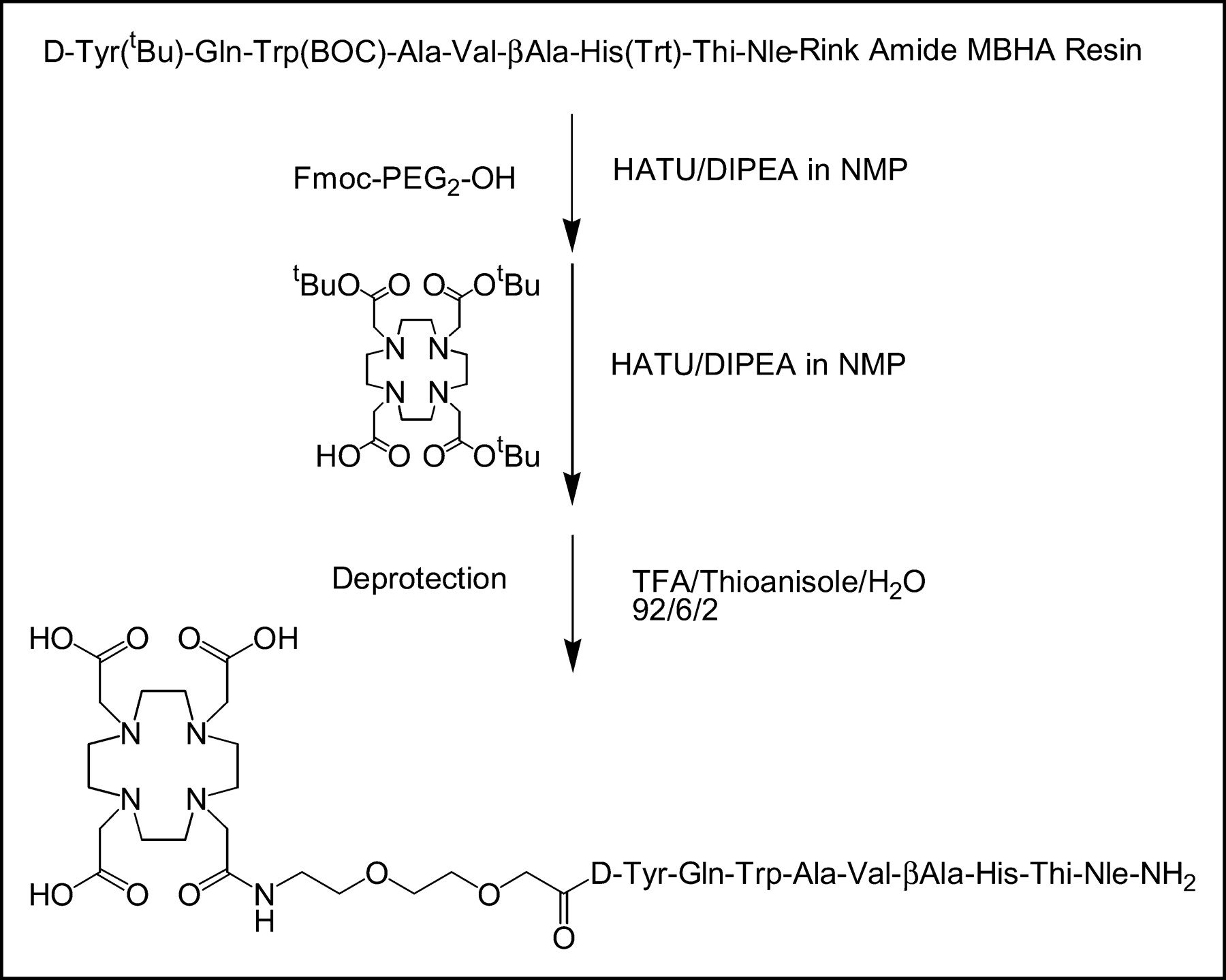
All chemicals were purchased from commercial sources and used without further purification. DOTA-Tris (tBu ester) was obtained from Macrocyclics. Rink amide MBHA (4-methylbenzhydrylamine) resin and all Fmoc-protected amino acids were available from Novabiochem and Neosystem, respectively. Peptide synthesis was performed on a semiautomatic peptide synthesizer (Rink Combichem Technologies) according to general Fmoc chemistry. The peptide was assembled on a Rink amide MBHA resin. Trityl (Trt) and tBu were used as protecting groups of His and d-Tyr, respectively, and Boc was used for Trp. The spacer (Fmoc-PEG2-OH [PEG is ethyleneglycol (2-aminoethyl)carboxymethyl ether]) was coupled to the chelator DOTA-Tris (tBu ester) as described previously (21). The peptide chelator conjugate was cleaved from the resin and deprotected by incubation with trifluoroacetic acid (TFA)/thioanisole/water, 92:6:2 for 4–6 h at room temperature, and precipitated in isopropyl ether/petroleum ether (1:1). The crude peptide-chelator conjugate was purified by preparative high-performance liquid chromatography (HPLC) on a Nucleosil 100-5 C18 column (Macherey–Nagel) at a flow rate of 15 mL/min (eluents: A = 0.1% TFA in water and B = acetonitrile; nonlinear gradient: 0 min, 70% A; 10 min, 50% A). Mass spectometry (MS-MALDI) was used to characterize the composition of the conjugate.
