


Abstract
RS7 is an internalizing anti-Trop-2 pancarcinoma antibody capable of targeting most epithelial cancers. Because pretargeting strategies could improve the tumor localization of radionuclides, a new anti-Trop-2 × antihapten bispecific antibody for pretargeting, based on humanized RS7, was prepared and evaluated with a radiolabeled hapten-peptide in vitro and in vivo to determine whether its internalization properties would interfere with pretargeting. Methods: The anti-Trop-2 × antihapten bispecific antibody, TF12, was prepared using the modular dock-and-lock method. TF12 and humanized RS7 binding was assessed by cell binding assays and fluorescence-activated cell sorting analysis in a variety of human carcinoma cell lines. The internalization of TF12 was evaluated in vitro using a fluorescent TF12 conjugate or hapten-peptide and 111In-labeled TF12 and RS7. The biodistribution of TF12 and its use as a pretargeting agent with an 111In-labeled hapten-peptide were assessed in several human epithelial cancer xenografts. Dose optimization was examined in 2 tumor models. Results: TF12 internalizes, but a substantial fraction remained accessible on the tumor surface. Fluorescence-activated cell sorting analysis showed only a minor change in fluorescent signal when the tumor was probed with a fluorescent hapten-peptide over 4 h, and microscopy showed substantial membrane staining when reassessed at 24 h after TF12 exposure. Only 40.1% of 111In-TF12 was internalized after 24 h. In vivo, excellent tumor localization of the 111In-labeled peptide was observed in several tumor models. Conclusion: TF12 was retained sufficiently on the cell surface in several epithelial cancers, thereby making it suitable for pretargeted imaging and therapy of various Trop-2–expressing carcinomas.
- bispecific antibody
- epithelial cancers
- epithelial glycoprotein-1 (EGP-1)
- pretargeting
- radioimmunodetection
- Trop-2
Although radioimmunotherapy is an effective therapeutic option for follicular non-Hodgkin lymphoma (1–3), solid tumors pose challenges, and efforts continue in the search for new targets and procedures to improve imaging and therapeutic prospects (3). In this search, Trop-2 (also known as epithelial glycoprotein-1, gastric antigen 7331, and tumor-associated calcium signal transducer 2) is of interest (4). Trop-2 is a 47.8-kD integral membrane glycoprotein that is highly expressed on most epithelial cancers originating from the breast, prostate, lungs, ovaries, gastrointestinal tract, pancreas, and skin and exhibits selective expression in some normal tissues (5,6). Trop-2 expression is associated with poor prognosis, tumor aggressiveness, and metastasis (4). RS7 is an anti-Trop-2 monoclonal IgG antibody that targets epithelial cancers, including those of the lung, breast, colon, pancreas, and prostate (6–13). Previous studies showed that RS7 is internalized rapidly after binding Trop-2 on the target cell surface, with an estimate of 50% internalized within 70 min in a breast cancer cell line, whereas even faster internalization rates were determined in a lung cancer cell line (6,8). In vitro and in vivo studies showing that radiometal-labeled RS7 (e.g., 111In or 90Y) is retained at a much higher level than radioiodinated RS7 also are consistent with RS7’s internalization (10,11). More recently, this property led to an evaluation of RS7 as a targeting agent for drugs (14,15). However, unconjugated humanized RS7 (hRS7) showed potent antibody-dependent cell-mediated cytotoxicity activity in gynecologic cancers, suggesting that some portion of hRS7 is retained on the surface to allow immune effector cell interaction.
Numerous studies have shown that various pretargeting strategies are superior to directly radiolabeled antibodies in generating high tumor-to-nontumor ratios quickly, with tumor uptake frequently rivaling that of a directly radiolabeled F(ab′)2 or even an IgG (16). With its pancarcinoma-targeting capability, the anti-Trop-2 antibody RS7 is an interesting antibody for pretargeting, but with previous reports indicating that RS7 is internalized (6,8), its utility in pretargeting—for which the primary targeting agent must remain accessible for some time to allow binding of the secondary compound—was in question. Nevertheless, with the ease of making bispecific antibodies (bsmAbs) with the dock-and-lock technology (DNL) (17), an RS7-based anti-Trop-2 × antihapten Tri-Fab bsmAb, designated TF12, was prepared (Fig. 1). In this report, the potential for using TF12 in a pretargeting setting with a radiolabeled hapten-peptide was evaluated in vitro and in vivo in various epithelial cancer cell lines. The results show that TF12 provides excellent localization of the radiolabeled hapten-peptide in vivo, illustrating that the internalization properties of this bsmAb–target combination do not impede its development for pretargeting applications.