68Ga for PET was obtained from a 68Ge/68Ga generator, which consists of a column containing a self-made phenolic ion-exchanger loaded with 68Ge and coupled in series with a small-sized anion-exchanger column (AG 1×8 Cl−, mesh 200–400; Bio-Rad) to concentrate 68Ga during elution (22). This generator provides 68Ga with an average yield of ∼60% for >1.5 y. For peptide labeling, the eluate containing ∼0.5 GBq of 68Ga in 0.2 mL 0.5 mol/L HCl was evaporated to dryness and redissolved in 0.2 mL 0.1 mol/L acetate buffer (pH 4.8). After addition of 5 μL 1 mmol/L aqueous solution of BZH3, the mixture was kept for 10 min at 90°C. Uncomplexed 68Ga was separated by adsorption onto a C18-coated silica gel cartridge (Sep-Pak; Waters Corp.) that was equilibrated with 0.1 mol/L acetate buffer (pH 6.2), whereas 68Ga-BZH3 could be eluted with 1.5 mL ethanol. After evaporation of the organic solvent, the compound was redissolved in 0.01 mol/L phosphate-buffered saline ([PBS] pH 7.) containing 0.5 mg/mL human serum albumin. The preparations were checked for bound and free 68Ga by paper chromatography using Whatman no. 1 and a mixture of methanol and 0.01 mol/L acetate buffer (pH 6.2) at a ratio of 55:45. 68Ge contamination of the 68Ga-BZH3 preparations was determined by γ-counting after a waiting period of ≥30 h, which ensures complete 68Ga decay. 67GaCl3 and 177LuCl3 used for in vitro and biodistribution experiments were obtained in dilute HCl from Mallinckrodt and Perkin Elmer, respectively. All labeling steps with these radionuclides were identical to that of 68Ga. The radiochemical purity of 67Ga-BZH3 was analyzed by reversed-phase HPLC using a Nucleosil 120 C18 column (Macherey–Nagel) at a flow rate of 0.75 mL/min (eluents: A = 0.1% TFA in water; B = acetonitrile; nonlinear gradient: 0 min, 80% A; 20 min, 50% A).
Cell Lines
For all in vivo and in vitro experiments, the AR42J cell line, derived from a rat exocrine pancreas tumor, was used. Cells were obtained from the European Collection of Cell Cultures and were grown in RPMI 1640 medium supplemented with 2 mmol/L glutamine and 10% fetal calf serum. Adherent cells were dislodged with trypsin/ethylenediaminetetraacetic acid (0.02%:0.05%, w/v) and allowed to recover for 2 h. Nonspecific binding of 67Ga-BZH3 was tested with the GRP-r–negative human breast carcinoma cell line AR-1, obtained from the Department of Gynecological Oncology (University Hospital Heidelberg, Heidelberg, Germany).
Receptor Binding
The equilibrium binding constant, Ka, of 67Ga-BZH3 to the GRP-rs of AR42J cells was determined using a fixed number of 4 × 105 cells in 150 μL RPMI 1640 medium and increasing amounts, 0.033–1.056 pmol (0.055–1.76 ng), of the peptide. Cells were placed in a U-shaped 96-well plate and agitated on a gyratory shaker for 1 h in an incubator (37°C, 5% CO2). Cells were washed 3 times with ice-cold RPMI 1640 medium and pellets were counted in a γ-counter. A least-squares fit from a Scatchard plot (bound activity/free activity [B/F] vs. bound activity [B]) results in a straight line, the slope of which indicates -Ka. The intercept of B/F with B represents the maximum concentration of peptide (Bmax) bound to the GRP-rs of cells at infinite peptide excess.
Internalization
Internalization of 67Ga-BZH3 was studied at low and high peptide-to-receptor ratios. Twenty-seven million cells in 8.1 mL medium were incubated (37°C, 5% CO2) with either 0.45 or 45 pmol (0.75 or 75 ng) of the peptide. During incubation, cells were kept in suspension by gyratory shaking. After 15, 30, 60, and 120 min of incubation, six 300-μL samples, containing 1 × 106 cells and 0.0166 or 1.66 pmol of peptide, were taken, mixed with 700 μL ice-cold RPMI 1640 medium in a conical tube, and centrifuged (3 min, 500g). Three samples were washed twice with 1 mL ice-cold RPMI 1640 medium, and cell pellets were counted in a γ-counter. Radioactivity in the cell pellets represents surface bound together with internalized peptide. The remaining 3 samples were further incubated (5 min, 22°C) with 1 mL of an acidic buffer (pH 4.3) containing 0.15 mol/L NaCl, 0.02 mol/L NaOAc, and 2 mg/mL human serum albumin to remove surface-bound peptide (23). Subsequently, cells were washed twice with the acidic buffer. Radioactivity in the cell pellet was assumed to represent internalized peptide
Efflux
Since an acid treatment of cells might unfavorably affect cell metabolism (23), we used an excess of unlabeled BZH3 to remove surface-bound radioactive peptide before efflux determination, which additionally blocks rebinding of internalized radioactivity released from the cells during incubation. Sixteen million cells in 4.8 mL RPMI 1640 medium were preloaded with 10 pmol of either 67Ga- or 177Lu-labeled BZH3 for 1 h at 37°C, 5% CO2. Cells were washed 3 times and then resuspended in 4.8 mL medium; two 300-μL samples were taken as a control for total cell-associated activity (cell surface bound and internalized). Subsequently, 50 μL of 1 mmol/L unlabeled BZH3 were added to the remaining incubation mixture. Ten, 20, 30, 60, 90, and 120 min after the addition of unlabeled peptide, two 303-μL samples (1 × 106 cells) were taken, diluted with 700 μL ice-cold RPMI 1640 medium, and centrifuged. Pellets were washed twice with cold medium and counted for radioactivity.
Receptor Density
Since the receptor density of AR42J cells might decrease with increasing passages, accompanied by a reduction in cellular size and tumorigenic activity, the binding capacity of cells was tested before each tumor inoculation. One million cells in 150 μL RPMI 1640 medium were incubated with 1.5 pmol of 67Ga-BZH3, kept in suspension for 1 h at 37°C, 5% CO2, washed 3 times with cold medium, and counted for radioactivity.
Biodistribution
Animal experiments were performed in accordance with the German laws for the protection of animals. Female Swiss CD1 nu/nu mice (8-wk old; Iffa Credo) had subcutaneous inoculation of 5 × 106 AR42J tumor cells (in 0.2 mL of RPMI 1640 medium) into the right thoracic wall near the shoulder. Eleven to 13 d later, tumors that weighed 100–600 mg were selected for the experiments. Mice received a tail vein injection of 0.2 mL 0.01 mol/L PBS containing 100 μg human serum albumin and the labeled compounds under investigation. Animals were anesthetized with ether, bled from the retroorbital plexus, and killed by cervical dislocation at the time points indicated. Organs were removed, weighed, and counted for radioactivity with a high-resolution germanium detector. For evaluation of a dose dependence of the tumor uptake, mice were injected with 5, 15, 45, or 135 pmol of 67Ga-BZH3 and killed 1 h after injection. Biokinetics were studied in animals receiving a 15-pmol dose of either 67Ga- or 177Lu-labeled BZH3. Mice were killed 1, 2, 4, and 24 h after injection of 67Ga-BZH3 and 1 and 24 h after injection of 177Lu-BZH3. GRP-r–blocking studies were performed using AR42J tumor-bearing male Lewis rats (body weight, 160–200 g; Iffa Credo) as described previously (24). Briefly, 2 groups of rats, each comprising 4 animals, were injected with either 60 pmol (0.1 μg) 177Lu-BZH3 or a mixture containing 0.1 μg 177Lu- BZH3 and 50 μg of unlabeled BZH3. Rats were killed 4 h after injection. Pancreas and tumor were dissected and counted for 177Lu activity.
PET
AR42J tumor-bearing mice were injected with 15 pmol (0.49 MBq) 68Ga-BZH3, sacrificed 1 h after injection, and imaged with a 30-min emission scan and a 10-min transmission scan on an ECAT EXACT HR+ scanner (Siemens/CTI). Images were taken in the 3-dimensional (3D) mode and reconstructed iteratively with a fully 3D algorithm from a 256 × 256 matrix for viewing transaxial, coronal, and sagittal slices of 0.57-mm thickness (25). Pixel size was 1.14 mm and transaxial resolution obtained was 2.8 mm. Subsequently, tumors were removed and counted for 68Ga activity.
RESULTS
Synthesis and Radiolabeling
BZH3 (Fig. 1) was synthesized using the Fmoc strategy affording an overall yield of 30% based on the removal of the first Fmoc group; the purity analyzed by HPLC was ≥97%. MS-MALDI confirmed the calculated exact mass of 1,669.79: m/z (%): 1,670.0 (100, [M+H]+); 1,692.0 (19, [M+Na]+); 1,708.9 (10, [M+K]+). Labeling of BZH3 with M3+ radionuclides took 60 min. Starting with 0.5 GBq (13.5 mCi) of 68Ga, specific activities of 37–44 MBq/nmol (1–1.2 mCi/nmol) of peptide were obtained and preparations contained <3 ppm 68Ge contamination. Labeling with commercially available 67Ga and 177Lu resulted in 20–22 and 35–40 MBq/nmol, respectively. The radiochemical purity of 67Ga-BZH3, as assessed by reversed-phase HPLC, showed a single product with a slightly reduced retention time of 12.2 min versus 12.6 min for the unlabeled BZH3. Heating to 90°C during the labeling procedure caused no detectable degradation of the peptide, a finding that has also been reported by Breeman et al. (14). Thus, subsequent preparations were only checked by paper chromatography for the nonchelated fraction of the radionuclide that remains at the beginning. Typically, ≥98% of the activity migrated with an Rf of 0.6–0.7 corresponding to the labeled peptide.

Synthesis of DOTA-PEG2-[d-Tyr6,βAla11,Thi13,Nle14] BN(6–14) amide (BZH3). Thi = 3-(2-thienyl)-l-alanine; MBHA = 4-methylbenzhydrylamine; HATU = O-(7-azabenzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; DIPEA = diisopropylethylamine; NMP = N-methylpyrrolidone.