Schematic representation of TF12 DNL construct. (A) hRS7-Fab-DDD2 (DDD is dimerization and docking domain) module forms homodimers (B). h679 anti-HSG Fab is fused with AD2 sequence (AD is anchoring domain) (C). When these 2 proteins are brought together, AD2 portion docks to docking domain of DDD2 module, forming tri-Fab anti Trop-2 × anti HSG bsmAb TF12 (D). Disulfide bridges form between strategically placed cysteines to link 2 modules covalently.
MATERIALS AND METHODS
Cell Lines
MDA-MB-468 (breast cancer), SK-OV-3 (ovarian cancer), PC3 (prostate cancer), Calu-3 (lung cancer), HT29 (colon cancer), LS174T (colon cancer), Capan-1 (pancreatic cancer), and Raji (Burkitt lymphoma) were purchased from American Type Culture Collection.
Antibodies and Other Agents
The TF12 Tri-Fab bsmAb (molecular weight, 157 kDa) was prepared by the DNL procedure (17), using a humanized version of the murine RS7 as the partner with the humanized anti-HSG (histamine-succinyl-glycine) hapten antibody, 679 (Fig. 1) (18). Purified TF12, hRS7, and other antibodies that were used as controls, including veltuzumab (humanized anti-CD20 IgG (19)) and TF8 (humanized anti-CD22 [based on epratuzumab (20)] × anti-HSG DNL Tri-Fab bsmAb construct), were all provided by Immunomedics, Inc., or IBC Pharmaceuticals, Inc.
Unconjugated AffiniPure goat-antihuman (GAH) IgG (heavy- and light-chain–specific) and fluorescein isothiocyanate–conjugated AffiniPure GAH IgG (Fc-specific; fluorescein isothiocyanate-GAH IgG) were purchased from Jackson ImmunoResearch. GAH IgG, TF12, hRS7, and veltuzumab were conjugated with AlexaFluor 488 (Invitrogen) according to the manufacturer’s instructions. The AlexaFluor conjugates were designated AF-[antibody name].
IMP288, the di-HSG hapten-peptide (DOTA-d-Tyr-d-Lys(HSG)-d-Glu-d-Lys(HSG)-NH2; molecular weight, 1,453 Da), was prepared and radiolabeled with 111In as described previously (21). Radioiodinated TF12 and hRS7 were made by the IODO-GEN method (22). 125I and 111In were provided by either Perkin Elmer or Covidien. Instant thin-layer chromatography showed less than 5% unbound radionuclide. The immunoreactivity of radiolabeled TF12 or hRS7 was determined by Lindmo assays (23) or by size-exclusion high-performance liquid chromatography using an excess of an anti-hRS7 idiotype antibody, as described previously (18).
A fluorescent-conjugated peptide, designated RDC017, was used to assess the accessibility of the bsmAb bound to the surface of several cell lines. RDC017 was prepared from a thiol-containing derivative of the IMP288 (supplemental data; available online only at http://jnm.snmjournals.org).
Fluorescence-Activated Cell Sorting (FACS) Analysis
FACS analysis was performed to determine hRS7 and TF12 binding to Trop-2–expressing tumor cell lines. Cell suspensions were incubated with 5 μg of RS7 and TF12 AF conjugates per milliliter for 30 min at room temperature and were analyzed by FACS (FACSCalibur; Becton Dickinson) after being washed and fixed with 10% formalin. For each cell line, AF-veltuzumab was used as a negative control with the same procedures.
Internalization Assays
MDA-MB-468 and PC3 were incubated with TF12 (5 μg/mL) for 1 h at 4°C. After being washed to remove the unbound antibody, fresh medium was added and incubation was continued at 37°C. At 1, 2, and 4 h, the presence of surface-bound TF12 was then probed using a molar excess of RDC017 for 30 min at 4°C before being washed and then fixed with 10% buffered formalin. As an internalizing control, Raji cells were incubated with the Tri-Fab bsmAb, TF8 (anti-CD22 × anti-HSG), using the same procedure and probing as was performed with the RDC017 peptide. Nonspecific binding was examined by incubating samples only with the fluorescent agent.
In a second approach, internalization assays using 111In-radiolabeled RS7 and TF12 with PC3 cells in culture were performed as described previously (24,25). Briefly, cells were grown to confluence in 6-well plates and incubated in triplicate with either 6.15 ng of 111In-radiolabeled hRS7, 4.02 ng of 111In-TF12, or a combination of 4.02 ng of 111In-TF12 and 1.74 ng of unlabeled IMP288 in 2 mL of medium plus 0.5% bovine serum albumin (all wells received 1,200 Bq in 100 μL of phosphate-buffered saline [PBS]). As a control, separate wells were incubated in triplicate in the presence of 2 μg of unlabeled antibody before the radiolabeled product was added. After 1, 2, 4, 9, and 24 h of incubation at 37°C, the cells were washed twice with cold PBS. To remove the membrane-associated fraction, the cells were incubated with an acid buffer (0.1 M acetic acid, 154 nM NaCl, pH 2.6) for 10 min. Then, after the cells were washed twice with PBS, they were removed from the plate with a cotton swab. The percentage of radioactivity in the 2 fractions, the membrane-associated and internalized fractions, with and without the addition of excess unlabeled antibody, was determined in triplicate.
Microscopic Evaluation of Internalization
An assessment of internalization of TF12 by fluorescent microscopy was performed in MDA-MB-468 cells following the procedure in the study by Pirker et al. (26). Briefly, 5 × 105 MDA-MB-468 cells were incubated with 5 μg of TF12 or control TF8 anti-CD22 per milliliter for 1 h at 4°C. CD22-expressing Raji human Burkitt lymphoma cells incubated with TF8 served as a positive control for internalization, because TF8 was based on epratuzumab, a rapidly internalizing, humanized anti-CD22 antibody (20). After cells were washed with ice-cold PBS, 0.1% bovine serum albumin, and 0.1% NaN3 to remove excess unbound bsmAb, the cells were resuspended in fresh medium. A sample was collected immediately after washing, with the remaining cells allowed to incubate at 37°C for 1 or 24 h. At the prescribed times, the cells were washed with cold medium and then probed using either the fluorescent di-HSG-peptide RDC 017 (3.2 nmol/mL) or the AF-conjugated AffiniPure GAH IgG (AF-GAH IgG). The set of cells probed with AF-GAH IgG was first fixed with 4% formalin in PBS and then permeabilized with 0.1% Triton-X-100 (5 min, room temperature) before the AF-GAH IgG was added. This procedure allowed the secondary agent to localize both surface and internalized TF12. Both the RDC017 and the AF-GAH IgG were incubated for 30 min at 4°C before the cells underwent a final wash in PBS, followed by a brief fixation in PBS–formalin. A sample was then placed on a slide for microscopic examination.
Biodistribution Studies
Studies were performed at either the Radboud University Nijmegen Medical Centre (The Netherlands) or the Center for Molecular Medicine and Immunology (United States) after these locations had received approval from their respective animal welfare committees. Mice were obtained from either Taconic or Janvier and were acclimated to laboratory conditions for at least 1 wk before tumors were implanted. The mice were housed under nonsterile standard conditions in cages with free access to food and water.
A typical study involved nude mice that were inoculated subcutaneously with 1 × 106 to 1 × 107 of MDA-MB-468, SK-OV-3, PC3, or Capan-1 cells for the assessment of targeting with radiolabeled TF12 or hRS7 IgG or pretargeting with TF12. Pretargeting was also assessed using TF8 anti-CD22 bsmAb with subcutaneously grown Raji human Burkitt lymphoma cells. Once tumors were palpable, the studies were initiated (tumors generally averaged 0.1–0.3 g at the time of necropsy). The specific details for each study are given in the “Results” section. Animals were anesthetized and bled intracardially before being euthanized by CO2/O2 asphyxiation at various time points. After necropsy, tumors and organs of interest were excised, weighed, and counted in a scintillation counter. The percentage injected dose per gram (%ID/g) was calculated on the basis of a standard of the injected product counted with the tissues.