Receptor Binding
The saturation binding curve of 67Ga-BZH3 to GRP-r of AR42J cells and its Scatchard transformation are shown in Figure 2. From the Scatchard plot, a high receptor affinity Ka of 2.19 × 109 L/mol was calculated, which corresponds to a dissociation constant Kd of 0.46 nmol/L. Determination of Bmax resulted in 9.7 × 104 receptors per cell. Nonspecific binding of the peptide was tested with the GRP-r–negative breast carcinoma cell line AR-1 and amounted to <3.5%.

Saturation curve (A) and Scatchard transformation (B) of binding of increasing amounts of 67Ga-BZH3 to 4 × 105 AR42J cells. Cells were incubated for 1 h. Data are means of triplicate samples and are corrected for nonspecific binding (≤3.5%).
Internalization
Internalization of 67Ga-BZH3 by AR42J cells at 2 different concentrations was followed for 2 h (Fig. 3). The fraction of internalized activity compared with total cell-associated activity (cell surface bound + internalized) was nearly identical for both the low and the high concentration and amounted to 71%–75% after a 15- and 30-min incubation and 85%–88% after a 1- and 2-h incubation. The kinetics of internalization during the first hour of incubation also were very similar. In contrast, using the low dose and a 2-h incubation, which reduced the free peptide concentration in the medium to ≤0.02 nmol/L, the internalization rate drastically decreased.

Internalization of 67Ga-BZH3 by 1 × 106 AR42J cells in 300 μL medium containing either 0.0166 pmol (low dose, dashed line) or 1.66 pmol (high dose, solid line) of peptide. +, Total activity (cell surface bound + internalized); •, internalized activity. Note different scaling of the y-axis. Data are means of triplicate samples.
Efflux
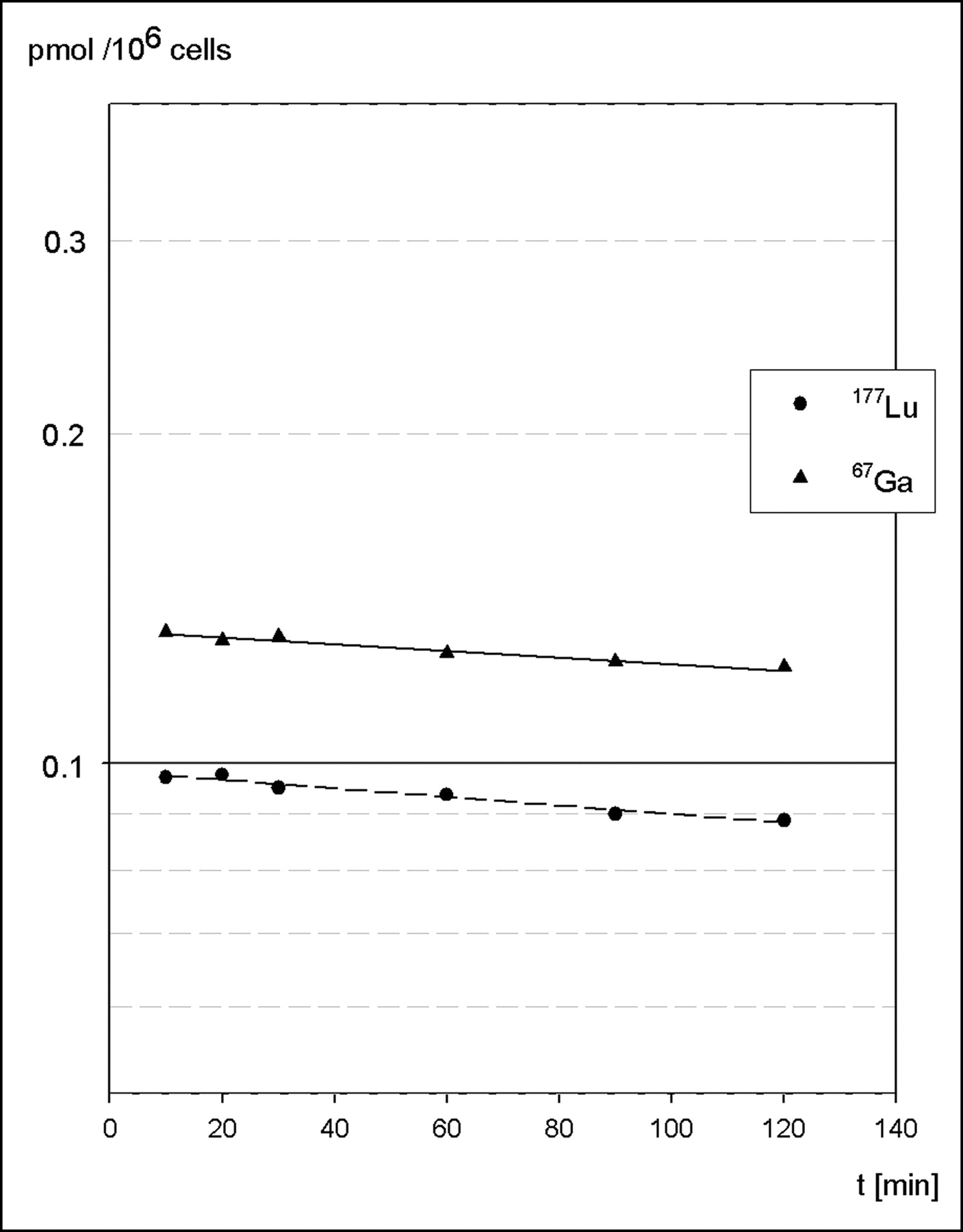
Efflux of internalized radioactivity was evaluated after preloading of AR42J cells with either 67Ga- or 177Lu-labeled BZH3. Cell surface–bound peptide was removed by excess unlabeled peptide within the first minutes of incubation and amounted to ∼10% of total activity, a fraction that is consistent with the results obtained in internalization experiments using an acid treatment of cells. Another ∼10% was excreted during the following 2-h incubation. From a semilogarithmic plot of data, half-lives of 16.5 ± 2.4 h and 12.8 ± 1.4 h for the excretion of 67Ga and 177Lu, respectively, were calculated (Fig. 4).
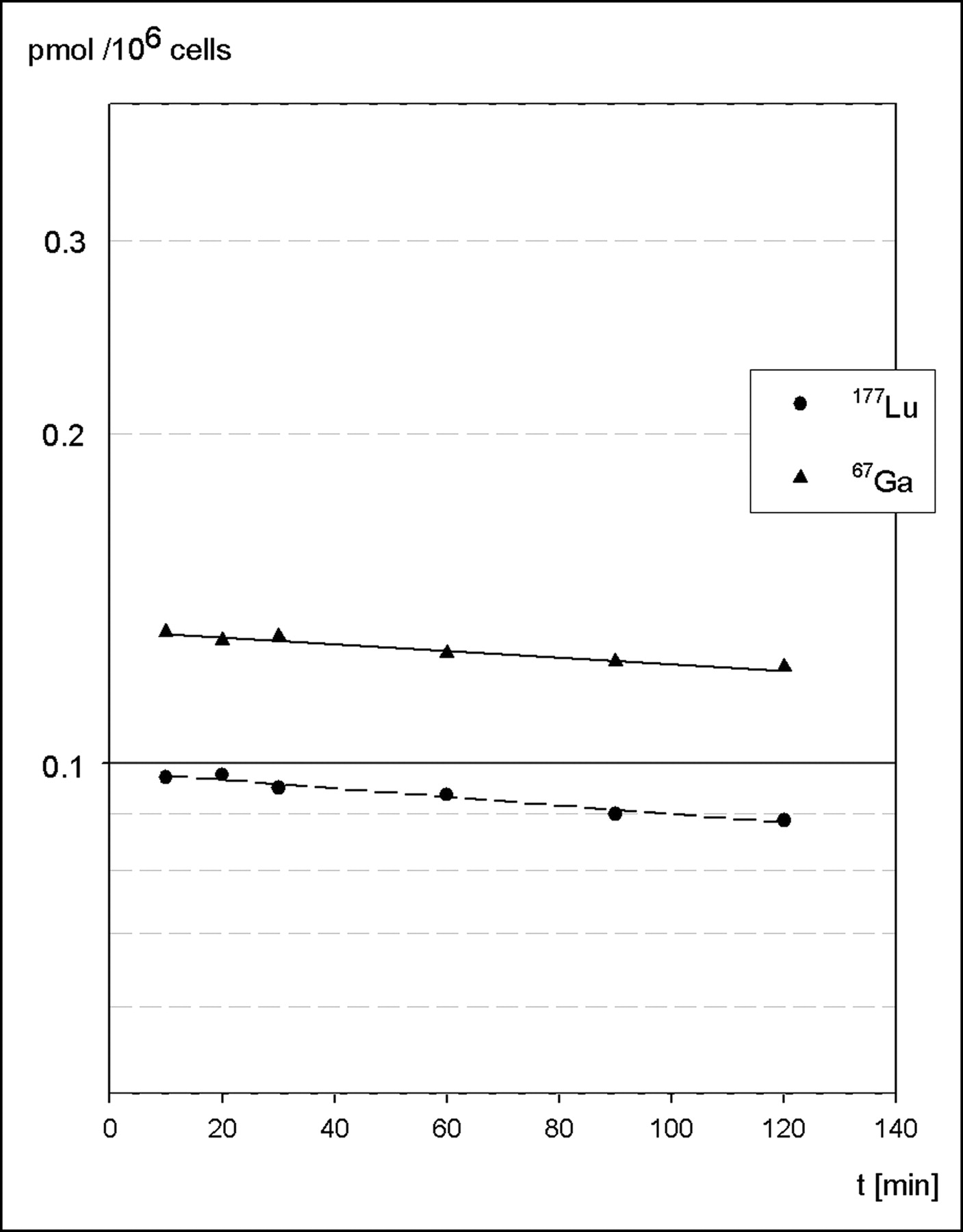
Semilogarithmic plot shows efflux (externalization) of radioactivity from AR42J cells preloaded with either 67Ga-BZH3 (solid line) or 177Lu-BZH3 (dashed line). Data are means of duplicate samples from 2 experiments.