RESULTS
In Vitro Characterization of TF12 Binding
The molecular characterization and binding properties of TF12 are given in Supplemental Figure 1. Size-exclusion high-performance liquid chromatography and sodium dodecyl sulfate polyacrylamide gel electrophoresis confirmed purity, and the binding properties of TF12 (using Biacore) to rat anti-RS7 idiotype antibody as a surrogate for Trop-2 and the hapten HSG were confirmed. The binding analysis of TF12 to MDA-MB-231 and SK-OV-3 breast and ovarian carcinoma cell lines showed a similar affinity. By flow cytometry, TF12 and hRS7 bound in a similar manner to 4 cell lines of different origin shown in Table 1. Trop-2 expression in 2 other cell lines using hRS7 also is shown.
FACS Analysis of Trop-2 Expression in Several Epithelial Cancer Cell Lines
In Vitro Assessment of Internalization
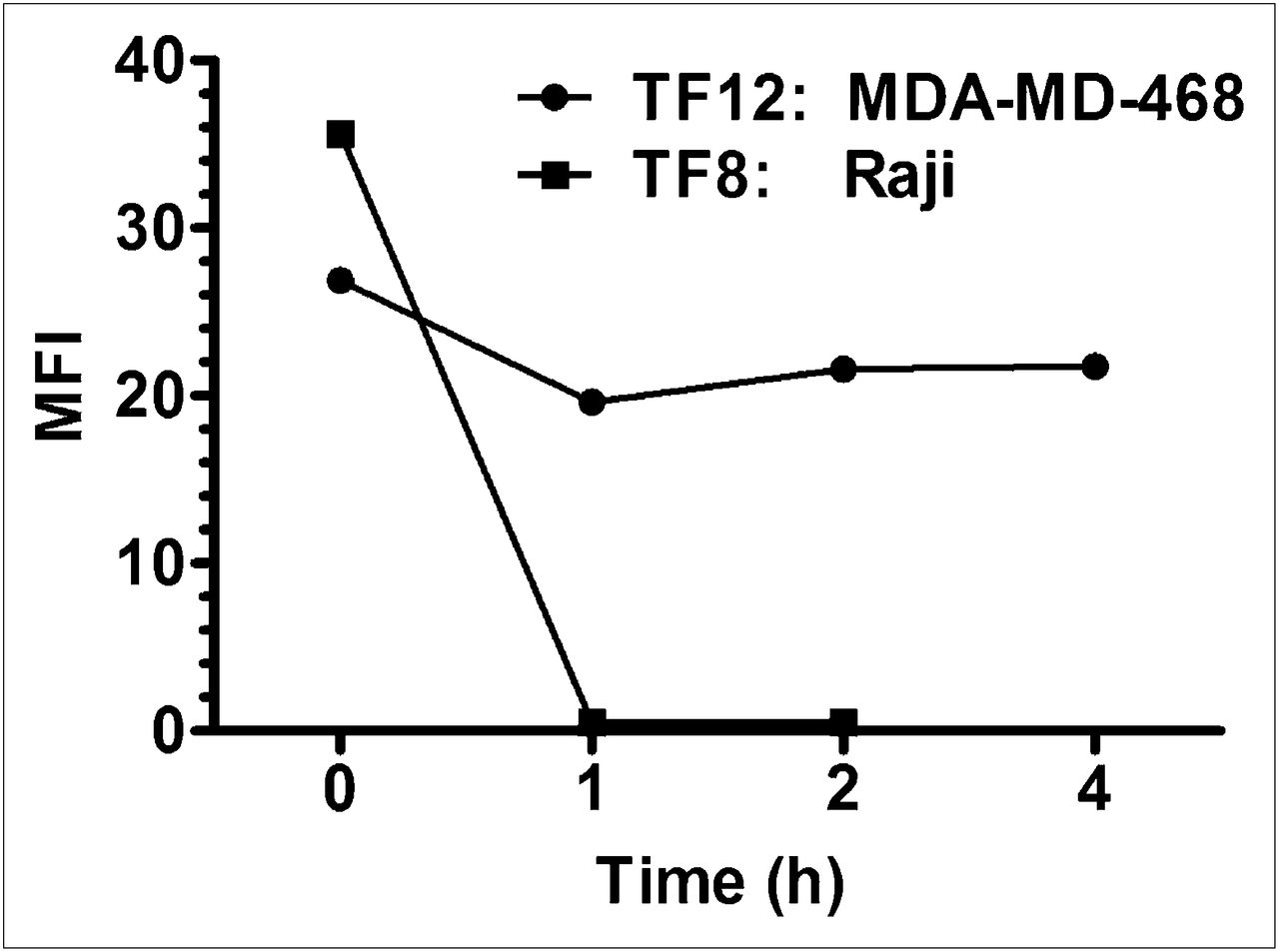
The internalization rate of murine RS7 in Calu-3 or MBA-MD-468 previously indicated 50% of the bound antibody was internalized in approximately 1 h or less (6,8). MDA-MB-468 cells that were preincubated with TF12 at 4°C before being washed and incubated at 37°C showed continued evidence of binding the fluorescent hapten-peptide RDC017 over 4 h (Fig. 2). This duration was in sharp contrast to Raji lymphoma cells incubated with TF8, an anti-CD22 bsmAb. Even after just 1 h, the RDC017 no longer bound to the cells, suggesting TF8 had internalized completely. It is important to emphasize that this assay does not account for bsmAbs that might otherwise be released from the cells into the medium rather than being internalized. However, given the reported rapid internalization of the epratuzumab anti-CD22 antibody (19,27,28), it is reasonable to assume that the inability of the peptide to bind to the Raji cells preincubated with TF8 was due to its internalization and not its release into the medium. By microscopy, MBA-MD-468 cells preincubated with TF12 and probed with RDC017 showed intense membrane localization through 24 h, indicating that TF12 was still present on the surface (Fig. 3). However, when the cells were probed with a fluorescent-conjugated antihuman IgG after being fixed and permeabilized to allow the fluorescent conjugate to penetrate inside the cells, staining was seen within the cells (Figs. 3D and 3E), indicating that some portion had internalized. Microscopic examination of Raji cells incubated with TF8 and probed with RDC017 immediately after the 4°C incubation showed a patchy distribution that became more diffuse and faint (nearly absent) when probed after a 1-h incubation at 37°C, again suggesting that most of the anti-CD22 bsmAb had internalized (not shown).