Receptor Density
Three separate tumor inoculations (dose dependence, biokinetics, and PET) were performed with AR42J cells that differ in the number of passages from 7 to 16 relating to the commercially obtained cells. No marked difference in peptide uptake was noted. The uptake amounted to 0.132, 0.145, and 0.159 pmol per 106 cells, respectively.
Biodistribution
Tumor and normal tissue uptake 1 h after the administration of increasing doses (5, 15, 45, and 135 pmol) of 67Ga-BZH3 are presented in Table 1. A dose dependence was noted in the AR42J tumor and the normal GRP-r–expressing tissues, including small intestine, colon, and pancreas. Small intestine and colon showed a continuously decreasing uptake with increasing doses. The uptake in pancreas and tumor showed a bell-shaped dose dependence. The maxima obtained were 10.9 percentage injected dose per gram (%ID/g) for tumor and 46.1 %ID/g for pancreas after administration of a 15- and a 45-pmol dose, respectively. No dose dependence was found in blood, liver, spleen, muscle, lung, and bone. In kidneys, a significantly reduced activity was noted after the administration of the highest dose (135 pmol = 0.23 μg).
Tissue Distribution of Increasing Doses of 67Ga-BZH3 at 1 Hour After Injection in AR42J Tumor-Bearing Nude Mice
The tissue distribution of 67Ga- or 177Lu-labeled BZH3 as a function of time was determined using the 15-pmol dose (Table 2). The radioactivity in the GRP-r–expressing tissues after administration of the 67Ga-BZH3 did not significantly decrease up to 4 h after injection. From data at 1 and 24 h after injection, the average half-lives of 67Ga-BZH3 retention in pancreas and tumor of ∼22 h and ∼16 h could be estimated, respectively, the latter being very similar to the excretion half-life of 67Ga activity in vitro after internalization of 67Ga-BHZ3 into AR42J cells. Low activity was found in blood, liver, lung, muscle, and bone, even at 1 h after injection, and tumor-to-tissue ratios did not markedly increase after 2 h following injection (Table 3).
Tissue Distribution of 67Ga- and 177Lu-BZH3 as Function of Time in AR42J Tumor-Bearing Nude Mice After Injection of 15 pmol of Peptide
Tumor-to-Tissue Ratios of 67Ga- and 177Lu-BZH3 in Tumor-Bearing Nude Mice
The difference in tumor uptake 1 h after injection between the 2 experiments using a 15-pmol dose, 10.9 %ID/g (dose dependence) versus 5.6 %ID/g (kinetics), is not readily understood because AR42J cells used for tumor inoculation showed no difference with regard to in vitro binding and internalization of 67Ga-BZH3. A possible explanation might be that the fraction of tumor cells in relation to the total tumor mass was different, since tumors were highly hemorrhagic.
As an estimate for therapeutic applications, BZH3 was labeled with 177Lu, which is a medium-energy β-emitter (Eβ-max = 497 keV) with a low abundance of γ-radiation at 113 and 208 keV, and biodistribution of 177Lu-BZH3 was determined at 1 and 24 h after injection of a 15-pmol dose (Table 2). Uptake in the tumor and pancreas 1 h after injection appears to be somewhat lower compared with that of 67Ga-BZH3, 4.5 versus 5.6 %ID/g and 31.8 versus 42.1 %ID/g, respectively. The in vivo half-life of 177Lu-BZH3 in the tumor, estimated from the 1- and 24-h biodistribution data, was ∼14 h and, thus, corresponds to the in vitro data of efflux. In contrast, the decline of 177Lu activity in the pancreas was significantly accelerated compared with that of the 67Ga-labeled BZH3 (t1/2, ∼7.5 vs. ∼22 h).
Coadministration of excess unlabeled BZH3 in the blocking experiments reduced uptake of 177Lu-BZH3 in the pancreas of rats from 2.69 ± 0.60 %ID/g to 0.06 ± 0.01 %ID/g and in the AR42J tumor from 1.11 ± 0.31 %ID/g to 0.09 ± 0.01 %ID/g, indicating a specific and GRP-r–mediated uptake of the labeled peptide.
PET
PET images of tumor-bearing mice obtained 1 h after injection of 15 pmol 68Ga-BZH3 are presented in Figure 5. Transaxial slices cutting the tumor and mediastinal region demonstrate a clear delineation of the tumor tissue and a low activity background in lungs, liver, and circulation. Coronal slices show the highest activity in pancreas and a nonuniform activity distribution in the duodenum, which complicates visualization of the kidneys. γ-Counting indicated an activity accumulation of 5.3 and 7.0 %ID/g in the resected tumors of the left and the right animal, respectively. Tumor weights were 200 mg (left animal) and 128 mg (right animal).

Iteratively reconstructed transaxial (left) and coronal (right) PET images of 2 AR42J tumor-bearing nude mice 1 h after injection of 15 pmol (0.49 MBq) 68Ga-BZH3. Tumors are located in thoracic wall. Tumor uptake and weight: left animal, 5.3 %ID/g, 200 mg; right animal, 7.0 %ID/g, 128 mg.