Analysis of internalization of TF12 anti-Trop-2 or TF8 anti-CD22 bsmAb in MDA-468 and Raji cells, respectively. Cells were probed with fluorescent hapten-peptide RDC017 after incubation with each bsmAb. Fluorescence, as determined by FACScan, is expressed as mean fluorescent intensity, normalizing data by subtracting mean fluorescent intensity for RDC 017 alone, which was minimal. MFI = mean fluorescent intensity.

Visualization of TF12 internalization. MDA-MB-468 cells were incubated for 1 h at 4°C with TF12 before being replaced with fresh medium and incubation temperature was raised to 37°C. (A–C) Cells were then probed with fluorescent RDC017 immediately after 1 h of incubation at 4°C (A), after 1 h at 37°C (B) or after 24 h at 37°C (C). D and E represent specimens of same MDA-MB-468 cells first incubated with TF12 for 1 h at 4°C, but in this case, TF12 binding was revealed using AF-GAH IgG after cells were fixed and permeabilized (binding immediately after 1 h of incubation at 4°C [D] and after 1 h at 37°C [E]). Images were brightened and contrast was enhanced using Photoshop (Adobe).
To provide a more quantitative estimate of how much TF12 is internalized, TF12 and hRS7 were radiolabeled with 111In, and the fractional amount internalized over 24 h was evaluated in the PC3 prostate cell line. One hour after cells were incubated with the antibodies, approximately 14% of the antibody had internalized (Table 2). Over the next 8 h, the percentage internalized increased nearly 2-fold, to 25.3% ± 0.18% for the hRS7 IgG and to 28.3% ± 2.54% for TF12. By 24 h, 40.1% ± 4.99% of the TF12 bsmAb had internalized, whereas about one third of the hRS7 IgG was inside the cell. Thus, in this cell line, approximately 60% of the bsmAb is retained on the surface of the cells. When divalent hapten-peptide IMP288 was coadministered with radiolabeled TF12, uptake over the first 2 h was higher than with the bsmAb alone. These data suggest that cross-linked TF12 initiates an earlier spurt of internalization that seems to plateau, achieving the same maximum uptake as TF12 alone (not cross-linked by the hapten-peptide). As expected, preincubating cells with unlabeled antibody blocked binding of the radiolabeled antibody to Trop-2, illustrating that internalization of the radiolabeled antibody was related to its binding to Trop-2.
Internalization of 111In-Labeled RS7, TF12, and TF12 Plus IMP288 in PC3 Cells in Culture
Biodistribution Studies with Antibody Alone
A biodistribution study performed in animals bearing the Capan-1 human pancreatic xenograft showed that radioiodinated TF12 cleared rapidly from the blood, with 0.15 ± 0.03 %ID/g in the blood at 24 h (Fig. 4), which is in agreement with the rapid clearance found with other tri-Fab bsmAbs (17). Tumor uptake peaked at 2 h after injection (5.6 ± 0.63 %ID/g), falling to 1.43 ± 0.09 %ID/g at 24 h. Similar levels of 125I-TF12 were found in MBA-MD-468 breast and SK-OV-3 ovarian cancer cell lines at 1 d after injection (0.91 ± 0.018 %ID/g and 1.29 ± 0.33 %ID/g, respectively), with blood concentrations averaging 0.4 %ID/g or less. The spleen was the only tissue that showed an unusual early uptake, averaging approximately 20%–25 %ID/g at 2 and 5 h after injection, but like all the other normal tissues, TF12 cleared quickly, being equal to that in the liver (∼0.5 %ID/g) at 24 h.

Biodistribution of 125I-TF12 in nude mice bearing Capan-1 human pancreatic cancer xenografts. %ID/g of tumor and blood (A) and other tissues (B).
The rapid clearance of the TF12 bsmAb and its relatively low tumor uptake were in sharp contrast to the slow blood clearance of the hRS7 IgG and its higher uptake. For example, in PC3-bearing nude mice, tumor uptake of 125I-hRS7 on day 3 was 7.3 ± 2.3 %ID/g, whereas the concentration in the blood was 8.1 ± 1.1 %ID/g. However, in PC3-bearing mice given 111In-hRS7, tumor uptake was 36.5 ± 13.3 %ID/g, whereas the blood concentration was similar to radioiodinated hRS7 IgG on day 3. These results are indicative of an internalizing antibody binding to a stable chelate–radiometal, where the percentage uptake accounts for the cumulative binding of hRS7, its internalization, and entrapment of the 111In activity over time. However, the uptake of radioiodinated IgG or bsmAb is expected to reveal only the amount of antibody that remains accessible (i.e., has not been internalized and catabolized).
Pretargeting Studies
Despite in vitro evidence that some portion of TF12 is internalized, biodistribution studies in mice bearing different subcutaneous tumor xenografts and pretargeted the day before with TF12 showed a reasonably high (∼10–15 %ID/g) uptake of the 111In-labeled peptide in the tumors (Table 3). Tissue uptake and blood concentrations were low, with tumor-to-blood ratios ranging from approximately 60:1 to 1,000:1.
Biodistribution of Pretargeted 111In-IMP288 in MDA-468, SK-OV-3, and PC3 at 3 Hours After Injection
Additional studies examined several doses of TF12 (40, 79, and 158 μg) given with a fixed amount of IMP288 (0.026 nmol; TF12-to-IMP288 mol ratios, 10, 20, and 40:1) in the SK-OV-3 and MDB-MD-468 models (Fig. 5). 111In-IMP288 uptake in SK-OV-3 at 3 h increased from 5.3 ± 1.2 %ID/g at the 10:1 ratio to as high as 21.3 ± 6.2 %ID/g at the 40:1 ratio. Tumor uptake for TF12 in these animals (as measured by 125I-TF12 added to TF12) was similar in the groups, ranging from 1.1 to 1.5 %ID/g. In contrast, MDA-MB-468 uptake was only modestly increased from 11.6 ± 1.9 to 15.2 ± 3.3 %ID/g over this same range of TF12. Interestingly, tumor uptake of the 125I-TF12 was nearly 2-fold higher in the MDA-MB-468 xenografts than in the SK-OV-3 tumors, yet the breast cancer xenograft did not have a higher IMP288 uptake commensurate with the higher TF12 level. By 24 h, tumor uptake for the 111In-IMP288 plateaued in both models, even though the uptake of the 125I-TF12 decreased nearly 2- to 4-fold.

Optimizing IMP288 uptake in SK-OV-3 human ovarian cancer and MDA-MB-468 human breast cancer xenografts in nude mice. Animals (n = 5/interval) were given 40, 79, or 158 μg of TF12 (containing trace 125I-TF12), and then 16 h later they received 0.026 nmol (∼40 μCi) of 111In-IMP288. Necropsy was performed at 3 and 24 h. Tumor and blood concentrations are shown for 111In-IMP-288 (bars) and 125I-TF12 (in parentheses) at 3 h (A) and 24 h (B). Tumors averaged between 0.1 and 0.3 g.