DISCUSSION
During the past decade, the successful application of radiolabeled somatostatin analogs in nuclear medicine for diagnostics and therapy of neuroendocrine tumors has stimulated the research in receptor targeting of additional tumor types (26). In a recent study, Van de Wiele et al. demonstrated the feasibility of scintigraphic imaging of GRP-r–positive prostate and breast carcinoma in patients with the 99mTc- labeled BN analog RP 527, indicating the potential of BN analogs for use in humans (13,27). In the present study, we investigated a novel BN analog, BZH3, for its in vitro and in vivo behavior under the aspects of improving targeting of GRP-r–positive tumors and introducing the short-lived positron emitter 68Ga for peptide labeling and, thus, to enable PET, which increases the spatial resolution and sensitivity of tumor detection compared with conventional γ-camera imaging (28–30). Additionally, scatter-corrected PET provides the ability to quantify biodistribution and, thus, may be favorably used for GRP-r density determination, which is a prerequisite for the appropriate selection of patients entering radiotherapy, and to control effectiveness of radiotherapy or surgical interventions.
The affinity of 67Ga-BZH3 to GRP-r of live AR42J cells showed a Kd of 0.46 nmol/L, which is a factor of 3 lower than that of 125I-Tyr4-BN (Kd = 1.6 nmol/L), which is commonly used as a standard for determination of GRP-r–specific binding. The affinity of 67Ga-BZH3 is thus comparable with that of 99mTc-demobesin 1 (Kd = 0.67 nmol/L) (18) and 111In-labeled DOTA-Aoc-BN(7–14) (50% inhibitory concentration = 0.6 nmol/L vs. 125I-Tyr4-BN) (15), which are the BN analogs with the highest GRP-r affinity reported so far. Though determination of Kd is independent of using live cells or cell membranes (31), the Bmax of 9.7 × 104 receptors per cell, obtained with live AR42J cells, overestimates the number of receptors actually present on the cell surface. This can be attributed to the recycling of part of the receptors to the cell surface after internalization of the GRP-r/BN complex (27,32), which increases the Bmax of live cells with incubation time.
Internalization of 67Ga-BZH3 into AR42J cells suggests an agonistic nature of the peptide. The rapid and high degree of internalization—70% of total cell-associated activity was endocytosed within 15 min using both the low and the high peptide concentration—fits well with the internalization rate of the agonist 125I-Tyr4-BN/GRP-r complex (t1/2 ∼ 6 min) and its independence from the peptide-to-receptor ratio (31).
Efflux of 67Ga and 177Lu activity from AR42J cells after internalization of labeled BZH3 showed half-lives of 13–16 h in vivo and in vitro, which are much longer than that of 125I-Tyr4-BN (t1/2 ∼ 3 h) (33). Since lysosomal degradation of BN analogs is very rapid—after a 15 min-incubation, 80% of internalized 125I-Tyr4-BN has already been metabolized (34)—the prolonged intracellular retention in tumor cells appears to be a specific feature of the M3+ DOTA chelate and presumably of the linker used for coupling rather than of the peptide degradation products. This is supported by similar efflux data of 177Lu from PC-3 tumor cells using a different, but DOTA-conjugated, BN analog (35).
Biodistribution studies with escalating doses of 67Ga-BZH3 in tumor-bearing mice indicated a dose-dependent uptake in GRP-r–positive tissues. Intestines showed a continuously decreasing %ID/g uptake with increasing amounts of peptide, which indicates an increasing saturation of the receptors and an uptake maximum equal to or less than the lowest dose of 5 pmol per animal. In contrast, tumor and pancreas uptake peaked at the 15- and 45-pmol dose, respectively. Breeman et al. (36) also described such a bell-shaped, dose-dependent uptake in the pancreas of rats. These authors suggest that the initial stimulation of the %ID/g uptake with increasing amounts of peptide might be related to a clustering of the receptor/BN complexes in the cell membrane, which might be required for internalization of the complexes. Since we found no dose dependence of internalization in vitro with doses much lower than that used in biodistribution experiments, in vivo a preferential trapping of the peptide at low doses in the intestines appears to be more likely. Assuming receptor densities of pancreas > tumor > intestine—but taking into account the high absolute amount of intestinal GRP-r and a much higher blood flow in the intestines compared with the tumor tissue (37)—an optimum tumor uptake may be achieved only after a sufficient saturation of the intestinal GRP-r. In GRP-r–negative tissues, only kidneys showed a significantly reduced activity uptake after administration of the highest dose. This is in contrast to published results reporting no change in the kidney uptake of BN analogs in mice and rats up to a 100-μg dose (36,37). Thus, a slightly prolonged retention time of the small doses of 67Ga-BHZ3 in the kidneys seems to be more likely than a saturation of renal excretion by the high dose.
The clearance of 67Ga-BZH3 from the circulation and normal GRP-r–negative tissues was fast and resulted in low background activity with maximum tumor-to-tissue ratios at 2 h after injection, which meets the requirements of PET with the short-lived positron emitter 68Ga. The low liver uptake of 0.5 %ID/g versus 3–8 %ID/g observed for 99mTc-labeled BN analogs (17,18,33,38) should facilitate tumor localization in the abdominal region compared with 99mTc imaging.