These targeting results are in sharp contrast to those obtained with the TF8 anti-CD22 bsmAb in BALB/c nude mice bearing CD22-expressing Ramos or Raji tumors. In this study, animals were given 0.25 nmol (40 μg) of TF8 followed 24 h later with 0.025 nmol of 111In-IMP288. Three hours later, uptake was just 0.17 ± 0.07 %ID/g in Raji tumors and 0.15 ± 0.08 in Ramos (n = 4 in each). The low tumor uptake was predicted by the earlier in vitro studies that indicated efficient internalization within 1 h. Thus, even though tumor-to-blood ratios were high (e.g., averaging 38:1 in Raji-bearing mice), the low tumor uptake indicated unfavorable pretargeting with this bsmAb.
DISCUSSION
Pretargeting procedures for localizing radionuclides are attractive, because they are able to localize the radiolabeled product (e.g., hapten-peptide or biotin) quickly, developing high tumor–to–non-tumor ratios rapidly, and often with a tumor uptake that can rival a directly radiolabeled IgG or F(ab′)2 fragment (16). Bispecific antibody pretargeting procedures have typically relied on the bsmAb’s natural clearance from the blood and tissues, requiring a delay of several days before the hapten-peptide is given (16). It is imperative that the bsmAb remain accessible in the tumor (e.g., not internalized) in order for the radiolabeled hapten-peptide to be localized. Thus, if the pretargeting agent binds an antigen that triggers internalization, this target likely would not be suitable for pretargeting. For example, anti-CD22 antibodies are known to internalize rapidly, making these antibodies attractive candidates for targeting drugs or toxins that exert their effects intracellularly (28–33). Pantelias et al. (34) compared the pretargeting of 3 different antigens associated with hematopoietic malignancies, anti-CD20, CD22, and HLA-DR, finding that the anti-CD22 pretargeting agent had the lowest uptake of the 3 conjugates. However, CD22 also had the lowest density of the 3 antigens in the various cell lines examined, and thus this result may have merely reflected the lower antigen density. Herein, we included an evaluation of the TF8 tri-Fab bsmAb that binds to CD22, suspecting its rapid internalization would affect its utility in pretargeting. We first noted that in vitro, the TF8 anti-CD22 bsmAb was no longer accessible within just 1 h of its binding, and then in vivo, no appreciable localization of the radiolabeled hapten-peptide was observed. Thus, we agree that bsmAbs to CD22 are not suitable for pretargeting, not because they are insufficiently expressed on target cells but because the bsmAbs are efficiently internalized. Instead, we previously recommended that the TF4 anti-CD20 tri-Fab bsmAb be used for pretargeting B-cell malignancies (35). In those studies, 111In-TF4 had a substantially higher uptake in tumor xenografts than radioiodinated TF4, suggesting some internalization over the 1-d pretargeting interval.
Although antibody-based therapeutics (unconjugated and conjugated) are widely studied in hematologic malignancies, it has been more challenging to find suitable targets for epithelial cancers (3). In this regard, Trop-2 represents a promising new target for cancer, being expressed on the surface of many carcinomas (4). Early in its development, the anti-Trop-2 antibody RS7 was reported to internalize, and therefore most studies have focused on developing targeting strategies that take advantage of RS7’s internalization properties (7,10,14,36). Shih et al. (8) examined internalization in MDA-MD-468 cells in vitro over 30 min and reported an extrapolated internalization rate of 0.0147 min−1, indicating that under saturating conditions, surface-bound antibody will be internalized over approximately 70 min. Other studies in the Calu-3 human lung cancer cell line suggested an even faster rate of internalization, with most of the antibody being internalized within 45 min (6). This rate compares with that of the anti-CD22 antibody, for which 50% of the antibody was reported to be internalized within 10 min, with data indicating that nearly all the antibody had internalized within 2 h (28). In vivo studies comparing RS7 labeled with a radiometal with RS7 labeled by the more traditional noncovalent radioiodination procedures showed a substantially higher tumor retention when the antibody was labeled with a residualizing radionuclide, supporting the internalization capability of the RS7 antibody (7,10,11). Thus, these data prompted interest in developing a pretargeting procedure for Trop-2 localization. However, given the presence of Trop-2 in many carcinomas, and because the modular DNL method greatly simplifies bsmAb preparation (17), TF12 was prepared for evaluation.
Our findings with the hRS7 IgG and TF12 bsmAb indicate that despite some initial internalization, sufficient residual bsmAb remains on the surface of tumor cells to capture the hapten-peptide after an approximate 1-d delay. Some of the in vitro studies were performed with the washed cells placed in fresh medium before probing with the fluorescent hapten-peptide. Thus, it is unlikely that reexpression of antigen with reengagement by the bsmAb contained in the medium explained the continued presence of TF12 on the cell surface. Because TF12 clears so quickly from the blood, it is equally unlikely that there would be considerable replenishing of the bsmAb after the internalization process had occurred. Therefore, although there have been reports that internalization is nearly complete within approximately 1 h, our studies with TF12 and hRS7 radioconjugates found that a smaller portion of the antibody, perhaps as low as 10%–20%, is internalized over the first hour and that internalization proceeds at a much slower rate thereafter (i.e., it is not linear). Indeed, it appears that after an initial spurt of internalization, some cell lines had only about 50% of the antibody internalized over 24 h.
These observations are important, because antibodies are often characterized as being internalizing or noninternalizing when in fact there are varying rates among antibodies that are actively internalized and rates for those passively taken into the cell. At least in the case of TF12, the in vitro studies using the fluorescent hapten-peptide were helpful for illustrating the continued capacity to bind the hapten-peptide after several hours and even 1 d. This type of assay should be useful for predicting which bsmAb would have suitable surface retention for in vivo targeting, but it is unlikely it will be useful for predicting the actual quality of a given pretargeting agent, because there are other parameters that affect tumor localization (37). With recent clinical studies using the TF2 anti-CEACAM5 tri-Fab bsmAb also finding that the bsmAb is cleared sufficiently within just 1 d to allow the efficient localization of the radiolabeled hapten-peptide (38,39), we are encouraged to continue developing the TF12 bsmAb for future clinical evaluation.
CONCLUSION
TF12 is a suitable bsmAb for pretargeting different cancers that express Trop-2, primarily because after an initial spurt of internalization, sufficient bsmAb remains on the surface to allow efficient capture of the hapten-peptide. With recent preclinical studies showing hRS7 targeting of prostate cancer (13), and with the need for developing better imaging procedures for this disease, this clinical indication should be considered. However, hRS7-SN-38 drug conjugates also have been proven to be effective in several cancer cell lines, including lung, pancreatic, colorectal, and breast (6,7,12,14,15). Thus, developing a highly sensitive imaging procedure for these cancers, perhaps to be used in concert with the therapeutic agent, is of interest as well.
DISCLOSURE STATEMENT
The costs of publication of this article were defrayed in part by the payment of page charges. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank Jayson Jebsen, Ali Mostafa, Lenka Muskova, Preeti Trisal, Anke Reuser, Gerben Franssen, Annemarie Eek, Kitty Lemmens, and Bianca Lemmers for their excellent technical assistance. This study was supported in part by NJ Cancer Commission grant 10-8-CCR-EO and the Dutch Cancer Society grant KUN-2010-4820. Edmund A. Rossi, Celeste Regino, Thomas M. Cardillo, William J. McBride, Chien-Hsing Chang, and David M. Goldenberg are employed or have financial interests in Immunomedics, Inc., or IBC Pharmaceuticals, Inc. No other potential conflict of interest relevant to this article was reported.
Footnotes
Published online Sep. 5, 2012.
- © 2012 by the Society of Nuclear Medicine and Molecular Imaging, Inc.
REFERENCES
- Received for publication February 13, 2012.
- Accepted for publication May 11, 2012.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Current Landscape in Clinical Pretargeted Radioimmunoimaging and Therapy
- Pretargeted Imaging and Therapy
- {alpha}- Versus {beta}-Emitting Radionuclides for Pretargeted Radioimmunotherapy of Carcinoembryonic Antigen-Expressing Human Colon Cancer Xenografts
- Redirected T-Cell Killing of Solid Cancers Targeted with an Anti-CD3/Trop-2-Bispecific Antibody Is Enhanced in Combination with Interferon-{alpha}