Similar to somatostatin analogs, BN analogs labeled with a therapeutic radionuclide might have a potential for radiotherapy. Using the diagnostic radiolabel for calculation of the absorbed therapeutic radiation dose, a comparison of the biokinetics of the therapeutic and the diagnostic radiolabel is necessary, since a change in the M3+ radiometal used for DOTA labeling can alter the biodistribution of a DOTA-conjugated peptide (39). For the couple of 67/68Ga and 177Lu (the latter has been used successfully in therapy of neuroendocrine tumors (40)), we found a slightly reduced (∼20%) uptake of 177Lu-BZH3 in the tumor and a ∼20% shortened half-life of tumor retention, whereas biodistribution and biokinetics in normal tissues were quite similar to that of 67Ga-BZH3. An exception, which might be beneficial for therapy, is the observed 3-fold accelerated clearance of pancreatic 177Lu activity. Presently, no reasonable explanation for the different kinetics of the 67Ga- and 177Lu-labeled peptide could be found. However, the data are in agreement with those reported by Smith et al. (35) for the washout of 177Lu activity from the pancreas of mice after administration of the 177Lu DOTA-8-Aoc-BN(7–14) analog. Nevertheless, the high initial, receptor-mediated 177Lu uptake in the pancreas has to be considered for limiting-dose calculations.
PET clearly indicated the potential of 68Ga-BZH3 for a sensitive localization of GRP-r–positive tumors in the mediastinal area. Compared with a 64Cu-labeled DOTA-BN analog (t1/2, 12.7 h; β+, 19%), recently proposed for PET (37), 68Ga-BZH3 appears to be much more favorable with regard to in vivo stability and biodistribution, in vitro data, and simplicity of radionuclide production. Detection of metastatic prostate carcinoma, especially of an invasion of pelvic lymph nodes, remains less predictable because of the enhanced intestinal and bladder background activity. On the other hand, the high GRP-r density found in most of the prostatic neoplasms and metastases (8) might partially compensate these limitations.
CONCLUSION
The M3+ radiolabeled BN analog BZH3 evaluated in this study has shown many of the prerequisites for a successful targeting of GRP-r–positive tumors, such as a high affinity in the subnanomolar range and a rapid internalization with a prolonged intracellular retention of the M3+ radionuclide in vitro. In vivo, high tumor-to-tissue ratios 1 h after injection and a rapid clearance of unbound peptide from GRP-r–negative, normal tissues was observed. PET has demonstrated the diagnostic potential in the tumor-bearing mouse model with 68Ga-BZH3. Additionally, the ability of PET to quantify biodistribution data should enable more accurate dose calculations for radiotherapy with M3+ therapeutic radionuclides—for example, 90Y or 177Lu. The physiologic features together with PET and the simplicity of 68Ga production from a generator recommend 68Ga-BZH3 as a promising, versatile tool for an improved localization of GRP-r–positive tumors also in patients.
Because our generator is not commercially available, we refer the reader to a TiO2-based 68Ge/68Ga generator, which is commercially available from the Cyclotron Co. Ltd. Obninsk. Meyer et al. (41) have published a comparison, showing a similar suitability and efficiency of both generators.
Acknowledgments
We thank the Swiss National Science Foundation, the Swiss Commission for Technology and Innovation (KTI project 4668-1), the Deutsche Forschungsgemeinschaft (DFG grant 2901/3-1), and Mallinckrodt Medical for financial support of this work and Novartis for analytic support.
Footnotes
Received Jun. 15, 2004; revision accepted Oct. 25, 2004.
For correspondence or reprints contact: Jochen Schuhmacher, PhD, Department of Diagnostic and Therapeutic Radiology, German Cancer Research Center, Im Neuenheimer Feld 280, D-69120 Heidelberg, Germany.
E-mail: j.schuhmacher{at}dkfz.de
REFERENCES
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Increasing molar activity by HPLC purification improves 68Ga-DOTA-NAPamide tumor accumulation in a B16/F1 melanoma xenograft model
- Variation of Specific Activities of 68Ga-Aquibeprin and 68Ga-Avebetrin Enables Selective PET Imaging of Different Expression Levels of Integrins {alpha}5{beta}1 and {alpha}v{beta}3
- PET of Tumors Expressing Gastrin-Releasing Peptide Receptor with an 18F-Labeled Bombesin Analog
- Bombesin functionalized gold nanoparticles show in vitro and in vivo cancer receptor specificity
- Evaluation of a 1,4,7,10-Tetraazacyclododecane-1,4,7,10-Tetraacetic Acid-Conjugated Bombesin-Based Radioantagonist for the Labeling with Single-Photon Emission Computed Tomography, Positron Emission Tomography, and Therapeutic Radionuclides
- Preparation of a Promising Angiogenesis PET Imaging Agent: 68Ga-Labeled c(RGDyK)-Isothiocyanatobenzyl-1,4,7-Triazacyclononane-1,4,7-Triacetic Acid and Feasibility Studies in Mice
- 68Ga-Labeled Bombesin Studies in Patients with Gastrointestinal Stromal Tumors: Comparison with 18F-FDG
- 18F-Labeled Bombesin Analogs for Targeting GRP Receptor-Expressing Prostate Cancer